

Abstract
Objective:
To investigate inflammatory processes after aneurysmal subarachnoid hemorrhage (aSAH) with network models.
Methods:
This is a retrospective observational study of serum samples from 45 participants with aSAH analyzed at multiple predetermined time points: <24 hours, 24 to 48 hours, 3 to 5 days, and 6 to 8 days after aSAH. Concentrations of cytokines were measured with a 41-plex human immunoassay kit, and the Pearson correlation coefficients between all possible cytokine pairs were computed. Systematic network models were constructed on the basis of correlations between cytokine pairs for all participants and across injury severity. Trends of individual cytokines and correlations between them were examined simultaneously.
Results:
Network models revealed that systematic inflammatory activity peaks at 24 to 48 hours after the bleed. Individual cytokine levels changed significantly over time, exhibiting increasing, decreasing, and peaking trends. Platelet-derived growth factor (PDGF)-AA, PDGF-AB/BB, soluble CD40 ligand, and tumor necrosis factor-α (TNF-α) increased over time. Colony-stimulating factor (CSF) 3, interleukin (IL)-13, and FMS-like tyrosine kinase 3 ligand decreased over time. IL-6, IL-5, and IL-15 peaked and decreased. Some cytokines with insignificant trends show high correlations with other cytokines and vice versa. Many correlated cytokine clusters, including a platelet-derived factor cluster and an endothelial growth factor cluster, were observed at all times. Participants with higher clinical severity at admission had elevated levels of several proinflammatory and anti-inflammatory cytokines, including IL-6, CCL2, CCL11, CSF3, IL-8, IL-10, CX3CL1, and TNF-α, compared to those with lower clinical severity.
Conclusions:
Combining reductionist and systematic techniques may lead to a better understanding of the underlying complexities of the inflammatory reaction after aSAH.
Aneurysmal subarachnoid hemorrhage (aSAH) affects ≈30,000 people annually and accounts for 10% to 15% of all strokes.1 Despite improvements in clinical management, morbidity after aSAH remains high. Studies have shown that early elevation of central and peripheral inflammatory cytokines is associated with delayed neurologic deteriorations.2 However, such studies typically use methods to examine differences in concentrations of individual cytokines across disease severity or clinical outcomes. They fail to account for the complexities and redundancies that arise from cytokine interactions inherent to the immune reaction.3,4 In addition to examining individual cytokines, investigating associations between them can lead to a better understanding of the inflammatory process after aSAH. A systematic approach based on network theory can offer insights into the multifactorial relationships and can delineate the complexities underlying inflammatory processes.5
We propose a data-driven model of immune response at different acute time periods after aSAH. The initial time period after aneurysm rupture (<72 hours), also known as the early brain injury phase, has recently become the focus of investigation6 because it is implicated in delayed neurologic deterioration and poor outcomes. Uncontrolled inflammation is posited to contribute to clinical worsening during the early brain injury phase, manifesting clinically as poor neurologic status.7,8 Hence, in addition to the examination of inflammation at different times, we investigate systematic differences across clinical status at admission. While some studies have used similar techniques to model the inflammatory condition in chronic neurologic conditions,9 these techniques have not been fully explored in acute aSAH research.
METHODS
Study population, patient criteria, and clinical variables.
This is a retrospective, observational, single-center study of patients with aSAH admitted to the Neuroscience Intensive Care Unit at the Memorial Herman Hospital–Texas Medical Center between July 2013 and August 2014. Serum samples were collected at 4 predetermined time points after admission: <24 hours (T1), 24 to 48 hours (T2), 3 to 5 days (T3), and 6 to 8 days (T4) after aSAH. To maintain homogeneity, only participants from whom at least 3 serum samples were drawn at any of the above-mentioned time points were included. Participants with SAH from nonaneurysmal causes, including trauma, arteriovenous malformation, and mycotic aneurysms, were excluded. Patients with known autoimmune disease and other conditions that may affect baseline inflammation, including history of malignancy, and current pregnancy were excluded. All clinical and radiographic variables used in the study (the supplemental material at Neurology.org gives the definitions of clinical variables), including Hunt-Hess (HH) score, intraventricular hemorrhage score, Fisher score, and delayed cerebral ischemia (DCI), were adjudicated by at least 2 attending neurointensivists.
Standard protocol approvals, registrations, and patient consents.
This study was conducted with Institutional Review Board approval (No. HSC-MS-12-0637). Written informed consent was obtained from the patient or surrogate.
Sample collection and analysis.
