

Abstract
Background
The capacity of Aedes mosquitoes to resist chemical insecticides threatens the control of major arbovirus diseases worldwide. Until alternative control tools are widely deployed, monitoring insecticide resistance levels and identifying resistance mechanisms in field mosquito populations is crucial for implementing appropriate management strategies. Metabolic resistance to pyrethroids is common in Aedes aegypti but the monitoring of the dynamics of resistant alleles is impeded by the lack of robust genomic markers.
Methodology/Principal findings
In an attempt to identify the genomic bases of metabolic resistance to deltamethrin, multiple resistant and susceptible populations originating from various continents were compared using both RNA-seq and a targeted DNA-seq approach focused on the upstream regions of detoxification genes. Multiple detoxification enzymes were over transcribed in resistant populations, frequently associated with an increase in their gene copy number. Targeted sequencing identified potential promoter variations associated with their over transcription. Non-synonymous variations affecting detoxification enzymes were also identified in resistant populations.
Conclusion /Significance
This study not only confirmed the role of gene copy number variations as a frequent cause of the over expression of detoxification enzymes associated with insecticide resistance in Aedes aegypti but also identified novel genomic resistance markers potentially associated with their cis-regulation and modifications of their protein structure conformation. As for gene transcription data, polymorphism patterns were frequently conserved within regions but differed among continents confirming the selection of different resistance factors worldwide. Overall, this study paves the way of the identification of a comprehensive set of genomic markers for monitoring the spatio-temporal dynamics of the variety of insecticide resistance mechanisms in Aedes aegypti.
Author summary
The ability of mosquitoes to resist insecticides represents a serious threat to the prevention of arbovirus diseases such as dengue, zika and chikungunya. The detection and monitoring of the resistances developed by natural mosquito populations is essential to enabling their management in the field for as long as there are no operating alternatives to the use of insecticides. However, the genetic factors for the biodegradation of insecticides by mosquito detoxification enzymes are still poorly understood prejudicing their early-detection and monitoring in natural populations. Here we used a combination of next-generation sequencing approaches to identify the genetic bases for this resistance mechanism in the mosquito Aedes aegypti. Our data confirmed that an increase in the activity of detoxification enzymes in resistant mosquitoes is often triggered by a rise in their gene copy number. We also highlighted mutations likely affecting the regulation and the protein structure of these enzymes in resistant mosquitoes. These findings provide new opportunities to develop simple molecular tests for the early-detection and monitoring of insecticide resistance in the field.
Introduction
Mosquitoes are considered by the World Health Organization (WHO) as the most dangerous animals in the world, transmitting numerous infectious diseases and causing millions of death worldwide every year. Vector-borne diseases account for 16% of the estimated global burden of communicable diseases. Among these diseases, dengue, chikungunya and zika viruses transmitted by the mosquito Aedes aegypti represent a burden in more than 100 countries with over 2.5 billion people at risk [1–3]. Although progresses are being made on vaccine development [4], several limitations remains and vector control by removing larval habitats and using insecticides still remain the first line of defense against these arbovirus diseases [5]. Chemical insecticides are widely used for vector control but the rapid adaptation of Ae. aegypti to chemical insecticides is now threatening vector control programmes [6, 7]. Resistance affects most insecticides including pyrethroids, the most recent class of insecticides used against mosquitoes [5]. Although alternative vector control tools are being developped [8–12], their global implementation will not be effective until the next decade and managing insecticide resistance represents an important health challenge in the context of the emergence and re-emergence of arbovirus diseases worldwide [13]. With this in mind, characterizing molecular mechanisms underlying insecticide resistance is crucial for tracking down insecticide resistance alleles and improving resistance management strategies [14, 15].
Resistance of mosquitoes to pyrethroid insecticides can be the consequence of various mechanisms, such as non-synonymous mutations affecting the voltage-gated sodium channel targeted by these insecticides (knock down resistance mutations, known as kdr mutations), a lower insecticide penetration, its sequestration, and its biodegradation (metabolic resistance) [16, 17]. Knock down resistance mutations and metabolic resistance are known as the two main pyrethroid resistance mechanisms in mosquitoes. While kdr mutations are well known in Ae. aegypti and can easily be tracked using PCR tools [18], molecular mechanisms underlying metabolic resistance are not yet fully understood with a limited set of robust molecular markers [16, 19]. Metabolic resistance is caused by an increased activity of detoxification enzymes such as cytochrome P450 monooxygenases (P450s or CYPs for genes), carboxyl/cholinesterases (CCEs), glutathione S-transferases (GSTs) and UDP-glycosyl-transferases (UDPGTs) although other families can be involved [17, 20, 21]. Theoretically, metabolic resistance can be the consequence of an increased expression of one or multiple detoxification enzymes capable of metabolizing the insecticide and/or the selection of variants showing a higher insecticide metabolism rate due to modifications of their protein structure [16]. As over expression is frequently associated with over transcription, most candidate genes have been identified by comparing gene expression profiles between susceptible and resistant populations with expression microarrays. These studies highlighted many candidate genes over-expressed in resistant populations, some of them being later validated as encoding enzymes capable of metabolizing pyrethroids [20, 22]. However, studies performed in different geographical areas often highlighted different candidate genes [20, 22], questionning the universality of genes associated with metabolic resistance. In addition, microarray approaches failed to identify the genomic changes causing the over-production of detoxification enzymes and particular variants associated with increased insecticide metabolism, impairing the identification of DNA markers of metabolic resistance.
Thanks to technical advances, microarrays are now being replaced by next generation sequencing approaches such as RNA-seq and DNA-seq. RNA-seq presents many advantages over microarrays in term of sensitivity and specificity of expression data and allows investigating concomitantly gene expression and polymorphism variations associated with a given phenotype across the whole transcriptome [23, 24]. To date, only few studies used RNA-seq for investigating pyrethroid resistance mechanisms in mosquitoes and most of them compared the expression profiles of a few populations collected from a single geographic area or laboratory-selected strains [25–27]. Recently, Faucon et al. [28] used deep targeted DNA sequencing for identifying genomic changes associated with pyrethroid resistant in multiple Ae. aegypti populations collected from South America and South-East Asia together with a laboratory-selected strain. This study evidences several gene copy number variations (CNVs) and point mutations affecting detoxification enzymes associated with resistance to the pyrethroid deltamethrin.
In this context, the present study aims at complementing the genomic data generated by Faucon et al. [28] by combining a RNA-seq approach with a targeted DNA-seq approach focused on regions upstream ATG in order to better understand the genomic bases of metabolic resistance to pyrethroids in Ae. aegypti. The same biological samples as those used in Faucon et al [28] were used in order to combine results obtained from both studies. The importance of CNVs as a cause of over-production of detoxification enzymes was investigated by comparing RNA-seq expression data with targeted DNA-seq data. Polymorphism of upstream regions identified by DNA-seq were cross-linked with RNA-seq expression data and upstream variations associated with the up regulation of detoxification enzymes involved in metabolic resistance were identified. Polymorphism variations identified within detoxification genes were used to identify structural variants potentially involved in increased insecticide metabolism. These results are discussed in regards to the identification of novel genomic markers of pyrethroid resistance in Ae. aegypti.
Methods
Ethics statement
Blood feeding of adult mosquitoes was performed on mice. Mice were maintained in the animal house of the federative structure Environmental and Systems Biology (BEeSy) of Grenoble-Alpes University agreed by the French Ministry of animal welfare (agreement n° B 38 421 10 001) and used in accordance to European Union laws (directive 2010/63/UE). The use of animals for this study was approved by the ethic committee ComEth Grenoble-C2EA-12 mandated by the French Ministry of higher Education and Research (MENESR).
