

Abstract
Purpose
Hermansky-Pudlak syndrome (HPS) is a rare inherited disorder with ten reported genetic types; each type has defects in subunits of either Adaptor Protein-3 complex or Biogenesis of Lysosome-related Organelles Complex (BLOC)-1, -2, or -3. Very few patients with BLOC-1 deficiency (HPS-7, -8, and -9 types) have been diagnosed. We report results of comprehensive clinical testing and molecular analyses of primary fibroblasts from a new case of HPS-7.
Results
A 6-year old Paraguayan male presented with hypopigmentation, ocular albinism, nystagmus, reduced visual acuity, and easy bruising. He also experienced delayed motor and language development as a very young child; head and chest trauma resulted in intracranial hemorrhage with subsequent right hemiparesis and lung scarring. There was no clinical evidence of immunodeficiency or colitis. Whole mount transmission electron microscopy revealed absent platelet delta granules; platelet aggregation testing was abnormal. Exome sequencing revealed a homozygous nonsense mutation in the Dystrobrevin binding protein 1 (DTNBP1) gene [NM 032122.4: c.307C>T; p.Gln103*], previously reported in a Portuguese adult. The gene encodes the dysbindin subunit of BLOC-1. Dysbindin protein expression was negligible in our patient’s dermal fibroblasts, while his DTNBP1 mRNA level was similar to that of a normal control.
Conclusions
Comprehensive clinical evaluation of the first pediatric case reported with HPS-7 reveals oculocutaneous albinism and platelet storage pool deficiency; his phenotype is consistent with findings in other patients with BLOC-1 disorders. This patient’s markedly reduced Dysbindin protein expression in HPS-7 resulted from a mechanism other than nonsense mediated decay.
Keywords: Biogenesis of Lysosome-related Organelles Complex-1, DTNBP1, Dysbindin, oculocutaneous albinism, platelet storage pool disorder
1.1 INTRODUCTION
1.2 Hermansky-Pudlak syndrome (HPS), a rare autosomal recessive disorder, is characterized by improper formation of lysosome-related organelles [1]. Ten types of HPS (HPS-1 to HPS-10) have been identified, and each type is associated with mutations in genes that encode proteins belonging to Biogenesis of Lysosome-related Organelles Complex (BLOC) -1, BLOC-2, BLOC-3, or the Adaptor Protein-3 complex [1–3]. These cellular defects in patients with HPS lead to heterogeneous disease manifestations, including oculocutaneous albinism, bleeding due to a platelet storage pool deficiency, and granulomatous colitis [4]. In addition, certain manifestations of disease are limited to specific HPS types. For example, progressive and generally fatal pulmonary fibrosis develops in patients with HPS-1, -2, and -4, and HPS-2 patients manifest immunodeficiency and neutropenia responsive to granulocyte colony-stimulation factor [1, 5–9].
1.3 HPS-7, -8, and -9 are uncommon types characterized by BLOC-1 subunit defects; these disorders are associated with mutations in DTNBP1, BLOC1S3, and BLOC1S6, respectively [2, 10, 11]. Patients with BLOC-1 disorders present with various clinical manifestations. Pulmonary fibrosis has not been described in the 12 other published cases with a BLOC-1 disease; only one patient had recurrent cutaneous infections [2, 10–15]. Two adult patients with HPS-7 have been reported [10, 13]. In this work, we expand the phenotypic characterization of Dysbindin-related HPS-7 by reporting the extensive clinical evaluation and molecular testing of the first child, and the third case, with HPS-7.
2.1 MATERIALS AND METHODS
2.2 Patient Consent, Ethics Approval, and Clinical Testing
2.2.1 The patient was enrolled in National Institutes of Health protocols 76-HG-0238 (ClinicalTrials.gov Identifier NCT00369421, “Diagnosis and Treatment of Inborn Errors of Metabolism and Other Genetic Disorders”) and 95-HG-0193 (ClinicalTrials.gov Identifier NCT00001456, “Clinical and Basic Investigations into Hermansky-Pudlak Syndrome”) approved by the Institutional Review Board of the National Human Genome Research Institute. Written informed consent was obtained from the patient’s parents.
2.2.2 Clinical and molecular evaluations of the patient were performed at the National Institutes of Health Clinical Center in Bethesda, Maryland. Whole mount preparations were made from platelet-rich plasma, isolated from fresh citrated peripheral blood, from the patient and a healthy European American research volunteer. The preparations were imaged by electron microscopy, as described [16]. Pulmonary function tests (i.e., spirometry) were performed in accordance with guidelines from the American Thoracic Society as described [17]. High-resolution computed tomography scan images were obtained without intravenous contrast in the prone position as described [18].