Blood samples were collected in K2 EDTA vacutainer tubes and were centrifuged within an hour of draw (1,460g for 10 minutes at 4°C), generating plasma that was centrifuged a second time (1,460g for 10 minutes at 4°C) to generate platelet-poor plasma. Platelet-poor supernatant was collected and stored at −80°C until ready for use. Cytokine sample concentrations were determined with a MAGPIX magnetic bead–based ELISA 41-plex assay (EMD Millipore, Billerica, MA) to test for the following cytokines: CC chemokines (CCL2, CCL5, CCL7, CCL11, and CCL22), CX chemokines (CX3CL1 and CXCL1P1), macrophage inflammatory proteins (MIP-1α and MIP-1β), the interleukin (IL)-1 superfamily (IL-1A, IL-1R1, IL-4, IL-5 and IL-6), IL-17 family (IL-17a), IL-2, IL-3, IL-7, IL-8, IL-9, IL-10, IL-13, IL-15, platelet-derived growth factors (PDGF-AA and PDGF-AB/BB), epidermal growth factor (EGF), fibroblast growth factor-2 (FGF-2), colony-stimulating factors (CSF3 and CSF2), tumor necrosis factors (TNF-α/TNF-β), vascular endothelial growth factor (VEGF) A, IL-12p40, IL-12p70, soluble CD40 ligand (sCD40L), IL-1b, interferon-inducible protein 10, type-II interferon family (IFNG), interferon- α2 (IFN-α2), FMS-like tyrosine kinase 3 ligand (FLT3L), and transforming growth factor-α (TGF-α). Plasma was analyzed according to the manufacturer's protocol (supplemental material).
Statistical analysis.
To describe differences in demographics across HH grades, the χ2, Student t, Fisher exact, and Mann-Whitney U tests were used when appropriate. To test cytokine expression levels across HH grades and DCI status, the Mann-Whitney U test was used. To test for cytokine expression levels across time, the generalized estimated equations were used because they account for within-subject correlation. To compare networks at different time points, the random skewers algorithm was used.10 To adjust for multiple comparisons, the Benjamini-Hochberg method11 with a false discovery rate of 10% was applied. The open-source statistical packages in R (version 3.1.3) were used to perform statistical analysis. Cytoscape (version 3.2.1) was used for network visualization.
Data transformation.
For network models, Pearson correlation coefficients (Pccs) between all possible cytokine pairs were computed. Because most raw cytokine values are not normally distributed and highly skewed, the Box-Cox transformation12 was used to normalize each cytokine distribution. The Kolmogorov-Smirnov test was used to test for normality of the transformed distribution at a 5% significant level.
Clustering.
A correlation matrix was constructed with each cell entry representing a Pcc between a cytokine pair. For a 40-plex array, there are 780 unique pairwise correlation coefficients. In the matrix, the lower triangle is a duplicate of the upper triangle, and the diagonal elements are self-correlations (equal to one). The hierarchical clustering algorithm with average-linking criteria was used to group correlated cytokines on the basis of euclidean distances between cytokine correlations.13
Network model.
A network has nodes and edges. The edges represent a relationship between the nodes. We modeled the immune response as a network with the cytokines as nodes and the Pccs as edges between them. Because a high number of correlations are possible among the 40 cytokines, a hard threshold of 0.75 was used to exclude correlations below this value and to focus only on strong correlations. To construct networks for the HH ≤ 3 and HH ≥ 4 groups, pairwise correlations for each group were ranked on the basis of significant p values, and only the top 5% (≈40) of the correlations were used.
RESULTS
Demographics.
During the study period, 124 patients with aSAH were admitted to our institution. Sixty-eight patients consented for blood samples to be drawn. We included 45 consecutively admitted participants who were able to provide samples at least 3 time points. A total of 151 samples were analyzed (45 samples at T1, 31 samples at T2, 38 samples at T3, and 37 samples at T4). There were more female participants in the study population compared to those who were excluded (80% vs 60%, p = 0.04). There were no other differences in characteristics (table e-1).
Participants included in the study were stratified on the basis of their clinical status at admission into good (HH ≤ 3) and poor (HH ≥ 4) groups. There were no significant differences in age, sex, smoking habits, history of hypertension, incidence of DCI, intraventricular hemorrhage score, hospital length of stay, and Fisher score across injury grades. As expected, the HH ≥ 4 group had significantly higher mortality rate and worse modified Rankin score at discharge (table e-2). The differences in demographics were similar when participants were dichotomized as HH ≤ 2 vs HH ≥ 3 (table e-3).
Individual trends.
The cytokine CCL22 (MDC) was undetectable at all time points and was excluded from the analysis. Three distinct trends in cytokine expression were observed; increasing, decreasing, and peaking (figure 1). PDGF-AA, PDGF-AB/BB, sCD40L, and TNF-α increased significantly over time (figure 1A). CSF3, IL-13, and FLT3L significantly decreased over time (figure 1B). IL-6, IL-5, and IL-15 had peaking trends over time (figure 1C). IL-6 peaked at T2 (36.4 ± 82.9 pg/mL) and returned to baseline values by T4 (15.9 ± 29.8 pg/mL) (figure 1C).
Figure 1. Median (interquartile range) cytokine plot across time; for each cytokine, each of the 4 box plots represents expression levels from T1 to T4.