Mosquito populations
Eight Ae. aegypti populations of distinct geographical origins were used in the study. These populations were identical (same generation and egg batches) as those used in Faucon et al. 2015 [28] in order to cross link genomic data previously obtained with transcriptomic data generated by the current study. These included three laboratory strains fully susceptible to all insecticides (S populations) and five populations showing elevated resistance to the pyrethroid insecticide deltamethrin (R populations). Field resistant populations were collected in Thailand (NakhR and PhetR) and French Guiana (CaynR and StGeR) during 2014–2015 and maintained in insectaries for 2 generations. In addition to field resistant populations, one laboratory strain selected with deltamethrin for five generations at the adult stage was included in the study (DeltaR population selected from the ‘Bora-Bora’ strain, 80% mortality at each generation). Susceptible strains included the ‘Liverpool’ strain (LivpS) initially colonized from Africa and used for Ae. aegypti genome sequencing, the ‘Bora-Bora’ strain (BoraS) initially colonized from French Polynesia and the ‘New-Orleans’ strain (NwOrS) initially collected from southern USA.
Previous work showed that the populations used in the current study display a broad range of resistance to deltamethrin with the lethal dose killing 50% of individuals (LD50) after one hour exposure varying up to 750 fold between susceptible strains and the most resistant populations from South-America (Table 1 and [28]). Resistant populations from Thailand showed an intermediate resistance level (~ 250 fold) while the laboratory-selected strain DeltaR showed a moderate resistance level (~ 5 fold). The time necessary to knock down 50% of individuals (KDT50) varied from 11 min for susceptible populations to more than 8 hours for the most resistant ones. As described in Poupardin et al. 2014 [29], the NakhR population also showed a significant resistance to the organophosphate temephos. Previous genotyping of kdr mutations revealed that resistant populations from South-America carried the V1016I and F1534C mutations at high frequencies (V1016I from 77 to 96% and F1534C from 75 to 100%) while the S989P mutation was not detected. Asian populations carried the S989P, 1016G and F1534C mutations at medium to high frequencies (14 to 68%; 21 to 58% and 22 to 75% respectively) [28].
Table 1. Deltamethrin resistance levels.
| Population | Origin | Type | Resistance | RR50 a | KDT50 b |
|---|---|---|---|---|---|
| LivpS | Benin | Lab strain | susceptible | 1 | 11.5 |
| BoraS | French Polynesia (Bora-Bora) | Lab strain | susceptible | 1 | 12.1 |
| DeltaR | French Polynesia (Bora-Bora) | Lab strain | slightly resistant | ~5 | 14.2 |
| PhetR | Thailand (Phetchaburi) | Field | resistant | ~250 | 98.2 |
| NakhRc | Thailand (Nakonsawan) | Field | resistant | ~250 | 92.9 |
| NwOrS | USA (New Orleans) | Lab strain | susceptible | 1 | 11.9 |
| CaynR | French Guiana, Cayenne) | Field | highly resistant | ~750 | >500 |
| StGeR | French Guiana, St-Georges) | Field | highly resistant | ~750 | >500 |
a: Resistance ratios (RR50) were estimated based on the dose of insecticide used for paper impregnation leading to 50% mortality (±10%) after 1h exposure.
b: KDT50: Time (min) necessary to knock down 50% of individuals using papers impregnated with 0.05 g/100 mL deltamethrin following WHO insecticide testing recommendations.
C: population showing significant cross-resistance to organophosphates. Data extracted from [28].
Samples preparation
All mosquitoes used in this study consisted in 2–4 days-old non-blood fed Ae. aegypti females grown in standard laboratory conditions as described in [28]. For RNA sequencing, three pools of 30 females from each populations not exposed to insecticide were collected and stored in RNAlater (Life technologies). RNA extractions were performed on each pool separately using the RNAqueous-4PCR kit (Life technologies) according to manufacturer’s instructions and followed by a DNase treatment to remove genomic DNA contaminants. For the deep targeted sequencing of promoter regions, mosquitoes were collected as described in Faucon et al. 2015 [28] and genomic DNA was extracted from two pools of 65 individuals using the PureGene Core Kit A (Qiagen). The two DNA extracts were then mixed in equal proportion in order to be representative of 130 individuals.
RNA sequencing
Three RNA-seq libraries per population (three biological replicates) were prepared using the TruSeq Stranded mRNA sample Prep kit (Illumina) and sequenced on an Illumina GAIIx sequencer as 75 bp paired reads by Hybrigenics-Helixio (Clermont-Ferrand, France). Sequenced reads were assigned to each sample (unplexing) and adaptors were removed. Reads quality was checked for each sample using FastQC. Reads were then filtered based on their length, pairing and quality using Trimmomatic [30] with parameters set as follows: Leading 25; Trailing 25; Minlen 60; Slidingwindow 4–25. Only paired reads were kept. Reads were then mapped to Ae. aegypti genome (AaegL3 assembly) using Tophat2 [31] with the following parameters: don’t report discordant pair alignments; final read mismatches = 3; intron length = 45–300000; use coverage search. Only read pairs mapping at a unique location (mapQ > 50) were retained. Quantification of transcription levels was performed using the Cuffdiff2 module of Cufflinks implemented into a Galaxy pipeline (http://galaxyproject.org) based on Fragment Per Kilobase exon Model (FPKM) values obtained for each gene across all samples.
Transcription ratios between each resistant and each susceptible strain were computed across all biological replicates using Cuff-diff. Genes showing an FC ≥ 3 (in either direction) and a q value ≤ 0.001 between a given resistant population and all three susceptible strains were considered differentially expressed (DE genes).
Polymorphism analysis was performed with Strand NGS V2.2 (Strand Life Sciences). SNPs were called against the reference genome with the following parameters: ignore spill overs at locations around homopolymer stretch greater than 10; ignore reference locations with homopolymer stretch greater than 10; ignore reference locations with coverage below 10; ignore reference locations with variants below 2; confidence score cutoff 50; error rate 0.0010. For each SNP, allele frequencies were estimated based on the number of read supporting each allele. Only substitutions being polymorphic across populations were retained (i.e. showing > 5% allele frequency variation). SNPs showing an allele frequency variation > 40% (in either direction) between any resistant population and all three susceptible strains were considered associated with resistance (Diff SNPs). Genic effects were computed based on reference genome annotation and non-synonymous differential SNPs were identified (NS Diff SNPs). Then the potential effect of NS Diff SNPs on detoxification enzyme binding sites was investigated. For P450s, variations were considered as potentially impacting substrate specificity if located within one of the six substrate recognition sites [32]. For CCEs, variations were considered as potentially impacting substrate specificity if located within 5 bp of amino-acids forming the active site after alignment with the LcαE7 esterase [33]. For GSTs, variations were considered as potentially impacting substrate specificity if located near any amino-acid forming the G-site and the H-site after alignment with agGSTD11 and agGSTD6 [34]. For UGTs, variations were considered as potentially impacting substrate specificity if located near any amino-acid forming the DBR1 and DBR2 domains after alignment with UDPGT1-10 [35].
Cross analysis of RNA-seq and DNA-seq data
RNA-seq data obtained from detoxification genes were cross-compared with targeted DNA-seq data previously generated for all detoxification genes from the same biological samples [28] together with novel targeted DNA-seq data covering up to 1kb upstream ATG of all detoxification genes.
First, expression data obtained from RNA-seq were cross-compared with exome sequencing data previously obtained by DNA-seq in order to confirm the role of copy number variations (CNVs) in the over-expression of detoxification enzymes associated with resistance. Log2 expression ratios and Log2 CNV ratios were correlated across all resistant populations and genes showing a significant positive correlation (Pearson’s r ≥ 0.7 and p ≤ 0.05) were considered up-regulated as a consequence of CNV through gene dosage effect [36].