2.3. Mutation Analysis
2.3.1 Single-nucleotide polymorphism (SNP) array for genotyping was performed on genomic DNA using HumanOmniExpress DNA Analysis BeadChip (Illumina). SNPs were analyzed using GenomeStudio software (Illumina, San Diego, CA) [19, 20]. Exome sequencing was performed by the National Institutes of Health Intramural Sequencing Center using the HiSeq2000 (Illumina) that employed 101 base pair paired-end read sequencing. Image analysis and base-calling were performed using Illumina Genome Analyzer Pipeline software (V.1.13.48.0) with default parameters [21]. Reads were aligned to a human reference sequence (University of California Santa Cruz assembly hg19, NCBI build 37) using the Efficient Large-scale Alignment of Nucleotide Databases package (Illumina). VarSifter was used to view, sort, and filter the variants, which were selected on the basis of autosomal recessive inheritance [22]. Di-deoxy Sanger sequencing was performed to confirm the mutation by polymerase chain reaction (PCR) amplification. Sequencing of PCR products was performed using a genetic analyzer (Applied Biosystems 3130x; Foster City, CA). Analysis was performed using the proband’s genomic DNA isolated from whole blood; parental DNA was not available for analysis.
2.4. DTNBP1 mRNA and Dysbindin Protein Expression in Human Dermal Fibroblasts
2.4.1 Dermal fibroblasts were isolated from a punch biopsy of the patient’s skin. Control fibroblasts from a normal donor were purchased from ATCC (Manassas, VA). Fibroblast cultures were cultured in Dulbecco’s Modified Eagle Medium supplemented with high-glucose (4.5 g/L), 10% fetal bovine serum, 2 mM L-glutamine and penicillin-streptomycin; mRNA and protein expression were measured in cells isolated from the same cultures.
2.4.2 Total RNA was isolated from fibroblasts using TRIzol Reagent (Life Technologies, Carlsbad, CA), and cDNA was synthesized using an RNA-to-cDNA kit (Applied Biosystems). TaqMan Gene Expression Master Mix reagent and Assays-On-Demand (Applied Biosystems) were obtained for DTNBP1 (Hs01105864_m1) and a control gene, POLR2A (Hs00172187_m1). Quantitative real-time PCR was performed using 100 ng cDNA on an ABI PRISM 7900 HT Sequence Detection System (Applied Biosystems) using the relative gene expression comparative CT method (ΔΔCT). Data shown are levels relative to normal volunteer fibroblast mRNA and normalized to POLR2A expression for 4 independent experiments.
2.4.3 Cultured dermal fibroblasts were washed twice with ice-cold PBS and scraped into buffer containing 50 mM Tris-HCl (pH 7.5), 50 mM sodium fluoride, 5 mM sodium pyrophosphate, 1 mM sodium orthovanadate, 1 mM EDTA, 1 mM EGTA, 0.27 M sucrose, 1% Triton X-100 and Complete Mini Protease Inhibitor Cocktail (Roche Diagnostics, Indianapolis, IN). For immunoblotting, 15 micrograms of protein lysate were electrophoresed on a 4–12% Bis-Tris gel with MOPS buffer and transferred onto a polyvinylidene difluoride membrane using the iBlot® transfer system (Life Technologies, Carlsbad, CA). The membrane was blocked using Odyssey Blocking Buffer (LI-COR, Lincoln, NE) and then incubated at 4°C overnight with the following primary antibodies: mouse anti-Dysbindin (anti-HPS7; epitope maps between amino acids 233–262 near the C-terminus of human Dysbindin) (sc-390626; Santa Cruz Biotechnology, Santa Cruz, CA) and rabbit anti-beta-tubulin (2128; Cell Signaling Technology, Danvers, MA). The membrane was then washed with phosphate buffered saline with 0.1% Tween 20 three times, incubated for one hour with the appropriate secondary antibodies (LI-COR), and washed three more times in phosphate buffered saline with 0.1% Tween 20. The bands were imaged using the IR Odyssey® Imaging System (LI-COR). Dysbindin levels were normalized to beta-tubulin expression; immunoblots were run in triplicate.
2.5 Statistical Analysis
Data are expressed as mean values ± standard error of the mean. Significance of difference between means was evaluated using a paired or unpaired Student’s t-test. Analyses were performed using GraphPad Prism 5 (GraphPad Software, San Diego, CA).
3.1 RESULTS
3.2 Clinical Findings
3.2.1 The patient was a 6-year old Paraguayan male evaluated at the National Institutes of Health Clinical Center. There was no known history of consanguinity. He was the product of an uncomplicated pregnancy, and his perinatal period was notable only for delayed separation of the umbilical cord. His skin and hair showed some pigmentation (Figure 1A), and are noticeably paler than that of his parents. Nystagmus was noted at 3 months of age, and an ophthalmology exam at 6 months of age revealed ocular albinism due to iris transillumination. Ophthalmologic evaluation at the National Institutes of Health at age 6 years showed nystagmus and hyperopic astigmatism. Best-corrected visual acuity was 20/200 in each eye. Anterior segment examination was significant for brown irides with transillumination (Figures 1B and 1C). Dilated examination revealed fundus hypopigmentation, foveal hypoplasia, and optic nerve hypoplasia.