Cytokine levels are scaled either down or up by a factor for easier visualization. The superscript number following the cytokine represents a factor by which the values were scaled down, and dagger denotes a factor by which it was scaled up. (A) Platelet-derived cytokines, including platelet-derived growth factor (PDGF)-AA, PDGF-AB/BB, soluble CD40 ligand (sCD40L), and tumor necrosis factor-α (TNFa), showed increasing trend. (B) Colony-stimulating factor-3 (CSF3), interleukin (IL)-13, and FMS-like tyrosine kinase 3 ligand (FLT3L) show decreasing trend. (C) IL-6 and IL-5 peak at T2 and return to baseline values by T4. IL-15 has a peaking trend at T3. *p < 0.05, **p < 0.01, ***p < 0.001 from generalized estimated equations analysis.
Cytokine correlations and clusters over time.
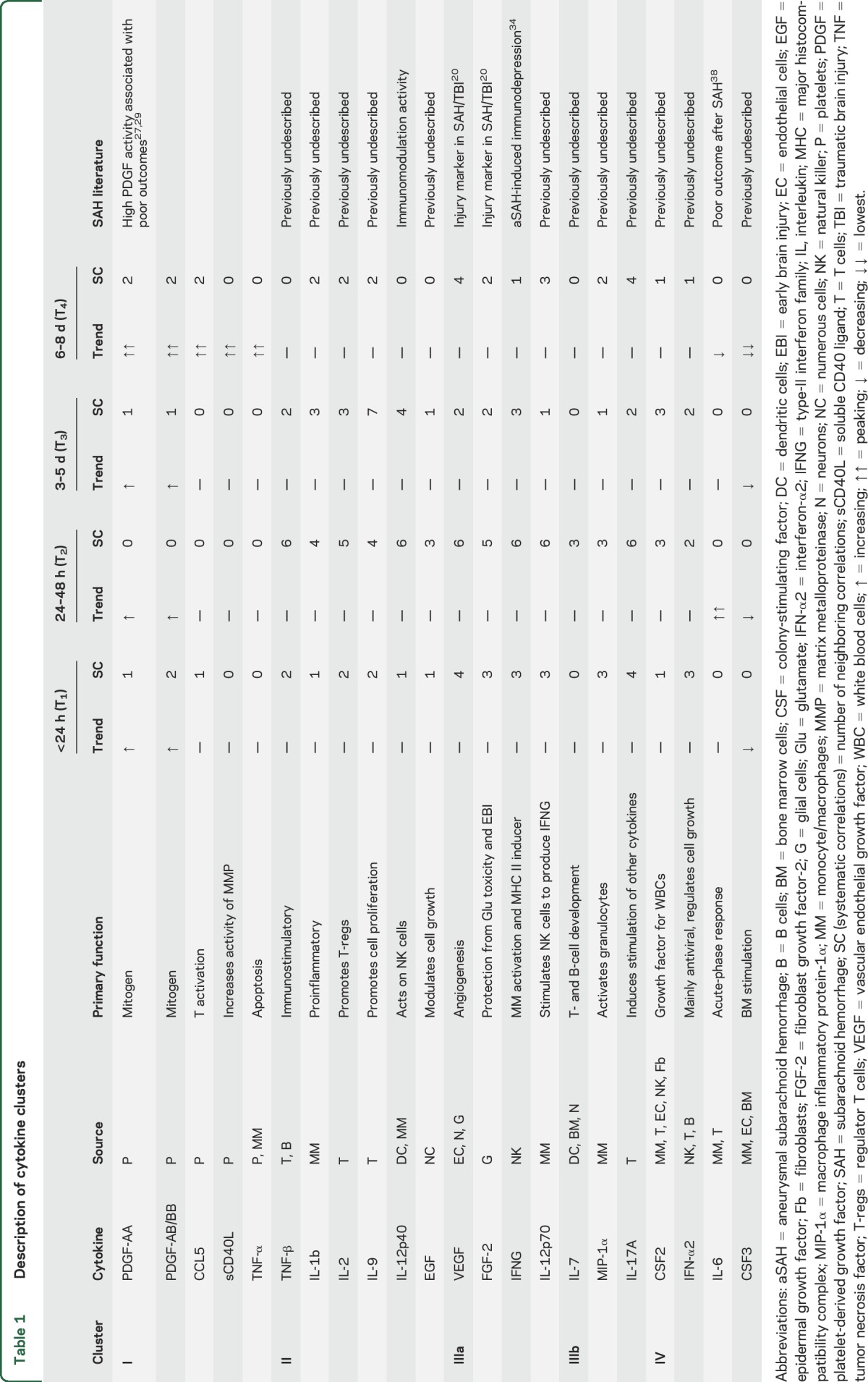
The random skewers methods computes a similarity index (SI) between the networks. The networks at all time points are significantly correlated (all p < 0.01). Among them, the most dissimilar were T2 and T4 (SI 0.834), T3 and T4 (SI 0.835), T2 and T3 (SI 0.843), and T1 and T2 (SI 0.867). The networks that were most similar were T1 and T4 (SI 0.891) and T1 and T3 (SI 0.893).To examine only strong correlations, a hard threshold of Pcc > 0.75 was applied to the network14; i.e., only edges with Pcc values >0.75 were considered. After the threshold was applied, there were 21, 40, 20, and 15 high-strength correlations observed at T1, T2, T3, and T4, respectively (figures 2 and 3). Clustering analysis reveals the presence of distinct correlated cytokine clusters. The source, function, trend, systemic activity, and clinical relevance in participants with aSAH of each cytokine in different clusters are outlined in table 1. Four cytokine clusters were observed at all times. The first cluster comprised cytokines generally related to platelet activity. The second cluster included IL-2, IL-9, IL-12p40, and TNF-β. The third cluster comprised endothelial growth factors, including VEGFA and FGF-2. The fourth cluster comprised CSF2 and IFN-α2.