Because CNV did not fully explain the over-expression of all detoxification genes associated with resistance, their upstream regions were sequenced in order to identify promoter variations potentially associated with their up-regulation. Sequencing was performed by deep targeted sequencing on genomic DNA extracted from 130 mosquitoes as described in [28] except that target genomic regions consisted in 1 Kb regions upstream ATG of all detoxification genes (S1 Table). Target regions were captured using the Agilent SureSelect target enrichment system and sequenced as 75 bp paired reads by Hybrigenics-Helixio (Clermont-Ferrand, France) on an illumina sequencing platform. Sequenced reads were filtered based on their length, pairing and quality using Trimmomatic [30] and mapped to Ae. aegypti genome (Aaegl3 assembly) using BWA algorithm [37] as previously described in [28]. SNPs were called against reference genome with a minimum coverage of 30 reads per locus and allele frequencies were computed for each population based on the number of read supporting each allele. Allele frequencies of SNPs located upstream detoxification genes over-transcribed in resistant populations were then correlated to their respective transcription levels (as FPKM or Log2 FPKM) across all populations. Variations upstream ATG showing a significant correlation (Pearson’s r ≥ 0.7 or ≤-0.7 and p ≤ 0.05) with the transcription level of their respective genes were identified.
Then, the effect of these upstream variations was examined in silico by comparing the occurrence of transcription factor binding sites (TFBS) at each locus between the reference and the variant alleles. For each gene, all possible variant haplotypes were compared with the reference haplotype. Transcription factor binding sites were screened with Promo V3.0 (http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3) using Transfac V8.3 database restricted to animal and factors of at least six bp. DNA patterns showing ≤ 5% matrix dissimilarity with known TFBS were retained and compared between reference and variant haplotypes. Differential TFBS were then manually curated and those likely having a major impact on gene regulation were identified based on i) their differential occurrence between reference and variant alleles, ii) their known enhancer effect on detoxification genes [38–42], iii) their expected effect in regards of the positive/negative correlation between the frequency of the variant allele and transcription level and iv) their position relative to core promoter elements and the putative transcription starting site. Putative transcription starting sites were manually identified based on the presence of core promoter elements previously identified in arthropods [42, 43].
Results
Sequencing and mapping metrics
RNA-seq analysis generated more than 928 million reads with an average of 116 million reads per population across the three biological replicates (Table 2). More than 92% of sequenced reads were of high quality (see methods). Further filtering reads based on their pairing status (only paired reads retained) and their mapping quality (map Q>50, ≤ 3 mismatch, unique mapping location) allowed retaining an average of 65.5 million reads per population (56.5%) for subsequent analyses.
Table 2. Sequencing and mapping statistics.
| Sequenced readsa | BoraS | LivpS | NwOS | DeltaR | NakhR | PhetR | CaynR | StGeR | % | |
|---|---|---|---|---|---|---|---|---|---|---|
| Sequenced | ||||||||||
| Total | 121.86 | 142.31 | 83.61 | 98.66 | 127.41 | 114.69 | 120.58 | 119.27 | 100 | |
| Mean per replicate | 40.62 | 47.44 | 27.87 | 32.89 | 42.47 | 38.23 | 40.19 | 39.76 | ||
| SE | 1.16 | 4.20 | 3.45 | 2.72 | 1.57 | 3.54 | 3.75 | 4.70 | ||
| Passing QC filtering | ||||||||||
| Total | 111.85 | 131.22 | 76.99 | 91.23 | 116.84 | 105.49 | 111.19 | 110.18 | 92.09 | |
| Mean per replicate | 37.28 | 43.74 | 25.66 | 30.41 | 38.95 | 35.16 | 37.06 | 36.73 | ||
| SE | 0.51 | 3.44 | 2.88 | 2.16 | 1.86 | 3.16 | 3.60 | 4.51 | ||
| Mapped | ||||||||||
| Total | 66.17 | 82.18 | 46.99 | 54.11 | 71.19 | 64.81 | 69.38 | 69.60 | 56.49 | |
| Mean per replicate | 22.06 | 27.39 | 15.66 | 18.04 | 23.73 | 21.60 | 23.13 | 23.20 | ||
| SE | 0.63 | 1.86 | 1.48 | 1.28 | 1.96 | 1.74 | 2.45 | 3.19 | ||
a: in million paired reads.
Differentially transcribed genes associated with deltamethrin resistance
Differential transcription analysis was performed on the 15491 transcripts showing a transcription level > 0.5 FPKM in each condition pair across all replicates; or > 1.0 FPKM across the two conditions in order to retain genes being silent in one condition but showing a significant transcription level in the other condition. Among these, 7,614 were annotated and manually associated to 15 biological categories (Fig 1 and S2 Table). A total of 118 annotated genes were found differentially transcribed (absolute FC > 3 and q-value < 0.001) in at least one resistant population as compared to all susceptible strains (DE genes). DE genes included 48 genes over-transcribed and 68 genes under-transcribed in at least one resistant population (Fig 1). Comparing the frequency of each biological category between DE genes and all detected genes revealed a strong enrichment of detoxification enzymes belonging to cytochrome P450s (P450s) and carboxyl/cholinesterases (CCEs) among over-transcribed genes. No such enrichment was detected among under-transcribed genes.
Fig 1. Gene families affected by DE genes in resistant populations.

Genes showing a fold change ≥ 3 in either directions, and q-value ≤ 0.001 in any resistant population as compared to all susceptible strains were considered differentially expressed. Biological categories mostly affected were identified by comparing their proportion between all genes detected by RNA-seq and those being differentially expressed using a one-sided Fisher’s exact test (*** p ≤ 0.001).
Comparing the transcription profiles of the 20 detoxification genes over-transcribed in resistant populations revealed their frequent conservation within regions while more divergence was observed between continents (Fig 2). Fourteen P450s were found over-transcribed in resistant populations, most of them belonging to the CYP6 and CYP9 families. Several CYP9Js located on chromosome 3 (supercontigs 1.1188 and 1.221) were over-transcribed in Asian populations with some of them also over-transcribed in South-America. Among CYP6s, the CYP6CB1-like AAEL009018 was over-expressed in all resistant populations while CYP6BB2 and the CYP6P12-like AAEL014891 were specifically over-transcribed in South-America. Finally, CYP4H28 appeared specifically over-transcribed in Asian population while CYP6AG7 was specific to the PhetR population. Two P450s slightly over-transcribed in the laboratory-selected strain DeltaR (CYP9J8 and CYP6CB1-like AAEL009018) were also found significantly over-transcribed in field resistant populations. Two epsilon GSTs (GSTe5 and GSTe7) were specifically over-transcribed in Asia while four CCEs located on chromosome 2 (CCEae3A, CCEae4A, CCEae6A and AAEL005123) were specifically over-transcribed in the Asian NakhR population showing cross resistance to temephos [29].
Fig 2. Transcription profiles of detoxification enzymes associated with deltamethrin resistance.

Color scale shows the mean Log2 Fold Change between resistant populations and all susceptible strains. Stars indicate a fold change q value ≤ 0.001. Black ‘+’ marks indicate a significant positively correlation between Log2 transcription ratios and Log2 CNVs (Pearson’s r ≥ 0.7 and p ≤ 0.05). Grey ‘+’ marks indicate genes belonging to genomic clusters displaying CNV associated with deltamethrin resistance as reported in [28].
Genomic variations associated with the over transcription of detoxification genes
Comparing the transcription profile of detoxification genes over-transcribed in resistant populations with their copy number variations (CNVs) previously inferred from targeted DNA-seq revealed that most detoxification genes differentially expressed in resistant populations belong to genomic clusters affected by CNV variations associated with resistance [28]. Among them, seven showed a significant positive correlation between their transcription level and their CNV (Fig 2 and S1 Fig). These genes include three CCEs (CCEae3A, CCEae4A and CCEae6A), two CYP9Js (CYP9J21-like AAEL014612 and CYP9J22-like AAEL014619) and two CYP6s (CYP6BB2 and CYP6P12-like AAEL014891). Except CYP4H28 and CYP6AG7, all over-transcribed detoxification genes, were located within gene clusters affected by CNV associated with insecticide resistance as described in Faucon et al. 2015 [28].