Figure 1.
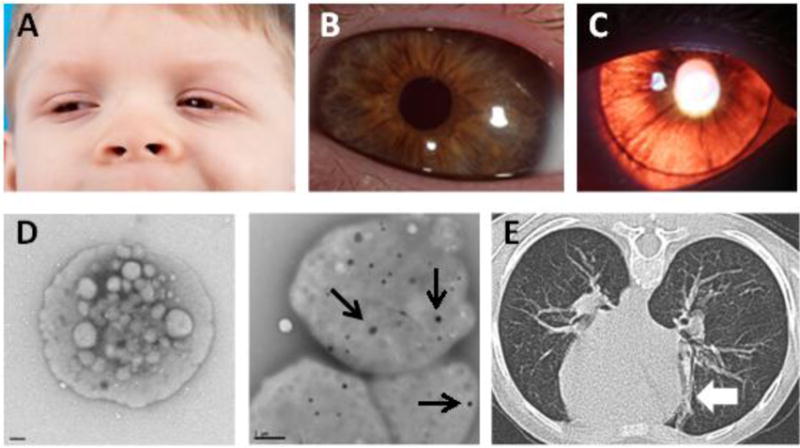
Clinical Findings of HPS-7.
(A) Reduced pigmentation of skin and hair is observed in this 6-year old male of Paraguayan descent. (B) The proband has a light brown iris color. (C) Iris transillumination results from reduced eye pigment. (D) Delta granules are absent in platelets from the proband (left panel; size bar = 0.5 um) compared to platelets from a healthy control (right panel; size bar = 1 um) (black arrows indicate delta granules). (E) Computed tomography of the chest shows focal linear lung parenchymal scarring (white arrow) but no evidence of interstitial lung disease or pulmonary fibrosis.
3.2.2 The patient had no history of recurrent or unusual infections that would suggest immunodeficiency. He bruised easily, particularly on his forehead and extremities, but he did not have epistaxis, gastrointestinal bleeding, intra-articular bleeding, or mucosal hemorrhage during tooth eruption. Prior hematologic evaluation revealed low ristocetin co-factor, and a diagnosis of von Willebrand disease was considered. Additional hematologic testing included platelet electron microscopy analysis showing absent delta granules, and this finding established a diagnosis of HPS. Antihemophilic factor/von Willebrand factor complex and platelet transfusion were recommended for minor bleeding. At the National Institutes of Health, platelet whole mount transmission electron microscopy was repeated at 6 years of age and confirmed absent delta granules (Figure 1D). Platelet aggregation testing was abnormal (Table 1). Prothrombin time, partial thromboplastin time, factor VIII activity, Willebrand factor activity and antigen, platelet count, hemoglobin, and total white blood count with differential testing were normal (Table 2).
Table 1.
Platelet Aggregation Study
| Test | Present | Absent |
|---|---|---|
| Low dose ADP aggregation | X | |
| High dose ADP aggregation | X | |
| Release of ATP from dense granules with low dose ADP | X | |
| Release of ATP from dense granules with high dose ADP | X | |
| Low dose collagen aggregation | X | |
| High dose collagen aggregation | X | |
| Release of ATP from dense granules with low dose collagen | X | |
| Release of ATP from dense granules with high dose collagen | X | |
| Release of ATP from dense granules with thrombin | X | |
| Arachidonic acid aggregation | X | |
| Release of ATP from dense granules with arachidonic acid | X | |
| Low dose ristocetin agglutination | X | |
| High dose ristocetin agglutination | X | |
| Low dose platelet rich plasma epinephrine aggregation | X | |
| Release of ATP from dense granules with low dose epinephrine | X | |
| Release of ATP from dense granules with high dose epinephrine | X |
ADP = adenosine diphosphate
ATP = adenosine triphosphate
Table 2.
Hematology Studies
| Test | Result | Normal Range |
|---|---|---|
| Prothrombin Time (sec) | 14.3 | 11.6–15.2 |
| Partial Thromboplastin Time (sec) | 31.8 | 25.3–37.3 |
| Factor VIII Activity (IU/dL) | 178 | 41–184 |
| von Willebrand Factor Activity (IU/dL) | 120 | 52–156 |
| von Willebrand Factor Antigen (IU/dL) | 100 | 50–197 |
| Total White Blood Cell (K/ul) | 8.4 | 5.0–11.0 |
| Polymorphonuclear Leukocyte (K/ul) | 2.68 | 1.50–8.50 |
| Lymphocyte (K/ul) | 4.99 | 2.00–8.00 |
| Monocytes (K/ul) | 0.44 | 0.20–0.90 |
| Eosinophil (K/ul) | 0.02 | 0.02–0.60 |
| Hemoglobin (g/dl) | 13.1 | 11.5–14.0 |
| Platelet (K/ul) | 317 | 206–369 |
3.2.3 The patient had delayed motor and language development; he began walking at approximately 2 years of age. The patient had no other neurological symptoms until he sustained blunt trauma to his head and torso at 4 years of age. Computed tomography scan of the head revealed an intraparenchymal hemorrhage involving the left frontal, temporal and parietal lobes. The patient underwent craniotomy to evacuate and drain an intracranial hematoma and cranioplasty. Numerous platelet transfusions were required before and after neurosurgery. Following this injury, the patient experienced chronic right hemiparesis with very limited spontaneous movement of his right hand. He was functionally one-handed, used a right ankle foot orthosis, and received physical and occupational therapy. Asymptomatic intrathoracic scarring also persisted (Figure 1E), but notably, the patient’s radiographic findings of focal lung scarring on computed tomography scan of the chest were not consistent with the interstitial lung disease or pulmonary fibrosis that develops in patients with certain HPS types. In agreement with these imaging findings, pulmonary function measurements revealed normal airflow without evidence of restriction.