Figure 2. Dynamics of correlation networks (A and B) and clustering patterns (C and D) of cytokine associations in all participants with aSAH at 0 to 24 and 24 to 48 hours.

Each edge in the cytokine network represents a Pearson correlation coefficient ≥0.75. Networks were constructed from 45 samples at 0 to 24 hours (A), 32 samples at 24 to 48 hours (B), and after admission. The nodes with the first and second most number of correlations are highlighted in red and green, respectively. The bottom row is the hierarchical clustering of pairwise cytokine correlations. Each pixel in the dendrograms is a correlation coefficient value between a cytokine pair corresponding to that row and column. Green, red, and black pixels indicate a positive, negative, or near-zero correlation. Because the correlational matrix is symmetric, the upper triangle is a duplicate of the lower triangle. The diagonal elements are self-correlations and are equal to one. The number of associations peaks at 24 to 48 hours. The pairwise comparison of all networks using random skewers method indicates that the network at 24 to 48 hours was dissimilar to other networks. Clustering dendrograms group cytokines that are most closely associated with each other. The network model and clustering analysis indicate a correlated cluster of the platelet-derived cytokines involving platelet-derived growth factor (PDGF)-AA, PDGF-AB/BB, soluble CD40 ligand (sCD40L), CXCL1P1, and CCL5 at all periods. aSAH = aneurysmal subarachnoid hemorrhage; CSF = colony-stimulating factor; EGF = epidermal growth factor; FGF2 = fibroblast growth factor-2; FLT3L = FMS-like tyrosine kinase 3 ligand; IFNA2 = interferon-α2; IFNG = type-II interferon family; IL = interleukin; IP10 = interferon-inducible protein 10; MIP1B = macrophage inflammatory protein-1β; TNF = tumor necrosis factor; VEGF = vascular endothelial growth factor.
Figure 3. Dynamics of correlation networks (A and B) and clustering patterns (C and D) of cytokine associations in all participants with aSAH at 3 to 5 and 6 to 8 days.

Each edge in the cytokine network represents a Pearson correlation coefficient ≥0.75. Networks were constructed from 38 samples at 3 to 5 days (C) and from 37 samples at 6 to 8 days. The nodes with the first and second most number of correlations are highlighted in red and green, respectively. The number of correlations returns to baseline levels at 6 to 8 days. The pairwise comparison of all networks using random skewers method indicates that the network at 3 to 5 days was similar to the network at 0 to 24 hours. The bottom row is the hierarchical clustering of pairwise cytokine correlations. The network model and clustering analysis indicate a correlated cluster of the platelet-derived cytokines involving platelet-derived growth factor (PDGF)-AA, PDGF-AB/BB, soluble CD40 ligand (sCD40L), CXCL1P1, and CCL5 at all periods. aSAH = aneurysmal subarachnoid hemorrhage; CSF = colony-stimulating factor; EGF = epidermal growth factor; FGF2 = fibroblast growth factor-2; FLT3L = FMS-like tyrosine kinase 3 ligand; IFNA2 = interferon-α2; IFNG = type-II interferon family; IL = interleukin; IP10 = interferon-inducible protein 10; MIP1B = macrophage inflammatory protein-1β; TNF = tumor necrosis factor; VEGF = vascular endothelial growth factor.
Table 1.
Description of cytokine clusters
Cytokine levels across injury severity.
Several cytokines differed in concentration levels across injury severity (figure 4). At T1, the HH ≥ 4 group had higher levels of IL-6 (p < 0.001), CCL2 (p < 0.001), CCL11 (p < 0.01), and CSF3 (p < 0.01). IL-8, IL-10, CX3CL1, and TNF-α were also significantly elevated in the HH ≥4 group; however, they were not significant when adjusted for multiple comparisons. High PDGF-AB/BB at T2 was observed in participants who subsequently developed DCI (1,113.6 ± 388 vs 472.8 ± 388.7 pg/mL, p < 0.05). The correlation matrices of different HH grades indicate a higher number of negative correlations in the HH ≥ 4 group (figure e-1). Negative correlations were observed between CX3CL1 and IL-15 (r = −0.6, p = 0.09), IL-10 and IL-5 (r = −0.65, p = 0.07), EGF and interferon-inducible protein 10 (r = −0.66, p = 0.07), and IL-5 and TNF-α (r = −0.67, p = 0.06). The only significant negative correlation in the HH ≤ 3 group was between FLT3L and CCL5 (r = −0.3, p = 0.02). A hierarchical clustering analysis revealed more clusters in the HH ≥ 4 group compared to the HH ≤ 3 group (figure e-1, A and B). The top 5% of the significant correlations (≈40) in both groups are shown (figure e-1, C and D). Cytokines were ranked on the basis of the number of correlations they extended to other cytokines in the group. IL-9 (9), IFN-α2 (6), IL-15 (5), and IL-12p70 (6) had the highest correlations in the HH ≤3 (figure e-1C). In the HH ≥4 group, CX3CL1 (5), FGF-2 (7), IFNG (7), and IL-17A (7) had the most correlations (figure e-1D).