The sequencing of 1 Kb upstream regions of all detoxification genes using targeted DNA-seq identified 188 variations located upstream detoxification genes over-transcribed in resistant populations (S3 Table). Among them, 23 variations showed a significant correlation between their allele frequency and the transcription level of the downstream gene (r ≥ 0.7 and p ≤ 0.05). These 23 variations were located upstream of two GSTs and six P450s (S2 Fig). All of them, except those detected upstream CYP9J26, were negatively correlated with transcription levels meaning that the increased frequency of the allele from the reference genome was correlated to the over-transcription of the downstream gene and reciprocally. Seven variations were potentially favoring the binding of transcription factors previously identified as enhancers of detoxification genes in resistant populations (Fig 3 and S4 Table). These included two variations likely to have a major effect on the binding of enhancers: the variation at position -642 (respective to ATG) affecting the gene CYP6P12-like AAEL014891 favoring the binding of the HNF-1A element and the variation at position -410 affecting the gene CYP6CB1-like AAEL009018 favoring the binding of the HNF-3 element.
Fig 3. Promoter elements potentially associated with the up regulation of detoxification genes in resistant populations.

Only variations correlated with transcription level and favouring the binding of potential enhancer elements are shown. Potential initiation sites (Inr) and TATA boxes are indicated. For each gene, variations detected in the upstream region are shown as empty and filled dots depending on the significance of the correlation between their allele frequency and gene transcription level.
Non-synonymous variations associated with deltamethrin resistance
High quality RNA-seq reads allowed calling a total of 749,596 substitutions against the reference genome, with an average of 300,000 substitutions per population (Table 3). Among them, 244,211 (32.58%) were considered polymorphic (allele frequencies variation > 5% across all populations). Among them, 12,087 were considered associated with resistance (Diff SNPs) as they showed an allele frequency variation > 40% between at least one resistant population and all susceptible strains together with a variation occurring in the same direction in populations from the same region (S5 Table). Nearly half of them (5,857) were located within annotated genes with 625 being non-synonymous (NS Diff SNPs) affecting 436 annotated genes. Comparing the frequency of biological categories between genes where SNPs were called and those affected by Diff SNPs and NS Diff SNPs revealed that biological categories associated with insecticide resistance were significantly more affected (Fig 4). Diff SNPs were significantly enriched among detoxification enzymes such as P450s (2.5 fold), CCEs (9.5 fold) and UGTs (1.9 fold). Although less significant, a slight enrichment was also detected in genes related to hormone signaling and neurotransmitters (1.3 fold). When focusing on NS Diff SNPs, only P450s, CCEs and UGTs remained significantly enriched. Among the transcripts affected by NS Diff SNPs, 41 encode detoxification enzymes, ABC transporters, cuticle proteins and proteins involved in nervous system functioning (S5 Table). Comparing allele frequencies estimated from RNA-seq and from DNA-seq across all detoxification genes revealed a good correlation between both techniques (Pearson’s r > 0.9, S3 Fig).
Table 3. Overview of SNP analysis.
| BoraS | LivpS | NwOrS | DeltaR | NakhR | PhetR | CaynR | StGeR | Total | % | |
|---|---|---|---|---|---|---|---|---|---|---|
| Total called | 393,506 | 393,187 | 189,369 | 317,695 | 292,280 | 328,594 | 346,445 | 294,774 | 749,596 | 100 |
| Polymorphic substitutions | 162,192 | 148,298 | 89,652 | 142,655 | 108,977 | 123,696 | 133,867 | 127,432 | 244,211 | 32.58 |
| Diff SNPsa | - | - | - | 2,430 | 6,024 | 1,348 | 2,165 | 2,049 | 12,087 | 1.61 |
| NS Diff SNPsb | - | - | - | 218 | 576 | 123 | 209 | 187 | 1,146 | 0.15 |
a: Differential SNPs.
b: Non-synonymous differential SNPs.
Fig 4. Gene families affected by polymorphism variations in resistant populations.
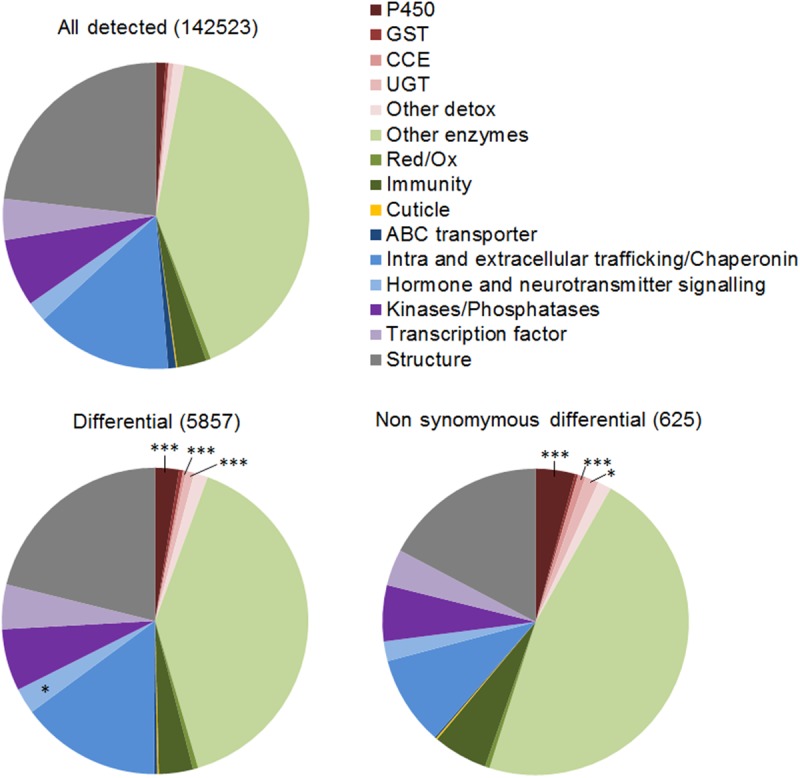
The frequency of each gene family was compared between all detected polymorphic SNPs (top) and both differential SNPs (Diff SNPs) and non-synonymous differential SNPs (NS Diff SNPs) using a one-sided Fisher’s exact test (*p ≤ 0.05,*** p ≤ 0.001).
Comparing the frequency of NS Diff SNPs affecting detoxification enzymes across populations (Fig 5) revealed that variations associated with deltamethrin resistance were frequently conserved within populations sharing the same genetic background but differed among continents. These NS Diff SNPs impacted fourteen P450s mainly of the CYP6 and CYP9 families together with three CCEs, two GSTs and four UGTs. Among these genes, ten were previously identified as impacted by non-synonymous variations associated with deltamethrin resistance based on targeted DNA-seq [28]. These included four P450s (CYP12F7, CYP6M10, CYP9J26 and CYP325V1), three CCEs (CCEAE4A, CCEAE3A and AAEL010592), one GST (GSTE6) and two UGTs (AAEL000559 and AAEL010366). Among all NS Diff SNPs affecting detoxification enzymes, none were located in the potential substrate binding sites of CCEs, GSTs and UGTs while seven were located in P450 substrate recognition sites (SRS). Most of them were detected in the laboratory-selected strain including CYP12F7 T501S in SRS6, CYP6M6 T210A in SRS2 and L239F in SRS3 and CYP6M10 Q414H/K in SRS6 while two were detected in the Asian PheR resistant population (CYP6M11 F478L in SRS6 and CYP9J9 I520M in SRS6).
Fig 5. Non-synonymous differential SNPs affecting detoxification genes.

Color scale shows the mean allele frequency variation between each resistant strain and all susceptible strains. Yellow/blue colors indicate an increase/decrease frequency of the variant allele in the resistant population respectively. Chromosome, supercontig, base position, nucleotide variation (ref>var), strand, position in cDNA, position in protein, amino-acid change (ref>var), gene accession and gene name are indicated. Stars indicate genes previously affected by NS Diff SNPs as reported in [28]. Boxed amino acid changes indicate variations likely affecting substrate binding site.