3.3 Identification of DTNBP1 Mutation
3.3.1 Molecular testing for mutations in GPR143, which is associated with ocular albinism type 1 (OA1), was negative at 9 months of age. Exome sequencing of the patient’s genomic DNA at 6 years of age at the National Institutes of Health identified a homozygous nonsense mutation in DTNBP1 (NM_032122.4; c.307C>T; p.Gln103*) encoding the Dysbindin subunit of BLOC-1 [1, 10]. SNP array analysis revealed two small regions of homozygosity (ROH) in the entire genome, and both of these were on chromosome 6: a 0.58 Mb region and a 2.02 Mb region which encompassed DTNBP1. No other genetic mutations known to be associated with HPS were found. Di-deoxy Sanger sequencing confirmed the nonsense mutation in exon 5 of DTNBP1 (Figure 2A).
Figure 2.
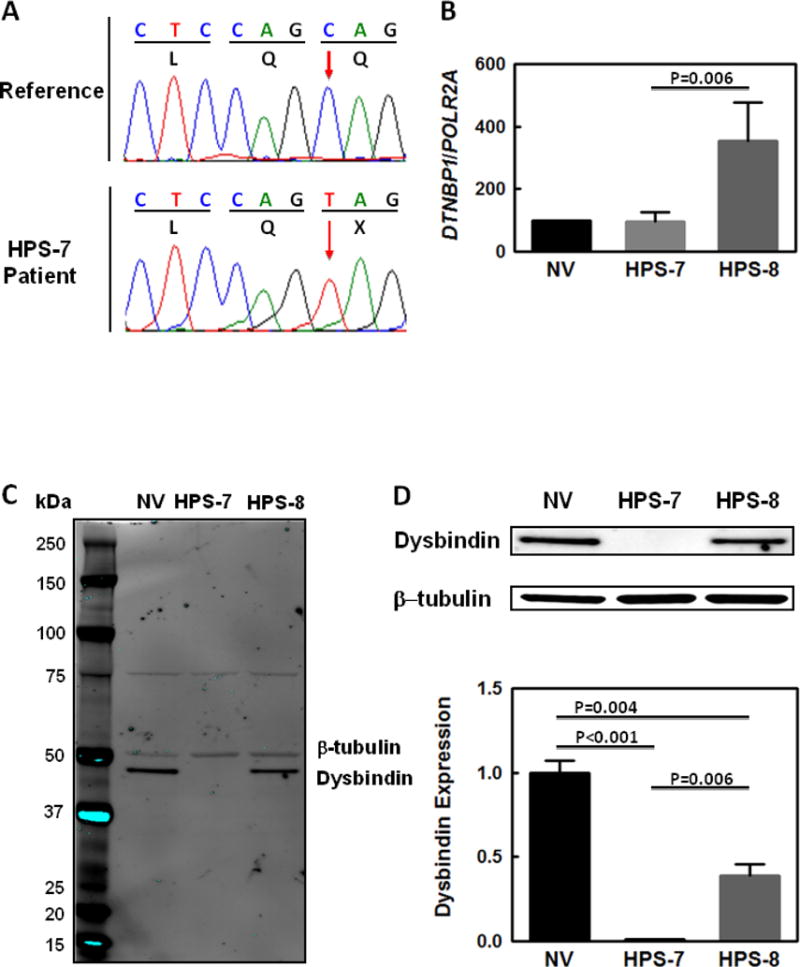
Genetic and Molecular Studies in HPS-7.
(A) Sequence chromatograms showing a homozygous c.307C>T mutation (red arrow) in DTNBP1 in the proband’s genomic DNA. This nucleotide change results in the introduction of a termination codon (denoted as “X” in figure) at the protein level (Gln103*). (B) DTNBP1 mRNA expression in the proband’s fibroblasts is similar to levels in cells from a normal volunteer control (NV). Fibroblast DTNBP1 mRNA levels are increased in a patient with HPS-8 compared with control and significantly higher than in the HPS-7 patient. (C and D) Immunoblots show negligible Dysbindin protein expression in the HPS-7 proband’s fibroblast lysates compared to levels in HPS-8 or normal cells. Dysbindin protein expression in HPS-8 fibroblasts was also significantly reduced compared to normal cells. The entire immunoblot is shown in Panel C; Dysbindin and β-tubulin bands from the same blot are shown in Panel D. Loading was controlled by normalizing for β-tubulin expression in the same immunoblot; normalized Dysbindin expression in the HPS-7 and HPS-8 patients is shown relative to normal volunteer control expression.