Figure 4. Difference in cytokine expression levels and correlations across HH grades (A) and difference in cytokine levels across patients with good and poor admission status (at T1) stratified by HH ≤ 3 (white) vs HH ≥ 4 (gray).

Interleukin (IL)-6 (A), CCL2 (B), CCL11 (C), and colony-stimulating factor-3 (CSF3) (D) were elevated in the Hunt-Hess (HH) ≥ 4 group. The unadjusted p values indicate possible elevation of IL-8 (E), IL-10 (F), CX3CL1 (G), and tumor necrosis factor (TNF)-α (H) in the HH ≥ 4 group (B and C). aSAH = aneurysmal subarachnoid hemorrhage; EGF = epidermal growth factor; FGF2 = fibroblast growth factor-2; FLT3L = FMS-like tyrosine kinase 3 ligand; IFNA2 = interferon-α2; IFNG = type-II interferon family; IL = interleukin; IP10 = interferon-inducible protein 10; MIP1B = macrophage inflammatory protein-1β; PDGF = platelet-derived growth factor; VEGF = vascular endothelial growth factor. *Significant p values represent the Benjamini-Hochberg–adjusted (false discovery rate 10%) values.
DISCUSSION
The main findings of this study are that (1) acute systematic inflammation peaks at 24 to 48 hours after the bleed and returns to baseline values by 6 to 8 days, (2) cytokines that do not significantly change over time may still play a dynamic role in the inflammatory processes, (3) cytokines that do not differ across injury severity may still play a role in disease severity, (4) platelet-derived cytokine levels increased in time and were highly correlated with each other, and (5) the severe injury grade group has a higher proportion of negative correlations suggesting the presence of regulatory mechanisms.
After the exclusion of Pcc values <0.75, the network at T1 and T2 had a similar number of cytokines (19 vs 22). However, the T2 network had twice the number of correlations (40 vs 21) compared to T1. This indicates that the correlations among the inflammatory markers examined peaks at the 24- to 48-hour postbleed window. The comparison of networks using random skewers method revealed that the network at T2 was most significantly different from the other time points. Several inflammatory events during this period, including increased leukocytosis,15,16 activation of mechanisms involving neutrophils17–19 and VEGF upregulation,20 have been reported in patients who proceed to develop DCI. A number of clinical events linked to inflammation, including cerebral edema,8,21 seizures,22 and high intracranial pressure,23 have also been reported during this period. The peak inflammatory interactions among the examined cytokines at 24 to 48 hours may be indicative of complications that occur later.
Network characterization indicates that some cytokines with no significant differences across time may play an important role in the inflammatory process. For instance, although VEGF, IFNG, FGF-2, IL-12p70, IL-12p40, TNF-β, and IL-9 do not vary significantly across time, they are highly correlated (figures 2 and 3) and likely to be involved in regulatory mechanisms involving other cytokines. Conversely, cytokines with significant changes across time, including sCD40L, TNF-α, CSF3, IL-13, IL-5, and IL-6, were not prominent in the networks. For instance, IL-6, a proinflammatory cytokine that has been implicated in vasospasm24 and delayed ischemia, peaked at T2 but correlated weakly with other cytokines in the system. This suggests that some cytokines are only marginally dominant at the systematic level, can exert effects on different physiologic processes independently of other cytokines, or affect the inflammatory processes via other mechanisms involving cytokines not investigated in this study. We hypothesize that the increased number of correlations during this phase and the peaking of the proinflammatory cytokine IL-6 could be indicative of a neutrophil-mediated response that begins immediately after the rupture and persists for the first 48 hours. In addition, the decrease in the associations at >48 hours and the decline in the expression levels of proinflammatory cytokines could be indicative of the activation of a monocyte-macrophage mediated response that increases after 48 hours.
