

Abstract
The development of clean and renewable energy materials as alternatives to fossil fuels is foreseen as a potential solution to the crucial problems of environmental pollution and energy shortages. Hydrogen is an ideal energy material for the future, and water splitting using solar/electrical energy is one way to generate hydrogen. Metal‐organic frameworks (MOFs) are a class of porous materials with unique properties that have received rapidly growing attention in recent years for applications in water splitting due to their remarkable design flexibility, ultra‐large surface‐to‐volume ratios and tunable pore channels. This review focuses on recent progress in the application of MOFs in electrocatalytic and photocatalytic water splitting for hydrogen generation, including both oxygen and hydrogen evolution. It starts with the fundamentals of electrocatalytic and photocatalytic water splitting and the related factors to determine the catalytic activity. The recent progress in the exploitation of MOFs for water splitting is then summarized, and strategies for designing MOF‐based catalysts for electrocatalytic and photocatalytic water splitting are presented. Finally, major challenges in the field of water splitting are highlighted, and some perspectives of MOF‐based catalysts for water splitting are proposed.
Keywords: catalysis, hydrogen evolution reaction, metal‐organic frameworks, oxygen evolution reaction, water splitting
1. Introduction
The global demand for energy has increased rapidly and continuously in recent decades due to the quickly expanding human population and industrialization; as a result, there has been a significant increase in the utilization of traditional fossil fuels, which has caused severe environmental problems, such as the greenhouse effect, air pollution and water pollution. The quickly expanded energy consumption has resulted in major concerns about energy crises due to the limited fossil fuels resources. For a sustainable future, the development of alternative energy material that is clean and sustainable is highly desirable but remains a major challenge. Among the various energy carriers (materials), hydrogen is one of the most ideal and cleanest energy materials for the future due to its high gravimetric energy density (120 vs. 44 MJ kg−1 for gasoline), high combustion efficiency, non‐toxicity, clean exhaust products, and renewable and storable nature. During the past two decades, tremendous attention has been given to the field of hydrogen energy by researchers and governments around the world. However, the success of the hydrogen economy is strongly determined by the availability of useful routes for the large‐scale generation of hydrogen. Currently, the production of hydrogen mainly relies on steam reforming and partial oxidation of fossil fuels (natural gas or other hydrocarbons), causing concerns about serious CO2 emissions and limited natural resources.1, 2, 3 Water, one of the most abundant resources on earth, is composed of hydrogen and oxygen atoms. Water splitting is one of the most effective ways to produce hydrogen. Among the various routes for hydrogen generation from water at low temperature, direct water splitting using solar/electrical energy over photocatalysts/electrocatalysts is highly promising because of its sustainability.4, 5, 6, 7, 8, 9
Water splitting (H2O → H2 + 1/2O2) consists of two half reactions, known as the oxygen evolution reaction (OER) and the hydrogen evolution reaction (HER). However, these reactions have sluggish kinetics and require catalysts. In the electrochemical process, the OER and the HER are generally catalyzed by precious metal (Ir/Ru and Pt, respectively) materials to achieve favorable reaction kinetics.10, 11, 12, 13 Unfortunately, noble metals suffer from low abundance and high cost, hindering their large‐scale use in water electrolysis. To ensure sustainable hydrogen production, it is of great importance to seek earth‐abundant alternatives to precious metal‐based catalysts with excellent activity and robust stability.14, 15, 16, 17, 18, 19, 20, 21, 22 For example, several electrocatalysts composed of earth‐abundant elements (e.g., Fe, Co and Ni) were found to be promising alternatives to precious catalysts, achieving high OER and HER activity.14, 15, 16, 17, 18 In addition, some carbon‐based or heteroatom‐doped carbon materials have been evaluated as innovative options as electrocatalysts for the OER and HER.19, 20, 21, 22 The availability of different carbons (nanotubes, graphene, etc.) with adjustable compositions has markedly increased the number of candidates for OER/HER electrocatalysis.23, 24, 25 Solar‐driven H2 generation from water using semiconductor‐based photocatalysts is another attractive route to solve the energy and environmental problems.26, 27, 28, 29, 30 To date, a number of metal oxide‐based photocatalysts have been demonstrated to be effective for water splitting under UV light irradiation.31, 32, 33 In particular, TiO2 has been reported as a benchmark for the UV‐light‐driven water splitting reactions due to its good photo‐stability, low toxicity, large abundance and low cost. Unfortunately, TiO2 has a large band gap of 3.2 eV, which can only be used in the UV light range, which includes only 5% of all solar energy (solar conversion efficiency in UV light is only 2% compared with 16% when visible light up to 600 nm can be utilized). Thus, the development of new photocatalysts with high photocatalytic activity under visible light irradiation is one of the most attractive research topics in photocatalytic water splitting.34, 35, 36, 37 In addition to the material composition, the activity of catalysts for electrocatalytic/photocatalytic water splitting relies heavily on the morphology of the catalyst.38, 39, 40, 41 Thus, optimizing the catalyst composition and morphological structure is of critical importance to achieve highly efficient hydrogen production from water splitting.