3.4 DTNBP1 and Dysbindin Cellular Expression
3.4.1 To study the cellular effect of the DTNBP1 nonsense mutation, studies were performed in fibroblasts from the HPS-7 patient, a normal control and a previously published HPS-8 patient [12]. HPS-8 is another HPS type with a BLOC-1 defect due to mutation in BLOC1S3 [12].
3.4.2 Quantitative real-time PCR analysis demonstrated normal DTNBP1 mRNA expression in the HPS-7 fibroblasts (95 ± 16% of control) (Figure 2B). However, DTNBP1 mRNA appeared upregulated in the HPS-8 patient’s fibroblasts (355 ± 61% of control) compared with normal cells and significantly higher than DTNBP1 levels in the HPS-7 patient (P=0.006).
3.4.3 An immunoblot for Dysbindin showed very low to absent protein expression in the HPS-7 proband’s cells; this was significantly reduced compared to Dysbindin levels in HPS-8 fibroblasts (P=0.006) or normal cells (P<0.001) (Figures 2C and 2D). Dysbindin level in HPS-8 fibroblasts was significantly reduced to 39% of expression in control cells (P=0.004). The entire immunoblot using a primary antibody that recognizes an epitope near the C-terminus shows an absence of bands corresponding to truncated Dysbindin protein that may result from the DTNBP1 nonsense mutation or alternatively spliced forms in cells from the HPS-7 patient and controls (Figure 2C).
4.1 DISCUSSION
4.2 We report the clinical features and molecular phenotype of the first child with HPS-7, an unusual Hermansky-Pudlak syndrome type associated with a defect in BLOC-1. In contrast to the 2 other reported adult cases of HPS-7, there is no history of consanguinity. Although the mutant allele was found within a small ROH, consanguinity in this family is not likely. The size of ROH is less than 3 Mb, and only 2 small ROH, which were both on chromosome 6, were identified in the entire genome. Our proband with HPS-7 has a pigmentation defect of his skin, hair and eyes, a history of bleeding tendency with trauma-induced intracranial hemorrhage, and delayed motor and language development. Whole mount transmission electron microscopy of his platelets showed absent delta granules, and platelet aggregation studies were abnormal. He had no clinical or hematologic evidence of an immunological deficiency. Radiographic imaging of his lungs showed no evidence of interstitial lung disease, and lung function tests were normal. This boy’s manifestations of disease resemble those reported in most patients with BLOC-1-related HPS types. Including our case, 3, 7 and 3 patients are reported to have HPS-7, HPS-8 and HPS-9, respectively [2, 10–15]. Compared to patients with the more common HPS-1 type, the relatively mild pigmentation defect and tendency to bleed may contribute to missed diagnoses and underreporting of patients with these unusual genetic types. Our patient had delayed motor and language development, and it is possible that minor neurological manifestations are phenotypic features of BLOC-1 disease. Further neurological assessment of our patient was limited by his chronic neurological deficits due to trauma-induced intracranial hemorrhage. Improving the knowledge and understanding of the signs and symptoms of these uncommon HPS types as well as advances in clinical genetic testing may enhance the identification of additional patients with these unusual HPS types.
4.3 Notably, a previously reported HPS-7 Portuguese patient has the same homozygous mutation in DTNBP1 (c.307C>T; p.Gln103*) as our proband, who is Paraguayan (Table 3). This variant (rs104893945 in dbSNP and RCV000003601.2 in ClinVar) is reported to have a very low allele frequency in ExAC and was found in only 2 of 120818 alleles from multiple populations (allele frequency 1.655e-05). Specifically, this DTNBP1 variant was identified in 1 of 11486 Latino alleles (allele frequency 8.706e-05) and in 1 of 66496 non-Finnish European alleles (allele frequency 1.504e-05). The only other reported variant in DTNBP1 (c.177G>A; p.Trp59*) was a homozygous nonsense mutation in exon 4 identified in a 77-year old Caucasian woman [13].
Table 3.