Discussion
Detoxification enzymes over expressed in resistant populations
By comparing susceptible and resistant Ae. aegypti populations from various continents, the present study confirmed the importance of detoxification enzymes in pyrethroid resistance in this species. Indeed, although resistant populations are positive for kdr mutations known to confer pyrethroid resistance [44, 45], their transcriptome profiling clearly shows an over-transcription of multiple genes encoding detoxification enzymes previously associated with insecticide resistance [19, 20, 22]. This includes P450s of the CYP9J subfamily from which several members were shown to contribute to deltamethrin metabolism [46]. The gene CYP6BB2, strongly over transcribed in South-American populations, was also shown to encode a P450 capable of degrading deltamethrin [45]. Orthologs of CYP6P12-like AAEL014891 in Anopheles mosquitoes (i.e. CYP6P3, CYP6P7, CYP6P9 and CYP6M2) were also shown as encoding enzymes capable of degrading pyrethroid insecticides [47–50]. Recent data obtained by Ishak et al. [51] indicated that the P450 encoded by Aedes albopictus CYP6P12 degrades pyrethroids. Although the role of CYP6CB1 in pyrethroid resistance has not yet been functionally validated, this P450 showed an over transcription in multiple resistant populations and was frequently associated with pyrethroid resistance in Ae. aegypti [20, 52, 53]. Four CCEs were strongly over-transcribed in the Asian resistant population NakhR. However, the over expression of these CCEs is unlikely conferring pyrethroid resistance as this population shows a strong cross resistance to the organophosphate temephos [29] and CCEAE3A has been validated as able to degrade and sequester temephos in both Ae. aegypti and Ae. albopictus [54, 55]. Finally, the genes GSTE5 and GSTE7 were specifically over-transcribed in Asian resistant populations. The over-expression of epsilon GSTs has frequently been associated with insecticide resistance in mosquitoes [56]. The genes GSTE5 and GSTE7 are frequently found over transcribed in insecticide resistant populations and GSTE5 exhibits a DDT dehydrochlorinase activity while the partial silencing of GSTE7 was associated to an increased susceptibility to deltamethrin [57]. Recent results from Riveron et al. [58] suggest that a specific variant of Anopheles funestus GSTE2 is involved in permethrin metabolism, supporting the role of epsilon GSTs in pyrethroid resistance. Overall, the present study confirmed the role of particular detoxification enzymes in pyrethroid resistance and identified novel candidates deserving further investigations.
Genomic variations underlying the over expression of detoxification enzymes
Comparing transcription profiles obtained by RNA-seq and CNV profiles obtained by DNA-seq on the same populations confirmed the importance of CNV as a genomic cause of the over-expression of detoxification enzymes in Ae. aegypti. The importance of CNV in this species is not surprising as Ae. aegypti genome is highly infected by transposable elements favouring gene duplication events [59]. Such frequent duplication events affecting clusters of detoxification genes may then rapidly be selected in natural populations by the strong selection pressure caused by chemical insecticides [36].
However, the transcription profiles of detoxification enzymes were not systematically associated with CNV suggesting that the selection of cis- and/or trans-regulatory elements also contributes to the over transcription of particular detoxification genes in resistant populations. The deep sequencing of proximal upstream regions of all detoxification genes using targeted DNA-seq identified 188 variations over transcribed in resistant populations. Among them, only seven variations had their allele frequency correlated with gene transcription levels and were favouring the binding of enhancer elements previously associated with the regulation of detoxification enzymes [38–42]. In insects, mutations in the promoters of insecticide-metabolizing P450s have been associated with their up regulation in insecticide-resistant populations but nuclear factors involved in such process have rarely been characterized [review in 60]. Using luciferase reporter assays, Itokawa et al. [61] recently demonstrated that a mutation affecting the promoter of the duplicated CYP9M10 further enhances its expression and confers high resistance to permethrin in Culex quinquefasciatus. Although our sequencing approach may not cover the full promoter regions of all detoxification genes, it allowed identifying variations upstream two CYP genes that may control their up regulation in pyrethroid-resistant populations. Among them, the -410 T>A variation affecting CYP6CB1-like AAEL009018, considerably favour the binding of an HNF-3 element. This variation appears particularly interesting as the over-expression of this gene in resistant populations does not appear under the control of CNV and Hepatocyte Nuclear Factors (HNFs) have frequently been associated with the regulation of drug-metabolizing P450s [41, 62, 63]. Secondly, although the transcription level of CYP6P12-like AAEL014891 is correlated with CNV, the T>A variation at position -642 appears interesting as it considerably favours the binding of an HNF-1A element.
Point mutations associated with metabolic resistance to deltamethrin
Multiple non-synonymous variations were strongly enriched in resistant populations as compared to susceptible strains. Although a relatively small proportion of them affected detoxification enzymes, statistical analyses revealed that detoxification enzymes such as P450s, CCES and UGTs were significantly more affected than other biological categories confirming the link between detoxome polymorphism and insecticide resistance. As for gene transcription data, polymorphism patterns were frequently conserved within regions but differed among continents confirming the selection of different adaptive trajectories worldwide. Although most variations affecting detoxification enzymes are probably not functionally associated with resistance but rather the consequence of the strong selection pressure undertaken by detoxification genes, some of them may reflect the selection of particular variants showing an increased metabolic activity against insecticides. Such variation has been recently discovered in An. funestus where a point mutation in the gene GSTE2 was associated with increased DDT metabolism [58]. Similarly, a conserved mutation affecting the capacity of the esterase αE7 to degrade organophosphates has been identified in various insects [64, 65]. Interestingly other point mutations allowed this esterase to metabolise pyrethroids such as deltamethrin, confirming that the selection of different enzyme variants can confer resistance to multiple insecticides [66–68]. In our study, seven non-synonymous variations were located within the Substrate Recognition Site (SRS) of P450s and may affect the binding and/or the metabolism of insecticides [69]. Five were detected in the laboratory-selected strain DeltaR while only two were detected in field resistant populations and none were shared between populations showing different genetic backgrounds. Whether these mutations are only reflecting the selection imprint affecting these P450s, or are functionally involved in metabolic resistance can’t be ruled out yet. However, if their role in resistance is confirmed, such variability may result from the selection of different adaptive trajectories according to different genetic background and local selection pressures. In such situation, the presence of such mutations in the laboratory-selected strain DeltaR after only a few generations of selection might indicate that, in the absence of major resistance factors, the purification of P450 variants showing an increased insecticide metabolism can occur in the early steps of the selection process. Overall, although some point mutations identified in this study may represent false positives or may result from selection imprints, this data set represent a good basis for investigating the role of the purification of detoxification enzyme variants in insecticide resistance in mosquitoes.
Novel genomic markers for tracking down metabolic resistance in mosquitoes
Managing insecticide resistance in mosquito vectors represent a significant challenge until efficient alternative control strategies and vaccines will be implemented at a global scale. Such management includes the early detection and tracking of resistance alleles in order to implement adequate resistance breaking strategies in a timely manner.
In vectors of arbovirus diseases, the detection of target-site mutations can be easily achieved through simple PCR assays on individual mosquitoes and allows monitoring the spatio-temporal dynamics of resistant alleles within and among populations. However, such fine monitoring is not yet achievable for metabolic resistance alleles because most validated metabolic resistance markers consists of RNA markers. Indeed, RNA markers are submitted to difficulties and flawless inherent to RNA analysis (sample storage and degradation, high variability, limited biological material, high costs …) impairing the production of individual genotyping data at a high throughput. Recently, Faucon et al. [28] identified particular CNVs as good markers of the over expression of detoxification genes in pyrethroid-resistant Ae. aegypti populations. However, identifying additional genomic marker associated with the regulation of detoxification enzymes and the selection of particular variants will allow tracking the full spectra of metabolic resistance alleles in the field.