Metal‐organic frameworks (MOFs) are a new class of porous materials with unique electronic, optical and catalytic properties.42, 43 In addition, they can be used as precursors for the fabrication of various metal, metal oxide‐carbon composites or pure carbon materials with rich morphological structures and versatile properties.43 In applications as electrocatalysts or photocatalysts or their precursors, MOFs offer several advantages, such as high design flexibility, tunable pore channels, large surface‐to‐volume ratios, flexibility to be functionalized with various ligands and metal centers, and rich compositions.43 The metal centers separated by organic linkers in MOFs can be considered as quantum dots; consequently, short diffusion lengths of the charge carriers can be achieved during the electrocatalytic and photocatalytic reactions.44 The specific surface areas and band gaps of MOFs can be tailored by tuning the organic ligands and/or metal centers, so their electrocatalytic and photocatalytic activities can be tailored to maximize their performance. In recent years, MOFs have been exploited directly as photocatalysts or as their precursors for hydrogen generation from water splitting, the degradation of organic pollutants and the reduction of CO2 into useful fuels.45, 46, 47, 48, 49 Recently, MOF‐based materials have also proved to be particularly suitable for electrocatalytic water splitting.50, 51, 52 In the last five years, tremendous efforts have been made to apply MOFs as photocatalysts and electrocatalysts for water splitting, and interest in this research field is projected to continue increasing. Thus, a review of the recent advances and challenges of MOF‐based materials in photocatalytic and electrocatalytic water splitting is highly desirable.
Herein, the recent development of MOF‐based materials for electrocatalytic and photocatalytic water splitting reactions is presented. Several critical factors that determine the activity for water splitting reactions are summarized, and strategies related to the design of catalysts are emphasized. Major challenges in the fields of photocatalytic and electrocatalytic water splitting are highlighted, and some perspectives from the current progress in the development of MOF‐based catalysts are given. Directions of the future research are also presented, with emphasis on achieving the desired MOF functionality and establishing structure‐property relationships to identify and rationalize the factors that determine the catalytic performance. This paper aims to provide a comprehensive review of the recent progress in this dynamic field, as well as some guidelines for the further development of highly efficient photocatalysts and electrocatalysts based on MOFs for water splitting.
2. Fundamentals of Water Splitting Reactions
2.1. Electrocatalytic Water Splitting
2.1.1. Basic Principles
Electrocatalytic water splitting involves two half reactions (OER and HER), and the mechanistic schemes of the OER and HER have been proposed in the literature.53, 54, 55, 56 The OER, which is a four‐electron process, is more complex than the HER and involves several surface‐adsorbed intermediates. In the following section, we mainly focus on the mechanistic study of the OER while that of the HER is described only briefly.