Genetic Mutations and Clinical Characteristics of 3 Reported Patients with HPS-7
| Pt | Gene | Mutations | Gender | Age | Ethnicity/Nationality | Consanguinity | Clinical Features | Reference |
|---|---|---|---|---|---|---|---|---|
| 1 | DTNBP1 | c.307C>T; p.Gln103* homozygous | female | 48 yrs | Portuguese | Yes (parents are first cousins) | Oculocutaneous albinism, bleeding tendency | 10 |
| 2 | DTNBP1 | c.177G>A;p.Trp59* homozygous | female | 77 yrs | Caucasian | Yes (parents are first cousins) | Oculocutaneous albinism, bleeding tendency, colitis | 13 |
| 3 | DTNBP1 | c.307C>T; p.Gln103* homozygous | male | 6 yrs | Paraguayan | No | Oculocutaneous albinism, easy bruising, mild delay in motor and language development | current paper |
Mutation nomenclature is based on accession NM_032122.4.
4.4 Our cellular analyses are the first reported molecular studies in human HPS-7 cells. Dermal fibroblasts were selected for our work, because these cells are readily cultured from a tissue sample obtained by skin biopsy. Immunoblotting using an anti-Dysbindin antibody recognizing an epitope near the C-terminus did not detect truncated Dysbindin protein. Primary human dermal HPS-7 fibroblasts also expressed markedly reduced to absent wild-type isoforms of Dysbindin protein compared to control fibroblasts while mRNA levels in the same cells were similar to those of normal control fibroblasts. These analyses were performed only in dermal fibroblasts, and it is possible that there could be differences in mRNA and protein levels of Dysbindin in other HPS-7 cell types. Our data suggest that nonsense-mediated decay of DTNBP1 due to the nonsense mutation is not occurring, and that this is not an underlying mechanism contributing to the downregulated Dysbindin protein in the HPS-7 patient’s cells. Regarding nonsense-mediated decay, the DTNBP1 nonsense variant is centrally located in exon 5 (of the 8 exon gene), not close to an intron/exon boundary, and not predicted to create alternative splicing. Thus, known factors that can prevent nonsense-mediated decay are not found with this mutation, and it is unclear why this process is not occurring [23]. Similar results were reported for the HPS-7 mouse model, sandy, which carries an in-frame 1.5-kb Dtnbp1 deletion and has a normal amount of Dtnbp1 mRNA expression but absent Dysbindin protein expression [10].
4.5 Degradation of Dysbindin secondary to destabilized assembly of BLOC-1 due to the nonsense variant is a possible explanation for markedly reduced Dysbindin protein in the HPS-7 patient’s cells. We found that the Dysbindin protein level was also reduced in HPS-8 fibroblasts compared to normal control cells, which provides further evidence that Dysbindin protein expression is sensitive to BLOC-1 stability. Of note, we previously reported absent (instead of greatly reduced) Dysbindin protein expression in fibroblasts from this patient with HPS-8 [12]; differences in these data are likely due to use of different anti-Dysbindin primary antibodies for immunoblotting. It is established in HPS human and mouse cells that a defect in one subunit affects stability of the entire complex, which can lead to degradation of other subunits in the same complex. This phenomenon at the protein level was shown for HPS defects in BLOC-1 [10, 12, 24], BLOC-2 [25, 26], BLOC-3 [27, 28], and the Adaptor-Protein-3 complex [8, 29].
5.1 CONCLUSIONS
5.2 Our report expands the understanding of Dysbindin-related HPS-7. We present the comprehensive clinical phenotype of the first child with HPS-7 and the only reported molecular analysis of primary human cells with a defect in HPS-7. The clinical manifestations of this patient with an uncommon HPS type are consistent with other patients with BLOC-1 disorders. His homozygous nonsense genetic mutation in DTNBP1 is associated with near absence of cellular Dysbindin protein, which is likely not due to nonsense mediated decay.
Highlights.
The first pediatric patient was identified with Hermansky-Pudlak syndrome type 7 (HPS-7).
Exome sequencing revealed a homozygous nonsense mutation in the Dystrobrevin binding protein 1 (DTNBP1) gene [NM 032122.4: c.307C>T; p.Gln103*], previously reported in a Portuguese adult.
Molecular analyses of human HPS-7 fibroblasts showed negligible Dysbindin protein expression and DTNBP1 mRNA level similar to that of a normal control.
This HPS-7 patient’s markedly reduced Dysbindin protein expression resulted from a mechanism other than nonsense mediated decay.
Acknowledgments
We thank our patients who participated in our research program. The authors thank Richard A. Hess, Roxanne Fischer and Carla Ciccone for their expert assistance in the laboratory and Gretchen Golas for excellent patient care. This study was supported in part by the Intramural Research Programs of the National Human Genome Research Institute, the National Eye Institute, the National Institute of Biomedical Imaging and Bioengineering, and the Office of the Director, National Institutes of Health, Bethesda, Maryland.