Although the present study identified novel putative genomic markers of deltamethrin resistance, their functional validation is still required which represents a significant research effort. Indeed, though high-throughput sequencing strategies allow studying the molecular bases of insecticide resistance with an unprecedented level of details, improving experimental designs is necessary for narrowing the number of candidate markers to be functionally validated. In addition, reverse genetic approaches such as TALEN and CRISPR/Cas9 appear highly efficient against mosquito detoxification genes [70] and their improvement may greatly accelerate the functional validation of resistance markers in the next few years. In this context, the development of novel molecular resistance diagnostic tools allowing the tracking of the variety of insecticide resistance mechanisms becomes at reach. However, such development will require combining the skills and knowledge of a broad research community through large research networks such as the Worldwide Insecticide resistance Network (WIN) recently developed for vectors of abrovirus diseases [15].
Supporting information
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Correlations were tested for each overexpressed detoxification gene by comparing Log2 CNV versus Log2 expression ratios. Correlations with Pearson’s r ≥ 0.7 and p ≤ 0.05 were considered significant. CNV data were extracted from [28].
(TIF)
Only variations located within a 1 kb upstream of detoxification genes overexpressed in resistant populations were considered. Among them, variations showing a significant correlation (Pearson’s r ≥ 0.7 and p ≤ 0.05) between their allele frequency and gene expression level (FPKM or Log2 FPKM) across all populations were considered significantly associated with gene expression. For each variation, position on supercontig, nucleotide change (ref>var) and position relative to coding sequence (brackets) are indicated.
(TIF)
Only SNPs affecting detoxification genes and detected by both approaches are shown.
(TIF)
Acknowledgments
The authors thank Dr. F. Chandre from the Institut de Recherche pour le Développement of France for supporting the French-Thai partnership and helpful discussions. The authors also thank Dr. P. Sirisopa from Kasetsart University Thailand and Dr. K. Thanispong from the Bureau of Vector Borne Diseases of the Ministry of Health of Thailand for their support regarding mosquito collections.
Data Availability
The RNA-seq sequence data from this study have been deposited at the European Nucleotide Archive (ENA; http://www.ebi.ac.uk/ena) under the accession number PRJEB15531. DNA-seq data from this study have been deposited at the European Nucleotide Archive under accession number PRJEB7976.
Funding Statement
This project was primarily funded by the French Institut de Microbiologie et Maladies Infectieuses (grant IMMI 109764). FF was supported by a PhD fellowship from the Grenoble-Alpes University (UGA). We acknowledge support from the federative structure Environmental and Systems Biology (BEeSy) of Grenoble-Alpes University. This work was also supported by the French-Thai cooperation programme-PHC Siam project (RESA 2013–2014) funded by the French embassy and the Office of the Higher Education Commission of Thailand and the Center for Advanced Studies for Agriculture and Food, Institute for Advanced Studies, Kasetsart University. WJ was supported by the Thailand Research Fund (senior research scholarship RTA 5558002 and grant for new researchers MRG 5380102) and Kasetsart University Research and Development Institute (KURDI). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.WHO. Dengue and dengue hemorrhagic fever. Fact Sheet N°117, WHO Fact sheet, updated February 2015, available from http://www.who.int/mediacentre/factsheets/fs117/en/.
- 2.WHO. Chikungunya. Fact Sheet N°327, WHO Fact sheet, updated February 2015, available from http://wwwwhoint/mediacentre/factsheets/fs327/en/.
- 3.WHO. Zika Virus Fact Sheet. WHO Fact sheet, updated march 2016, available from http://www.who.int/mediacentre/factsheets/zika/en/.
- 4.Vannice KS, Durbin A, Hombach J. Status of vaccine research and development of vaccines for dengue. Vaccine. 2016;34(26): 2934–2938. doi: 10.1016/j.vaccine.2015.12.073 [DOI] [PubMed] [Google Scholar]
- 5.Ranson H, Burhani J, Lumjuan N, Black WC. Insecticide resistance in dengue vectors. TropIKAnet. 2010;1(1): 1–12. [Google Scholar]
- 6.Dusfour I, Thalmensy V, Gaborit P, Issaly J, Carinci R, Girod R. Multiple insecticide resistance in Aedes aegypti (Diptera: Culicidae) populations compromises the effectiveness of dengue vector control in French Guiana. Mem I Oswaldo Cruz. 2011;106(3): 346–352. [DOI] [PubMed] [Google Scholar]
- 7.Marcombe S, Darriet F, Tolosa M, Agnew P, Duchon S, Etienne M, et al. Pyrethroid resistance reduces the efficacy of space sprays for dengue control on the island of Martinique (Caribbean). PLoS Neg Trop D. 2011;5(6):e1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scholte EJ, Knols BG, Samson RA, Takken W. Entomopathogenic fungi for mosquito control: a review. J Insect Sci. 2004;4: 19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lacey LA. Bacillus thuringiensis serovariety israelensis and Bacillus sphaericus for mosquito control. J Am Mosquito Contr. 2007;23(2 Suppl): 133–163. [DOI] [PubMed] [Google Scholar]
- 10.Hoffmann AA, Montgomery BL, Popovici J, Iturbe-Ormaetxe I, Johnson PH, Muzzi F, et al. Successful establishment of Wolbachia in Aedes populations to suppress dengue transmission. Nature. 2011;476(7361): 454–457. doi: 10.1038/nature10356 [DOI] [PubMed] [Google Scholar]
- 11.Walker T, Johnson PH, Moreira LA, Iturbe-Ormaetxe I, Frentiu FD, McMeniman CJ, et al. The wMel Wolbachia strain blocks dengue and invades caged Aedes aegypti populations. Nature. 2011;476(7361): 450–453. doi: 10.1038/nature10355 [DOI] [PubMed] [Google Scholar]
- 12.Harris AF, McKemey AR, Nimmo D, Curtis Z, Black I, Morgan SA, et al. Successful suppression of a field mosquito population by sustained release of engineered male mosquitoes. Nat Biotechnol. 2012;30(9): 828–830. doi: 10.1038/nbt.2350 [DOI] [PubMed] [Google Scholar]
- 13.Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, et al. The global distribution and burden of dengue. Nature. 2013;496(7446): 504–507. doi: 10.1038/nature12060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Corbel V, Nosten F, Thanispong K, Luxemburger C, Kongmee M, Chareonviriyaphap T. Challenges and prospects for dengue and malaria control in Thailand, Southeast Asia. Trends Parasitol. 2013;29(12): 623–633. doi: 10.1016/j.pt.2013.09.007 [DOI] [PubMed] [Google Scholar]
- 15.Corbel V, Achee NL, Chandre F, Coulibaly M, Dusfour I, Fonseca D, et al. Tracking insecticide resistance in mosquito vectors of arboviruses: The Worldwide Insecticide resistance Network (WIN). PLoS Neg Trop D. 2016; Forthcoming. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li XC, Schuler MA, Berenbaum MR. Molecular mechanisms of metabolic resistance to synthetic and natural xenobiotics. Annu Rev Entomol. 2007;52: 231–253. doi: 10.1146/annurev.ento.51.110104.151104 [DOI] [PubMed] [Google Scholar]
- 17.Hemingway J, Hawkes NJ, McCarroll L, Ranson H. The molecular basis of insecticide resistance in mosquitoes. Insect Biochem Mol Biol. 2004;34(7): 653–665. doi: 10.1016/j.ibmb.2004.03.018 [DOI] [PubMed] [Google Scholar]