In the HER, the chemical adsorption and desorption of H atoms are competitive processes. A good HER catalyst should have a bond with the adsorbed H* (the asterisk indicates a bond to the catalyst surface) that is sufficiently strong to enable the proton‐electron‐transfer process and also sufficiently weak to ensure easy bond breaking and release of the produced H2 gas.53 The change in the Gibbs free energy for H* adsorption on an electrocatalyst surface (Δ) can be applied to evaluate both H* adsorption and H2 desorption using the HER free energy diagram.54 The optimal Δ should be zero, under which condition the HER reaches the maximum rate.53 More importantly, a “volcano curve” correlation has been proposed between the experimental HER activity (HER exchange current density) and the quantum chemistry‐derived Δ for various catalyst surfaces.54 As a result, the relationship between the nature of the electrocatalyst surface and the HER kinetics can be established.
The OER pathways, in acidic or alkaline media, include elementary steps that differ according to different mechanisms, yet all involve the adsorption/desorption of intermediates, such as HO*, O* and HOO*.55, 56, 57 The free adsorption energies of the OER intermediates at selected potentials on Pt (111) and Au (111) and some other metals were studied in acidic environment by Rossmeisl et al. using density functional theory (DFT) calculations.58 The most difficult step in the OER is the formation of HOO* on the metal surface by splitting water on an adsorbed oxygen atom (O*). This step is downhill in free energy at high electrode potentials. At lower potentials, although water can dissociate to O*, the OER is initiated only on the oxidized surface, which makes this step slower than the O* formation process. In other words, the formation of OOH* from O* is uphill for the OER at the equilibrium potential of 1.23 V vs. reversible hydrogen electrode (RHE). Applying a voltage to move the potential positively away from 1.23 V (the difference defined as the overpotential) is thus necessary for spontaneous OER. The calculations show that the OER on Pt and Au surfaces should start at approximately 1.8 V. Simple linear relations between the stability of different intermediates and OER activity were found when the analysis was extended to other metals, which suggests that the oxygen adsorption energy is a good descriptor of the capability of a metal‐based electrocatalyst for the OER.58
In addition to metallic catalysts, the OER mechanism on oxide catalysts has also been studied using computational methods.59 Rossmeisl and co‐workers investigated the trends in the electrocatalytic properties of the most stable (110) surfaces of RuO2, IrO2 and TiO2. Similar to the findings on metal surfaces, the binding energies of O*, HO* and HOO* on the (110) surfaces of these rutile oxides showed universal linear relations. Based on this, a volcano plot was constructed to describe the trends in OER activity according to a simplified descriptor, the O* binding energy. It was found that RuO2 binds oxygen slightly too weakly, while IrO2 binds oxygen too strongly, leading to a higher overpotential, which was also observed in experiments.60 However, TiO2 binds O* too weakly, and it displays a low OER activity. These results suggest that a material that binds oxygen slightly more strongly than RuO2 is expected to exhibit even better OER activity.
The origin of the overpotential for OER catalysis was also studied using DFT calculations on various oxides.61 A universal scaling relation between the binding energies of the HOO* and HO* intermediates was identified, which defined the lowest theoretical overpotential for the OER on oxide surfaces. This led to a general description of OER activity with the introduction of a single descriptor (ΔG O*−ΔG HO*). For the oxides considered, the OER activity could not be greatly enhanced beyond RuO2 by tailoring the binding between the intermediates and the oxide surface. To avoid the limitations defined by the universal scaling relationship, relative stabilization of HOO* compared to HO* must be achieved. In this regard, three‐dimensional (3D) structures are likely to stabilize HOO*.
2.1.2. Factors to Determine the Electrocatalytic Activity
Generally, the catalytic activity of an electrocatalyst for water splitting is determined by the intrinsic activity and the number of active sites. For oxide‐based electrocatalysts, the intrinsic activity is often related to the material composition, mixed valence states of the compositional cations (redox couples), crystal structure, metal‐oxygen bond energy, oxygen vacancy concentration, electronic conductivity and charge transfer capability.56, 62, 63, 64, 65 The number of active sites can be increased by building high‐surface‐area structures, tuning the morphology and creating nanostructured catalytic systems. Compositing with other catalytic materials or conductive supports can result in hybrids with enhanced activity and more active sites, which is sometimes known as the synergistic effect. The most effective methods to maximize the HER/OER activity include tailoring the surface and/or bulk properties (by the selection of cations and anions), optimizing the morphology (by the use of advanced synthetic procedures), enhancing the charge transfer process (by the functional modification of the surface electronic structure) and forming composite or hybrid catalysts. These methods may produce more active sites for HER/OER and ideal pathways for the transportation of reactants and gaseous products (i.e., hydrogen and oxygen). The strategies for enhancing electrocatalytic activity are not limited to oxides and can be, in principle, applied to other types of electrocatalysts. Additionally, researchers often take advantage of several combined strategies to improve the efficiency of electrocatalysts in the HER/OER.