Clustering analysis revealed the presence of multiple correlated clusters. (1) Platelet factors: Platelet-derived cytokines are highly interactive at the systematic level from T1 to T4 (figures 2 and 3). This cluster included PDGF-AA, PDGF-AB/BB, TNF-α, CCL5 (a leukocyte attractant), sCD40L (a proinflammatory glycoprotein), and CXCL1P1 (a chemoattractant for neutrophils). They are involved in a wide range of biochemical processes, including mitosis, would healing, vessel formation, and vasoconstriction.25–27 Previously, PDGF levels and outcomes after aSAH have been reported28,29; however, our findings pertaining to interactions between PDGFs, CCL5, sCD40L,30 and CXCL1P1 are previously undescribed and warrant further investigation. (2) IL-2, IL-9, IL-12p40, and TNF-β cytokines: From T1 to T4, a cluster including IL-2, IL-12p40, TNF-β, and IL-9 was observed. With the exception of IL-12p40, which is reported to play a role in the immunomodulation after aSAH, all other cytokines in this cluster are previously undescribed, and their roles require investigation. (3a) Endothelial growth factor cluster: A cluster including VEGF, EGF, FGF-2, IFNG, and IL-12p70 was observed at all time points. While none of the cytokines in this cluster show increasing or decreasing trends, systematically, these cytokines, VEGF and FGF-2 in particular, were active from T1 to T4 with peak interactions at 24 to 48 hours after aSAH. Elevated VEGF and FGF-2 have been reported in acute traumatic brain injury and aSAH previously.20 VEGF and EGF2 are typically involved in pathologic mechanisms pertaining to angiogenesis, neurovascular disorders, endothelial repair, and neuroprotection.31 In an experimental aSAH model, VEGF, via the activation of mitogen-activated protein kinase pathways, has been implicated in increased blood-brain permeability and edema.32 A recent study has shown that administration of VEGF receptor-2 (an anti-VEGF antibody) to target VEGF-associated pathways reduced edema and improved outcomes in animal model.33 The results presented in our study corroborate these findings and suggest that these mechanisms also could be relevant in clinical aSAH. While IFNG has previously been reported in aSAH-induced immune depression,34 the role of IL-12p70 in aSAH is unclear. The high correlation between IFNG and IL-12p70 in our participant population suggests the presence of a mechanism involving IL-12p70 stimulation of natural killer cells, which in turn express IFNG.35 (3b) MIP-1α, IL-7, and IL-17A: MIP-1α, IL-7, and IL-17A were loosely correlated in the cluster with endothelial factors, and their roles warrant further investigation. (4) CSF2 and IFN-α2: We additionally report that CSF2 and IFN-α2 were highly correlated at all times after aSAH. The activities of both these cytokines in aSAH are unclear.
Our finding that participants with higher injury severity have elevated levels of IL-6, CCL2, and CCL11 at <24 hours of rupture is consistent with earlier reports.36–38 In addition, CSF3 was elevated in the HH ≥ 4 group. CSF3 is a glycoprotein that stimulates bone marrow to produce granulocytes and stem cells. CSF3 expression immediately after the bleed (<24 hours) is twice as high as it is at T4 (figure 1B), and the HH ≥ 4 group had almost 4 times the expression levels of CSF3 compared to the HH ≤ 3 group at T1, indicating that the production of granulocytes and stem cells increases over time in proportion to injury severity (figure 4). Network model indicates that CSF3 has a strong correlation with IL-6 and FGF-2 in the HH ≥ 4 group (figure e-1D). The correlation between FGF-2 and CSF3 was almost 10 times stronger in the HH ≥ 4 group. The coexpression of CSF3 and FGF-2 is associated with new vessel formation.39,40 Mechanisms involving CSF3 and FGF-2 production after aSAH are unclear. However, the dominance of FGF-2 in the HH ≥ 4 group network indicates a possible vital role in the injury process. IL-13 strongly correlated with FGF-2 and decreased over time. FGF-2 was the most correlated cytokine in the HH ≥ 4 group. FGF-2 is also implicated in different biological processes, including wound healing. This is consistent with earlier reports of FGF-2 after aSAH.20 Finally, the numbers of inverse correlations were higher in the HH ≥ 4 group compared to the HH ≤ 3 group, indicating the presence of regulatory pathways in poor-grade patients.
Limitations.
First, being a single-center observational study, it is bound by inherent biases. Every institution has a unique patient population and has specific protocols for aSAH management. This may limit the generalizability of our findings. Second, despite corrections for false positives, complete elimination of spurious correlations is not guaranteed. Third, the study design only permits correlational observations and does not describe putative mechanisms. To determine whether these changes contribute to clinical worsening or are simply an epiphenomena, interventional trials are needed. Fourth, the sample size is relatively small, and comparisons between clinical outcomes were not possible. Finally, in addition to peripheral inflammation, the examination of central inflammation through CSF or microdialysate may offer further insight into injury mechanisms.
This study models peripheral inflammation after aSAH using correlational networks. Systematic models of cytokines examined indicate a peak in their interactions at 24 to 48 hours. Several clusters of correlated cytokines were observed at different times. Cytokines that do not show trends or differ across groups may play dynamic roles systematically and vice versa. Severity of injury was associated with negative correlations, indicating the presence of regulatory mechanisms. In addition to traditional statistical methods, informatics tools may provide deeper insight into the immune response after aSAH.
Supplementary Material
ACKNOWLEDGMENT
The investigators thank the Vivian L. Smith Center for Neurologic Research and the Neuroscience Research Repository for assistance with obtaining the specimens.