- 18.Black WC, Vontas JG. Affordable assays for genotyping single nucleotide polymorphisms in insects. Insect Mol Biol. 2007;16(4): 377–387. doi: 10.1111/j.1365-2583.2007.00736.x [DOI] [PubMed] [Google Scholar]
- 19.Nkya TE, Akhouayri I, Kisinza W, David JP. Impact of environment on mosquito response to pyrethroid insecticides: facts, evidences and prospects. Insect Biochem Mol Biol. 2013;43(4): 407–416. doi: 10.1016/j.ibmb.2012.10.006 [DOI] [PubMed] [Google Scholar]
- 20.David JP, Ismail HM, Chandor-Proust A, Paine MJ. Role of cytochrome P450s in insecticide resistance: impact on the control of mosquito-borne diseases and use of insecticides on Earth. Philos T Roy Soc B. 2013;368(1612): 20120429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.David JP, Faucon F, Chandor-Proust A, Poupardin R, Riaz MA, Bonin A, et al. Comparative analysis of response to selection with three insecticides in the dengue mosquito Aedes aegypti using mRNA sequencing. BMC Genomics. 2014;15: 174 doi: 10.1186/1471-2164-15-174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vontas J, Kioulos E, Pavlidi N, Morou E, Torre AD, Ranson H. Insecticide resistance in the major dengue vectors Aedes albopictus and Aedes aegypti. Pestic Biochem Physiol. 2012;104(2): 126–131. [Google Scholar]
- 23.Wilhelm BT, Landry JR. RNA-Seq-quantitative measurement of expression through massively parallel RNA-sequencing. Methods. 2009;48(3): 249–257. doi: 10.1016/j.ymeth.2009.03.016 [DOI] [PubMed] [Google Scholar]
- 24.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7): 621–628. doi: 10.1038/nmeth.1226 [DOI] [PubMed] [Google Scholar]
- 25.Lv Y, Wang W, Hong S, Lei Z, Fang F, Guo Q, et al. Comparative transcriptome analyses of deltamethrin-susceptible and -resistant Culex pipiens pallens by RNA-seq. Mol Genet Genomics. 2016;291(1): 309–321. doi: 10.1007/s00438-015-1109-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bonizzoni M, Ochomo E, Dunn WA, Britton M, Afrane Y, Zhou G, et al. RNA-seq analyses of changes in the Anopheles gambiae transcriptome associated with resistance to pyrethroids in Kenya: identification of candidate-resistance genes and candidate-resistance SNPs. Parasite vector. 2015;8: 474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Despres L, Stalinski R, Tetreau G, Paris M, Bonin A, Navratil V, et al. Gene expression patterns and sequence polymorphisms associated with mosquito resistance to Bacillus thuringiensis israelensis toxins. BMC Genomics. 2014;15(1): 926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Faucon F, Dusfour I, Gaude T, Navratil V, Boyer F, Chandre F, et al. Identifying genomic changes associated with insecticide resistance in the dengue mosquito Aedes aegypti by deep targeted sequencing. Genome Res. 2015;25(9): 1347–1359. doi: 10.1101/gr.189225.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poupardin R, Srisukontarat W, Yunta C, Ranson H. Identification of carboxylesterase genes implicated in temephos resistance in the dengue vector Aedes aegypti. PLoS Neg Trop D. 2014;8(3): e2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15): 2114–2120. doi: 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4): R36 doi: 10.1186/gb-2013-14-4-r36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gotoh O. Substrate recognition sites in cytochrome P450 family 2 (CYP2) proteins inferred from comparative analyses of amino acid and coding nucleotide sequences. J Biol Chem. 1992;267(1): 83–90. [PubMed] [Google Scholar]
- 33.Jackson CJ, Liu JW, Carr PD, Younus F, Coppin C, Meirelles T, et al. Structure and function of an insect alpha-carboxylesterase (alphaEsterase7) associated with insecticide resistance. Proc Natl Acad Sci USA. 2013;110(25): 10177–10182. doi: 10.1073/pnas.1304097110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wongsantichon J, Robinson RC, Ketterman AJ. Structural contributions of delta class glutathione transferase active-site residues to catalysis. Biochem J. 2010;428(1): 25–32. doi: 10.1042/BJ20091939 [DOI] [PubMed] [Google Scholar]
- 35.Radominska-Pandya A, Bratton SM, Redinbo MR, Miley MJ. The crystal structure of human UDP-glucuronosyltransferase 2B7 C-terminal end is the first mammalian UGT target to be revealed: the significance for human UGTs from both the 1A and 2B families. Drug Metab Rev. 2010;42(1): 133–144. doi: 10.3109/03602530903209049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kondrashov FA. Gene duplication as a mechanism of genomic adaptation to a changing environment. Proc Biol Sci. 2012;279(1749): 5048–5057. doi: 10.1098/rspb.2012.1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14): 1754–1760. doi: 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dai G, Chou N, He L, Gyamfi MA, Mendy AJ, Slitt AL, et al. Retinoid X receptor alpha Regulates the expression of glutathione s-transferase genes and modulates acetaminophen-glutathione conjugation in mouse liver. Mol Pharmacol. 2005;68(6): 1590–1596. doi: 10.1124/mol.105.013680 [DOI] [PubMed] [Google Scholar]
- 39.Johnson PF. Transcriptional activators in hepatocytes. Cell Growth Differ. 1990;1(1): 47–52. [PubMed] [Google Scholar]
- 40.Gonzalez FJ, Lee YH. Constitutive expression of hepatic cytochrome P450 genes. Faseb J. 1996;10(10): 1112–1117. [DOI] [PubMed] [Google Scholar]
- 41.Honkakoski P, Negishi M. Regulation of cytochrome P450 (CYP) genes by nuclear receptors. Biochem J. 2000;347(Pt 2): 321–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Juven-Gershon T, Kadonaga JT. Regulation of gene expression via the core promoter and the basal transcriptional machinery. Dev Biol. 2010;339(2): 225–229. doi: 10.1016/j.ydbio.2009.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cherbas L, Cherbas P. The arthropod initiator: the capsite consensus plays an important role in transcription. Insect Biochem Mol Biol. 1993;23(1): 81–90. [DOI] [PubMed] [Google Scholar]
- 44.Hu Z, Du Y, Nomura Y, Dong K. A sodium channel mutation identified in Aedes aegypti selectively reduces cockroach sodium channel sensitivity to type I, but not type II pyrethroids. Insect Biochem Mol Biol. 2011;41(1): 9–13. doi: 10.1016/j.ibmb.2010.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kasai S, Komagata O, Itokawa K, Shono T, Ng LC, Kobayashi M, et al. Mechanisms of pyrethroid resistance in the dengue mosquito vector, Aedes aegypti: target site insensitivity, penetration, and metabolism. PLoS Neg Trop D. 2014;8(6): e2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stevenson BJ, Pignatelli P, Nikou D, Paine MJ. Pinpointing P450s associated with pyrethroid metabolism in the dengue vector, Aedes aegypti: developing new tools to combat insecticide resistance. PLoS Neg Trop D. 2012;6(3): e1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muller P, Warr E, Stevenson BJ, Pignatelli PM, Morgan JC, Steven A, et al. Field-caught permethrin-resistant Anopheles gambiae overexpress CYP6P3, a P450 that metabolises pyrethroids. PLoS Genet. 2008;4(11): e1000286 doi: 10.1371/journal.pgen.1000286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Duangkaew P, Kaewpa D, Rongnoparut P. Protective efficacy of Anopheles minimus CYP6P7 and CYP6AA3 against cytotoxicity of pyrethroid insecticides in Spodoptera frugiperda (Sf9) insect cells. Trop Biomed. 2011;28(2): 293–301. [PubMed] [Google Scholar]