The morphology and microstructure are crucial characteristics for electrocatalysts because they have a direct correlation with the number of active sites and, therefore, the catalytic activity.66, 67 For example, a simple self‐template strategy was developed to fabricate hollow Co‐based bimetallic sulfide (MxCo3−xS4, M = Zn, Ni and Cu) polyhedra from homogenous bimetallic MOFs.66 The combination of polyhedral morphology, hollow structure, homo‐incorporation of a second metal element and high Brunauer‐Emmett‐Teller (BET) surface area significantly enhanced the HER activity of Co3S4. The hollow Zn0.30Co2.70S4 exhibited the highest catalytic activity among the four electrocatalysts, indicating that cation selection is very important to achieve high electrocatalytic activity for water splitting.
In addition, nanostructured electrocatalysts generally benefit from increased specific surface area and, therefore, have more active sites for the electrocatalysis, which can be tailored by the preparation methods and annealing conditions.68, 69 For example, Shi et al. utilized an in situ carburization method to prepare MoC encapsulated by a graphitized carbon shell (nanoMoC@GS) electrocatalyst from a Mo‐based MOF.68 The nanoMoC@GS showed favorable activity in acidic media as an electrocatalyst for HER, which stemmed from the synergistic effects of the ultrafine MoC, ultrathin and conductive GS, high porosity and high surface area.68
Other methods to improve the surface area, such as synthesizing nanoparticles (NPs) and combining NPs with high‐surface‐area supports, have been used to enhance the activity of electrocatalysts for water splitting.70, 71 For example, Li et al. synthesized a nitrogen‐doped Fe/Fe3C@graphitic layer/carbon nanotube hybrid (Fe/Fe3C@NGL‐NCNT) using MIL‐101 (Fe) MOF as the precursor.70 This Fe/Fe3C@NGL‐NCNT hybrid showed superior OER activity and stability compared with the commercial Pt/C, which may originate from the abundant active sites and the synergistic effect of the unique architecture.
The charge transfer capability is also essential for achieving high electrocatalytic activity for water splitting, and the coupling of some functional materials, such as reduced graphene oxide (RGO), to MOF‐based electrocatalysts can improve the charge transfer capability (conductivity).72, 73 For example, Tang et al. used a simple pyrolyzing method to synthesize a porous Mo‐based hybrid from a polyoxometalate‐based MOF and graphene oxide (POMOFs/GO), which showed improved performance for the HER.73
2.2. Photocatalytic Water Splitting
2.2.1. Mechanism and Reaction Steps
Studies on splitting water into hydrogen and oxygen using light (photons) originated from the discovery of the Honda‐Fujishima effect in 1967. Water splitting using photocatalysts has since been widely investigated.74, 75, 76, 77, 78 Previous reviews of water splitting using semiconductors as photocatalysts have demonstrated the basic principles of the water splitting process.76, 77, 78 The electrons in the valence band (VB) of the photocatalyst are transferred to the conduction band (CB), and holes are left in the VB after absorbing UV and/or visible light, creating electron‐hole pairs. The photogenerated electron‐hole pairs can induce redox reactions similar to water electrolysis. Specifically, water molecules are reduced by the electrons to generate H2 and are oxidized by the holes to produce O2, completing the water splitting reactions.