GLOSSARY
- aSAH
aneurysmal subarachnoid hemorrhage
- CSF
colony-stimulating factor
- DCI
delayed cerebral ischemia
- EGF
epidermal growth factor
- FGF-2
fibroblast growth factor-2
- FLT3L
FMS-like tyrosine kinase 3 ligand
- HH
Hunt-Hess
- IFN-α2
interferon-α2
- IFNG
type-II interferon family
- IL
interleukin
- MIP
macrophage inflammatory protein
- Pcc
Pearson correlation coefficient
- PDGF
platelet-derived growth factor
- sCD40L
soluble CD40 ligand
- SI
similarity index
- TNF
tumor necrosis factor
- VEGF
vascular endothelial growth factor
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
J.P.J.S., H.A.C., D.H.K., and Y.L. were involved in the conception and design of the study. K.P., G.W.H., and S.A. assisted in the acquisition and analysis of data. L.Z. assisted in the analysis of data. J.P.J.S., H.A.C., S.S.B., T.C., and K.L. contributed substantially to the drafting of the manuscript and figures.
STUDY FUNDING
This work was partly funded by the Joe Niekro Foundation and the CCTS Scholar Program from the McGovern Medical School.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Hemorrhagic stroke [online]. 2014. Available at: stroke.org/understand-stroke/what-stroke/hemorrhagic-stroke. Accessed February 22, 2016. [Google Scholar]
- 2.McMahon CJ, Hopkins S, Vail A, et al. Inflammation as a predictor for delayed cerebral ischemia after aneurysmal subarachnoid haemorrhage. J Neurointerventional Surg 2013;5:512–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aderem A, Hood L. Immunology in the post-genomic era. Nat Immunol 2001;2:373–375. [DOI] [PubMed] [Google Scholar]
- 4.Zak DE, Tam VC, Aderem A. Systems-level analysis of innate immunity. Annu Rev Immunol 2014;32:547–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pavlopoulos GA, Secrier M, Moschopoulos CN, et al. Using graph theory to analyze biological networks. BioData Min 2011;4:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cahill WJ, Calvert JH, Zhang JH. Mechanisms of early brain injury after subarachnoid hemorrhage. J Cereb Blood Flow Metab 2006;26:1341–1353. [DOI] [PubMed] [Google Scholar]
- 7.Budohoski KP, Czosnyka M, Kirkpatrick PJ, Smielewski P, Steiner LA, Pickard JD. Clinical relevance of cerebral autoregulation following subarachnoid haemorrhage. Nat Rev Neurol 2013;9:152–163. [DOI] [PubMed] [Google Scholar]
- 8.Choi HA, Bajgur SS, Jones WH, et al. Quantification of cerebral edema after subarachnoid hemorrhage. Neurocrit Care 2016;25:64–70. [DOI] [PubMed] [Google Scholar]
- 9.Hornig M, Gottschalk G, Peterson DL, et al. Cytokine network analysis of cerebrospinal fluid in myalgic encephalomyelitis/chronic fatigue syndrome. Mol Psychiatry 2016;21:261–269. [DOI] [PubMed] [Google Scholar]
- 10.Cheverud JM, Marroig G. Research article comparing covariance matrices: random skewers method compared to the common principal components model. Genet Mol Biol 2007;30:461–469. [Google Scholar]
- 11.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol 1995;57:289–300. [Google Scholar]
- 12.Sakia RM. The Box-Cox transformation technique: a review. Statistician 1992;41:169–178. [Google Scholar]
- 13.Johnson SC. Hierarchical clustering schemes. Psychometrika 1967;32:241–254. [DOI] [PubMed] [Google Scholar]
- 14.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008;9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spallone A, Acqui M, Pastore FS, Guidetti B. Relationship between leukocytosis and ischemic complications following aneurysmal subarachnoid hemorrhage. Surg Neurol 1987;27:253–258. [DOI] [PubMed] [Google Scholar]
- 16.McGirt MJ, Mavropoulos JC, McGirt LY, et al. Leukocytosis as an independent risk factor for cerebral vasospasm following aneurysmal subarachnoid hemorrhage. J Neurosurg 2003;98:1222–1226. [DOI] [PubMed] [Google Scholar]
- 17.Provencio JJ, Fu X, Siu A, Rasmussen PA, Hazen SL, Ransohoff RM. CSF neutrophils are implicated in the development of vasospasm in subarachnoid hemorrhage. Neurocrit Care 2010;12:244–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Minami N, Tani E, Yokota M, Maeda Y, Yamaura I. Immunohistochemistry of leukotriene C4 in experimental cerebral vasospasm. Acta Neuropathol (Berl) 1991;81:401–407. [DOI] [PubMed] [Google Scholar]
- 19.Yang MF, Sun BL, Xia ZL, Zhu LZ, Qiu PM, Zhang SM. Alleviation of brain edema by L-arginine following experimental subarachnoid hemorrhage in a rat model. Clin Hemorheol Microcirc 2003;29:437–443. [PubMed] [Google Scholar]
