
FIG 8 .
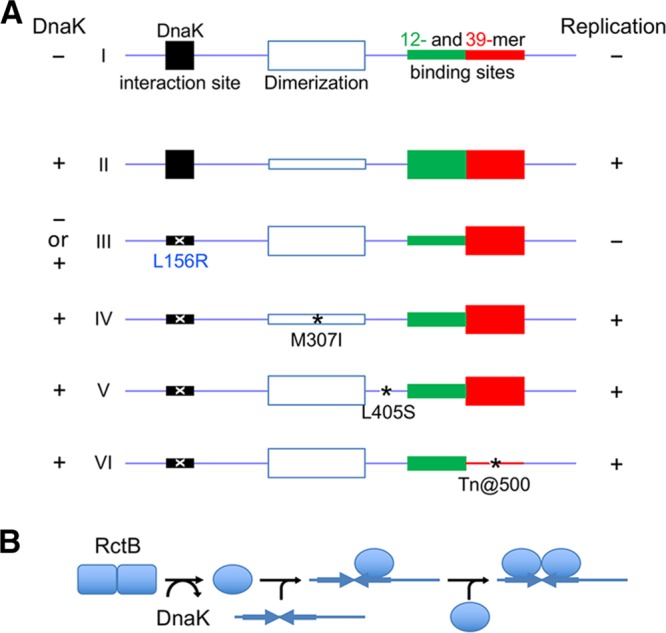
(A) A pictorial summary of activation of DNA binding of RctB by DnaK. Functional regions of RctB are represented as rectangles on a linear map of the initiator. Thinner rectangles indicate reduced activities. (I) In the absence of DnaK, both the 12-mer and 39-mer binding sites remain inactive but for different reasons: the former site cannot fold properly without interaction with DnaK, and the latter is inhibited by the DnaK interaction site (K-I site), possibly by direct contacts (autoinhibition). The improperly folded 12-mer region cannot support replication. (II) The autoinhibition is released by shielding of the K-I site by DnaK binding. DnaK interaction remodels RctB, which reduces dimerization and activates the monomers for DNA binding. (Note, however, that reduction in dimerization by mutating the dimerization domain does not obviate remodeling of monomers [not shown]). (III) The autoinhibition is also released by changes such as that represented by L156R in the K-I site. The mutation prevents DnaK interaction, and, without DnaK participation, the 12-mer region remains inactive. (IV to VI) The initiation defect due to the L156R change can be suppressed by second-site changes (*) that partially activate the 12-mer binding region but primarily reduce negative regulatory activities such as dimerization (IV), an uncharacterized activity from a WH domain (V), and 39-mer binding (VI). Full 12-mer activity, as well as a reduction in one of the negative activities, causes the copy-up phenotype (not shown). (B) Sequential binding of remodeled RctB monomer to a 12-mer. DnaK remodels RctB into a form that reduces its dimerization. The monomers bind to one half of an inverted repeat present in 12-mer sites. Upon an increase of remodeled RctB levels, both half-sites are occupied either by binding of a second monomer or binding of a dimer formed in solution (not shown). The details of protein-protein and DNA-protein interactions remain speculative.