Water splitting into H2 and O2 is an energetically uphill reaction with a standard Gibbs free energy change (ΔG) of +237 kJ mol−1 (corresponding to 1.23 eV). Therefore, the band gap of the photocatalysts and the edges of the CB and VB must be suitable for water splitting. The bottom level of the CB should be more negative than the redox potential of H+/H2 (0.0 V vs. normal hydrogen electrode (NHE)), while the top level of the VB should be more positive than the redox potential of O2/H2O (1.23 V vs. NHE).76 The theoretical minimum band gap for water splitting is therefore 1.23 eV, which is equivalent to a light wavelength of approximately 1100 nm. However, not all semiconductors meet the requirements for water splitting. For metal oxide‐based photocatalysts, the VB mainly consists of O 2p orbitals, and the top level of the VB is much higher than 1.23 V vs. NHE. Therefore, the oxidation‐reduction potentials (ORPs) of O2/H2O and H+/H2 are positioned between the top level of the VB and the bottom level of the CB. Higher photon energy than the band gap of the photocatalyst is needed due to an activation barrier in the charge transfer process in the water splitting reactions. At the same time, the much wider band gaps of these materials make them only photoactive in UV light. As approximately 50% of the solar spectrum consists of visible photons (400 < λ < 800 nm), it is critical to develop active photocatalysts with high activity under visible light for photocatalytic water splitting.
The development of photocatalysts is very important to enable water splitting with visible light. The main steps in the photocatalytic water splitting reactions should be tailored to meet the requirements for photocatalysts capable of water splitting. There are three steps in the photocatalytic water splitting reactions, which has been demonstrated in some informative reviews.76, 77, 78 The first step is the formation of electron‐hole pairs by incident photons. When the energy of incident light is greater than the band gap energy, the electrons in the VBs can be excited and transferred into the CB. Meanwhile, holes are generated in the VB. However, the band structure is only a thermodynamic requirement. Other factors, such as charge separation, mobility and the lifetime of photogenerated electrons and holes can also affect the photocatalytic activity for water splitting. The second step is charge separation and diffusion to the catalyst surface without recombination of the photogenerated carriers, which is drastically affected by the crystal structure, particle size and crystallinity of the photocatalyst.79, 80 A higher crystallinity can lead to superior charge migration efficiency because the defects in a photocatalyst with lower crystallinity act as recombination centers for the photogenerated electron‐hole pairs, which decreases the photocatalytic activity. A smaller particle size of the photocatalyst also suppresses the possibility of electron‐hole pair recombination. The final step is the reduction and oxidation of surface‐adsorbed species by the photogenerated electrons and holes to generate H2 and O2, respectively. In this step, the surface active sites of the photocatalyst play vital roles in efficient water splitting. Co‐catalysts, such as Pt, are usually loaded onto the photocatalyst surface as active sites to reduce the activation energy for the HER.81 These processes affect the overall efficiency of water splitting based on a semiconductor‐based photocatalyst.
2.2.2. Factors to Determine the Photocatalytic Activity
The main factors that determine the photocatalytic activity of the photocatalysts for water splitting include the band gap energy/visible light absorption capability, active sites/co‐catalysts and charge transfer/separation efficiency. Covalent modification is a method to reduce the band gap energy of photocatalysts. For example, the band gap of MOFs can be reduced by a diazo coupling with amino‐substituted ligands and other molecules.82 The photocatalytic activity of these modified MOFs corresponds to a red shift of the absorption edge, suggesting that the band gap energy/visible light absorption capability plays a vital role in the photocatalytic activity.
The incorporation of active co‐catalysts is an effective way to increase the number of active sites for water splitting.83, 84, 85, 86, 87 For example, the well‐defined cages of MIL‐101(Cr) MOF were used to engage the molecules of a high‐valent di‐µ‐oxo dimanganese catalyst with high activity for photo‐electrochemical (PEC) water oxidation and the incorporation of MnTD ([(terpy)Mn(µ‐O)2Mn](terpy)]3+; terpy: 2,2′:6′,2′′‐terpyridine) improved the turnover number of MIL‐101(Cr) more than 20‐fold while maintaining the initial high rate in the PEC water oxidation reaction.83 In another study, Hansen and Das found that MnTD⊂MIL‐101(Cr) showed superior activity to MIL‐101(Cr) and MnO2 catalysts for the OER.84 These studies suggested that the incorporation of active co‐catalysts can greatly enhance the photocatalytic activity for water splitting, and the selection and incorporation method of the co‐catalysts should be optimized.