Abbreviations
- BLOC
Biogenesis of Lysosome-related Organelles Complex
- DTNBP1
Dystrobrevin binding protein 1
- HPS
Hermansky-Pudlak syndrome
- PCR
polymerase chain reaction
- ROH
regions of homozygosity
- SNP
single-nucleotide polymorphism
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Web References
dbSNP, https://www.ncbi.nlm.nih.gov/projects/SNP/, accessed November 11, 2016
Exome Aggregation Consortium (EXAC), http://exac.broadinstitute.org, accessed January 30, 2017
ClinVar, https://www.ncbi.nlm.nih.gov/clinvar/, accessed November 11, 2016
ClinicalTrials.gov, https://clinicaltrials.gov/, accessed November 11, 2016
References
- 1.Huizing M, Helip-Wooley A, Westbroek W, Gunay-Aygun M, Gahl WA. Disorders of lysosome-related organelle biogenesis: clinical and molecular genetics. Annu Rev Genomics Hum Genet. 2008;9:359–386. doi: 10.1146/annurev.genom.9.081307.164303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cullinane AR, Curry JA, Carmona-Rivera C, Summers CG, Ciccone C, Cardillo ND, et al. A BLOC-1 mutation screen reveals that PLDN is mutated in Hermansky-Pudlak syndrome type 9. Am J Hum Genet. 2011;88:778–787. doi: 10.1016/j.ajhg.2011.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 3.Ammann S, Schulz A, Krägeloh-Mann I, Dieckmann NM, Niethammer K, Fuchs S, et al. Mutations in AP3D1 associated with immunodeficiency and seizures define a new type of Hermansky-Pudlak syndrome. Blood. 2016;127:997–1006. doi: 10.1182/blood-2015-09-671636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gahl WA, Brantly M, Kaiser-Kupfer MI, Iwata F, Hazelwood S, Shotelersuk V, et al. Genetic defects and clinical characteristics of patients with a form of oculocutaneous albinism (Hermansky-Pudlak syndrome) N Engl J Med. 1998;338:1258–1264. doi: 10.1056/NEJM199804303381803. [DOI] [PubMed] [Google Scholar]
- 5.Brantly M, Avila NA, Shotelersuk V, Lucero C, Huizing M, Gahl WA. Pulmonary function and high-resolution CT findings in patients with an inherited form of pulmonary fibrosis, Hermansky-Pudlak syndrome, due to mutations in HPS-1. Chest. 2000;117:129–136. doi: 10.1378/chest.117.1.129. [DOI] [PubMed] [Google Scholar]
- 6.Anderson PD, Huizing M, Claassen DA, White J, Gahl WA. Hermansky-Pudlak syndrome type 4 (HPS-4): clinical and molecular characteristics. Hum Genet. 2003;113:10–17. doi: 10.1007/s00439-003-0933-5. [DOI] [PubMed] [Google Scholar]
- 7.Gochuico BR, Huizing M, Golas GA, Scher CD, Tsokos M, Denver SD, et al. Interstitial lung disease and pulmonary fibrosis in Hermansky-Pudlak syndrome-2, an AP-3 complex disease. Mol Med. 2012;18:56–64. doi: 10.2119/molmed.2011.00198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huizing M, Scher CD, Strovel E, Fitzpatrick DL, Hartnell LM, Anikster Y, et al. Nonsense mutations in ADTB3A cause complete deficiency of the beta3A subunit of adaptor complex-3 and severe Hermansky-Pudlak syndrome type 2. Pediatr Res. 2002;51:150–158. doi: 10.1203/00006450-200202000-00006. [DOI] [PubMed] [Google Scholar]
- 9.Gil-Krzewska A, Murakami Y, Peruzzi G, O’Brien KJ, Merideth MA, Cullinane AR, et al. Natural killer cell activity and dysfunction in Hermansky-Pudlak syndrome. Brit J Haematol. 2017;176:118–123. doi: 10.1111/bjh.14390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li W, Zhang Q, Oiso N, Novak EK, Gautam R, O’Brien EP, et al. Hermansky-Pudlak syndrome type 7 (HPS-7) results from mutant dysbindin, a member of the biogenesis of lysosome-related organelles complex 1 (BLOC-1) Nat Genet. 2003;35:84–89. doi: 10.1038/ng1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morgan NV, Pasha S, Johnson CA, Ainsworth JR, Eady RAJ, Dawood B, et al. A germline mutation in BLOC1S3/Reduced Pigmentation causes a novel variant of Hermansky-Pudlak syndrome (HPS8) Am J Hum Genet. 2006;78:160–166. doi: 10.1086/499338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cullinane AR, Curry JA, Golas G, Pan J, Carmona-Rivera C, Hess RA, et al. A BLOC-1 mutation screen reveals a novel BLOC1S3 mutation in Hermansky-Pudlak Syndrome type 8. Pigment Cell Melanoma Res. 2012;25:584–591. doi: 10.1111/j.1755-148X.2012.01029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lowe GC, Sanchez Guiu I, Chapman O, Rivera J, Lordkipanidze M, Dovlatova N, et al. Microsatellite markers as a rapid approach for autozygosity mapping in Hermansky-Pudlak syndrome: identification of the second HPS7 mutation in a patient presenting late in life. Thromb Haemost. 2013;109:766–768. doi: 10.1160/TH12-11-0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Badolato R, Prandini A, Caracciolo S, Colombo F, Tabellini G, Giacomelli M, et al. Exome sequencing reveals a pallidin mutation in a Hermansky-Pudlak-like primary immunodeficiency syndrome. Blood. 2012;119:3185–3187. doi: 10.1182/blood-2012-01-404350. [DOI] [PubMed] [Google Scholar]