- 49.Stevenson BJ, Bibby J, Pignatelli P, Muangnoicharoen S, O'Neill PM, Lian LY, et al. Cytochrome P450 6M2 from the malaria vector Anopheles gambiae metabolizes pyrethroids: Sequential metabolism of deltamethrin revealed. Insect Biochem Mol Biol. 2011;41(7): 492–502. doi: 10.1016/j.ibmb.2011.02.003 [DOI] [PubMed] [Google Scholar]
- 50.Riveron JM, Irving H, Ndula M, Barnes KG, Ibrahim SS, Paine MJ, et al. Directionally selected cytochrome P450 alleles are driving the spread of pyrethroid resistance in the major malaria vector Anopheles funestus. Proc Natl Acad Sci USA. 2013;110(1): 252–257. doi: 10.1073/pnas.1216705110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ishak IH, Riveron JM, Ibrahim SS, Stott R, Longbottom J, Irving H, et al. The Cytochrome P450 gene CYP6P12 confers pyrethroid resistance in kdr-free Malaysian populations of the dengue vector Aedes albopictus. Sci Rep. 2016;6: 24707 doi: 10.1038/srep24707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dusfour I, Zorrilla P, Guidez A, Issaly J, Girod R, Guillaumot L, et al. Deltamethrin Resistance Mechanisms in Aedes aegypti Populations from Three French Overseas Territories Worldwide. PLoS Neg Trop D. 2015;9(11): e0004226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bariami V, Jones CM, Poupardin R, Vontas J, Ranson H. Gene Amplification, ABC Transporters and Cytochrome P450s: Unraveling the Molecular Basis of Pyrethroid Resistance in the Dengue Vector, Aedes aegypti. PLoS Neg Trop D. 2012;6(6): e1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grigoraki L, Lagnel J, Kioulos I, Kampouraki A, Morou E, Labbe P, et al. Transcriptome Profiling and Genetic Study Reveal Amplified Carboxylesterase Genes Implicated in Temephos Resistance, in the Asian Tiger Mosquito Aedes albopictus. PLoS Neg Trop D. 2015;9(5): e0003771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grigoraki L, Balabanidou V, Meristoudis C, Miridakis A, Ranson H, Swevers L, et al. Functional and immunohistochemical characterization of CCEae3a, a carboxylesterase associated with temephos resistance in the major arbovirus vectors Aedes aegypti and Ae. albopictus. Insect Biochem Mol Biol. 2016;74: 61–67. doi: 10.1016/j.ibmb.2016.05.007 [DOI] [PubMed] [Google Scholar]
- 56.Ranson H, Hemingway J. Mosquito glutathione transferases. Gluthione Transferases and Gamma-Glutamyl Transpeptidases. San Diego: Elsevier Academic Press Inc; 2005. pp. 226. [DOI] [PubMed] [Google Scholar]
- 57.Lumjuan N, Rajatileka S, Changsom D, Wicheer J, Leelapat P, Prapanthadara LA, et al. The role of the Aedes aegypti epsilon glutathione transferases in conferring resistance to DDT and pyrethroid insecticides. Insect Biochem Mol Biol. 2011;41(3): 203–209. doi: 10.1016/j.ibmb.2010.12.005 [DOI] [PubMed] [Google Scholar]
- 58.Riveron JM, Yunta C, Ibrahim SS, Djouaka R, Irving H, Menze BD, et al. A single mutation in the GSTe2 gene allows tracking of metabolically-based insecticide resistance in a major malaria vector. Gen Biol. 2014;15(2): R27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nene V, Wortman JR, Lawson D, Haas B, Kodira C, Tu ZJ, et al. Genome sequence of Aedes aegypti, a major arbovirus vector. Science. 2007;316(5832): 1718–1723. doi: 10.1126/science.1138878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Feyereisen R, Dermauw W, Van Leeuwen T. Genotype to phenotype, the molecular and physiological dimensions of resistance in arthropods. Pestic Biochem Physiol. 2015;121: 61–77. doi: 10.1016/j.pestbp.2015.01.004 [DOI] [PubMed] [Google Scholar]
- 61.Itokawa K, Komagata O, Kasai S, Tomita T. A single nucleotide change in a core promoter is involved in the progressive overexpression of the duplicated CYP9M10 haplotype lineage in Culex quinquefasciatus. Insect Biochem Mol Biol. 2015;66: 96–102. doi: 10.1016/j.ibmb.2015.10.006 [DOI] [PubMed] [Google Scholar]
- 62.Jover R, Moya M, Gomez-Lechon MJ. Transcriptional regulation of cytochrome p450 genes by the nuclear receptor hepatocyte nuclear factor 4-alpha. Curr Drug Metab. 2009;10(5): 508–519. [DOI] [PubMed] [Google Scholar]
- 63.Pascussi JM, Gerbal-Chaloin S, Duret C, Daujat-Chavanieu M, Vilarem MJ, Maurel P. The tangle of nuclear receptors that controls xenobiotic metabolism and transport: crosstalk and consequences. Annu Rev Pharmacol Toxicol. 2008;48: 1–32. doi: 10.1146/annurev.pharmtox.47.120505.105349 [DOI] [PubMed] [Google Scholar]
- 64.Claudianos C, Russell RJ, Oakeshott JG. The same amino acid substitution in orthologous esterases confers organophosphate resistance on the house fly and a blowfly. Insect Biochem Mol Biol. 1999;29(8): 675–686. [DOI] [PubMed] [Google Scholar]
- 65.Newcomb RD, Campbell PM, Ollis DL, Cheah E, Russell RJ, Oakeshott JG. A single amino acid substitution converts a carboxylesterase to an organophosphorus hydrolase and confers insecticide resistance on a blowfly. Proc Natl Acad Sci USA. 1997;94(14): 7464–7468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Heidari R, Devonshire AL, Campbell BE, Dorrian SJ, Oakeshott JG, Russell RJ. Hydrolysis of pyrethroids by carboxylesterases from Lucilia cuprina and Drosophila melanogaster with active sites modified by in vitro mutagenesis. Insect Biochem Mol Biol. 2005;35(6): 597–609. doi: 10.1016/j.ibmb.2005.02.018 [DOI] [PubMed] [Google Scholar]
- 67.Devonshire AL, Heidari R, Huang HZ, Hammock BD, Russell RJ, Oakeshott JG. Hydrolysis of individual isomers of fluorogenic pyrethroid analogs by mutant carboxylesterases from Lucilia cuprina. Insect Biochem Mol Biol. 2007;37(9): 891–902. doi: 10.1016/j.ibmb.2007.04.011 [DOI] [PubMed] [Google Scholar]
- 68.Coppin CW, Jackson CJ, Sutherland T, Hart PJ, Devonshire AL, Russell RJ, et al. Testing the evolvability of an insect carboxylesterase for the detoxification of synthetic pyrethroid insecticides. Insect Biochem Mol Biol. 2012;42(5): 343–352. doi: 10.1016/j.ibmb.2012.01.004 [DOI] [PubMed] [Google Scholar]
- 69.Feyereisen R. Insect cytochrome P450 In: Gilbert LI, Iatrou K, Gill S, editors. Comprehensive Molecular Insect Science: Elsevier; 2005. pp. 1–77. [Google Scholar]
- 70.Itokawa K, Komagata O, Kasai S, Ogawa K, Tomita T. Testing the causality between CYP9M10 and pyrethroid resistance using the TALEN and CRISPR/Cas9 technologies. Sci Rep. 2016;6: 24652 doi: 10.1038/srep24652 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Correlations were tested for each overexpressed detoxification gene by comparing Log2 CNV versus Log2 expression ratios. Correlations with Pearson’s r ≥ 0.7 and p ≤ 0.05 were considered significant. CNV data were extracted from [28].
(TIF)
Only variations located within a 1 kb upstream of detoxification genes overexpressed in resistant populations were considered. Among them, variations showing a significant correlation (Pearson’s r ≥ 0.7 and p ≤ 0.05) between their allele frequency and gene expression level (FPKM or Log2 FPKM) across all populations were considered significantly associated with gene expression. For each variation, position on supercontig, nucleotide change (ref>var) and position relative to coding sequence (brackets) are indicated.
(TIF)
Only SNPs affecting detoxification genes and detected by both approaches are shown.
(TIF)
Data Availability Statement
The RNA-seq sequence data from this study have been deposited at the European Nucleotide Archive (ENA; http://www.ebi.ac.uk/ena) under the accession number PRJEB15531. DNA-seq data from this study have been deposited at the European Nucleotide Archive under accession number PRJEB7976.