- 20.Mellergård P, Sjögren F, Hillman J. Release of VEGF and FGF in the extracellular space following severe subarachnoidal haemorrhage or traumatic head injury in humans. Br J Neurosurg 2010;24:261–267. [DOI] [PubMed] [Google Scholar]
- 21.Claassen J, Carhuapoma JR, Kreiter KT, Du EY, Connolly ES, Mayer SA. Global cerebral edema after subarachnoid hemorrhage: frequency, predictors, and impact on outcome. Stroke 2002;33:1225–1232. [DOI] [PubMed] [Google Scholar]
- 22.Claassen J, Albers D, Schmidt JM, et al. Nonconvulsive seizures in subarachnoid hemorrhage link inflammation and outcome. Ann Neurol 2014;75:771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Graetz D, Nagel A, Schlenk F, Sakowitz O, Vajkoczy P, Sarrafzadeh A. High ICP as trigger of proinflammatory IL-6 cytokine activation in aneurysmal subarachnoid hemorrhage. Neurol Res 2010;32:728–735. [DOI] [PubMed] [Google Scholar]
- 24.Osuka K, Suzuki Y, Tanazawa T, et al. Interleukin-6 and development of vasospasm after subarachnoid haemorrhage. Acta Neurochir (Wien) 1998;140:943–951. [DOI] [PubMed] [Google Scholar]
- 25.Williams LT. Signal transduction by the platelet-derived growth factor receptor. Science 1989;243:1564–1570. [DOI] [PubMed] [Google Scholar]
- 26.Alvarez RH, Kantarjian HM, Cortes JE. Biology of platelet-derived growth factor and its involvement in disease. Mayo Clin Proc 2006;81:1241–1257. [DOI] [PubMed] [Google Scholar]
- 27.Frontera JA, Aledort L, Gordon E, et al. Early platelet activation, inflammation and acute brain injury after a subarachnoid hemorrhage: a pilot study. J Thromb Haemost 2012;10:711–713. [DOI] [PubMed] [Google Scholar]
- 28.Yanamoto H, Kataoka H, Nakajo Y, Iihara K. The role of the host defense system in the development of cerebral vasospasm: analogies between atherosclerosis and subarachnoid hemorrhage. Eur Neurol 2012;68:329–343. [DOI] [PubMed] [Google Scholar]
- 29.Gaetani P, Tancioni F, Grignani G, et al. Platelet derived growth factor and subarachnoid haemorrhage: a study on cisternal cerebrospinal fluid. Acta Neurochir (Wien) 1997;139:319–324. [DOI] [PubMed] [Google Scholar]
- 30.Chen XD, Sun J, Lu C, et al. The prognostic value of plasma soluble CD40 ligand levels following aneurysmal subarachnoid hemorrhage. Thromb Res 2015;136:24–29. [DOI] [PubMed] [Google Scholar]
- 31.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med 2003;9:669–676. [DOI] [PubMed] [Google Scholar]
- 32.Kusaka G, Ishikawa M, Nanda A, Granger DN, Zhang JH. Signaling pathways for early brain injury after subarachnoid hemorrhage. J Cereb Blood Flow Metab 2004;24:916–925. [DOI] [PubMed] [Google Scholar]
- 33.Liu L, Fujimoto M, Kawakita F, Ichikawa N, Suzuki H. Vascular endothelial growth factor in brain edema formation after subarachnoid hemorrhage. Acta Neurochir Suppl 2016;121:173–177. [DOI] [PubMed] [Google Scholar]
- 34.Sarrafzadeh A, Schlenk F, Meisel A, Dreier J, Vajkoczy P, Meisel C. Immunodepression after aneurysmal subarachnoid hemorrhage. Stroke 2011;42:53–58. [DOI] [PubMed] [Google Scholar]
- 35.Cassatella MA, Meda L, Gasperini S, D'Andrea A, Ma X, Trinchieri G. Interleukin-12 production by human polymorphonuclear leukocytes. Eur J Immunol 1995;25:1–5. [DOI] [PubMed] [Google Scholar]
- 36.Kim GH, Kellner CP, Hahn DK, et al. Monocyte chemoattractant protein-1 predicts outcome and vasospasm following aneurysmal subarachnoid hemorrhage. J Neurosurg 2008;109:38–43. [DOI] [PubMed] [Google Scholar]
- 37.Kampen GT, Stafford S, Adachi T, et al. Eotaxin induces degranulation and chemotaxis of eosinophils through the activation of ERK2 and p38 mitogen-activated protein kinases. Blood 2000;95:1911–1917. [PubMed] [Google Scholar]
- 38.Sarrafzadeh A, Schlenk F, Gericke C, Vajkoczy P. Relevance of cerebral interleukin-6 after aneurysmal subarachnoid hemorrhage. Neurocrit Care 2010;13:339–346. [DOI] [PubMed] [Google Scholar]
- 39.Layman H, Sacasa M, Murphy AE, Murphy AM, Pham SM, Andreopoulos FM. Co-delivery of FGF-2 and G-CSF from gelatin-based hydrogels as angiogenic therapy in a murine critical limb ischemic model. Acta Biomater 2009;5:230–239. [DOI] [PubMed] [Google Scholar]
- 40.Layman H, Li X, Nagar E, Vial X, Pham SM, Andreopoulos FM. Enhanced angiogenic efficacy through controlled and sustained delivery of FGF-2 and g-CSF from Fibrin hydrogels containing ionic-albumin microspheres. J Biomater Sci Polym Ed 2012;23:185–206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.