- 15.Yousaf S, Shahzad M, Kausar T, Sheikh SA, Tariq N, Shabbir AS, et al. Identification and clinical characterization of Hermansky-Pudlak syndrome alleles in the Pakistani population. Pigment Cell Melanoma Res. 2016;29:231–235. doi: 10.1111/pcmr.12438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Witkop CJ, Krumwiede M, Sedano H, White JG. Reliability of absent platelet dense bodies as a diagnostic criterion for Hermansky-Pudlak syndrome. Am J Hematol. 1987;26:305–311. doi: 10.1002/ajh.2830260403. [DOI] [PubMed] [Google Scholar]
- 17.Rouhani FN, Brantly ML, Markello TC, Hess R, O’Brien K, Huizing M, et al. Alveolar macrophage dysregulation in Hermansky-Pudlak syndrome type 1. Am J Respir Crit Care Med. 2009;180:1114–1121. doi: 10.1164/rccm.200901-0023OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cullinane AR, Yeager C, Dorward H, Carmona-Rivera C, Wu HP, Moss J, et al. Dysregulation of galectin-3: Implications for Hermansky-Pudlak syndrome pulmonary fibrosis. Am J Respir Cell Mol Biol. 2014;50:605–613. doi: 10.1165/rcmb.2013-0025OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peiffer DA, Le JM, Steemers FJ, Chang W, Jenniges T, et al. High-resolution genomic profiling of chromosomal aberrations using Infinium whole-genome genotyping. Genome Res. 2006;16:1136–1148. doi: 10.1101/gr.5402306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Steemers FJ, Gunderson KL. Whole genome genotyping technologies on the BeadArray platform. Biotechnol J. 2007;2:41–49. doi: 10.1002/biot.200600213. [DOI] [PubMed] [Google Scholar]
- 21.Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Teer JK, Green ED, Mullikin JC, Biesecker LG. VarSifter: visualizing and analyzing exome-scale sequence variation data on a desktop computer. Bioinformatics. 2012;28:599–600. doi: 10.1093/bioinformatics/btr711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brogna S, Wen J. Nonsense-mediated decay (NMD) mechanisms. Nat Struct Mol Biol. 2009;16:107–13. doi: 10.1038/nsmb.1550. [DOI] [PubMed] [Google Scholar]
- 24.Starcevic M, Dell’Angelica EC. Identification of snapin and three novel proteins (BLOS1, BLOS2, and BLOS3/reduced pigmentation) as subunits of biogenesis of lysosome-related organelles complex-1 (BLOC-1) J Biol Chem. 2004;279:28393–401. doi: 10.1074/jbc.M402513200. [DOI] [PubMed] [Google Scholar]
- 25.Nazarian R, Huizing M, Helip-Wooley A, Starcevic M, Gahl WA, Dell’Angelica EC. An immunoblotting assay to facilitate the molecular diagnosis of Hermansky-Pudlak syndrome. Mol Genet Metab. 2008;93:134–44. doi: 10.1016/j.ymgme.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gautam R, Chintala S, Li W, Zhang Q, Tan J, Novak EK, Di Pietro SM, Dell’Angelica EC, Swank RT. The Hermansky–Pudlak syndrome 3 (cocoa) protein is a component of the biogenesis of lysosome-related organelles complex-2 (BLOC-2) J Biol Chem. 2004;279:12935–42. doi: 10.1074/jbc.M311311200. [DOI] [PubMed] [Google Scholar]
- 27.Suzuki T, Li W, Zhang Q, Karim A, Novak EK, Sviderskaya EV, Hill SP, Bennett DC, Levin AV, Nieuwenhuis HK, Fong CT, Castellan C, Miterski B, Swank RT, Spritz RA. Hermansky-Pudlak syndrome is caused by mutations in HPS4, the human homolog of the mouse light-ear gene. Nat Genet. 2002;30:321–4. doi: 10.1038/ng835. [DOI] [PubMed] [Google Scholar]
- 28.Carmona-Rivera C, Simeonov DR, Cardillo ND, Gahl WA, Cadilla CL. A divalent interaction between HPS1 and HPS4 is required for the formation of the biogenesis of lysosome-related organelle complex-3 (BLOC-3) Biochim Biophys Acta. 2013;1833:468–478. doi: 10.1016/j.bbamcr.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dell’Angelica EC, Shotelersuk V, Aguilar RC, Gahl WA, Bonifacino JS. Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the beta 3A subunit of the AP-3 adaptor. Mol Cell. 1999;3:11–21. doi: 10.1016/s1097-2765(00)80170-7. [DOI] [PubMed] [Google Scholar]