The charge transfer/separation efficiency of the photocatalyst plays a critical role in photocatalytic water splitting. The construction of heterojunctions is an effective way to enhance the charge transfer/separation capability of electron‐hole pairs.88, 89 For example, a MOF‐derived Co3O4/TiO2 composite photocatalyst with 2 wt.% Co loading and a p‐n heterojunction, exhibited a much higher hydrogen evolution rate than the conventional Co3O4/TiO2 nanocomposite (≈ 7‐fold enhancement).88
The photocatalytic activity of MOF‐based photocatalysts is determined by several crucial factors, such as the band gap energy, active sites/co‐catalyst and charge transfer capability. These factors are often closely related. For example, an azo‐carboxylic acid can be used as an organic linker to construct a Gd‐based MOF with a reduced band gap.90 Gd‐MOF has high photocatalytic activity for the HER due to its high visible light absorption capability. The addition of Ag co‐catalyst improved the HER activity of Gd‐MOF by providing more active sites and improving the charge transfer capability.
3. Recent Advances in MOF‐Based Catalysts for Water Splitting
Because of the many outstanding features of MOFs, such as tunable pore channels, high specific surface area, easy tailoring of the material composition, rich morphological structure, and capability to act as precursors for the preparation of various metal/metal oxide/carbon composites and carbon materials of various properties, during the past five years, the applications of MOFs as catalysts or the precursors of catalysts for electrocatalytic and photocatalytic water splitting reactions for hydrogen generation have been extensively exploited. Both the direct application of MOFs for water splitting and application as a precursor for metal/metal oxide/carbon composites or porous carbon materials (by leaching of the metal/metal oxide from the composites), which were then applied as electrocatalysts or photocatalysts, have been reported. Additionally, MOFs were studied as catalysts for both the OER and HER, and the reactions were conducted in acidic and alkaline electrolytes.
3.1. Electrocatalytic Water Splitting
For electrocatalytic water splitting, the direct application of MOFs as electrocatalysts was first reported in 2011 by Nohra et al., who pioneered the use of polyoxometalate‐based MOFs (POMOFs) for the HER.91 The structural properties were investigated but their electrocatalytic activity was only briefly studied and the efficiency of POMOFs to replace Pt catalyst for the HER was not clearly demonstrated. In 2015, Qin et al. reported a type of POMOFs called [TBA]3[ε‐PMoV 8MoVI 4O36(OH)4Zn4][BTB]4/3·xGuest (NENU‐500, BTB = benzene tribenzoate, TBA+ = tetrabutylammonium ion) as an ultrastable electrocatalyst for the HER.92 It displayed a Tafel slope of 96 mV dec−1 and an overpotential of 237 mV at a current density of 10 mA cm−2 (a metric associated with solar fuel synthesis), which was inferior to Pt/C (Tafel slope of 30 mV dec−1 and overpotential of 52 mV at 10 mA cm−2). Very recently, Dai et al. demonstrated MoSx anchored on Zr‐MOF (UiO‐66‐NH2) prepared by a solvothermal method for the HER.93 The introduction of MoSx nanosheets to the MOFs dramatically enhanced the HER activity due to the improved electron transport, the increased number of active sites and the favorable delivery of local protons in the Zr‐MOF structure. By optimizing the MoSx amount, the MoSx‐MOF composite with a Mo/Zr ratio of 0.5 displayed remarkable HER activity, with a Tafel slope of 59 mV dec−1, which was only slightly higher than that of Pt/C (32 mV dec−1).93
In the study of MOFs as precursors for the preparation of electrocatalysts for water splitting reactions, Chaikittisilp and co‐workers were the first to use a Co‐based MOF (zeolitic imidazolate framework‐9, ZIF‐9) as a precursor for the preparation of a nanoporous CoxOy‐C hybrid as an electrocatalyst for the OER.94 The conversion of ZIF‐9 to the CoxOy‐C hybrid is shown in Figure 1 a. As depicted in Figure 1b, for the OER, the Z9‐700‐250 and Z9‐800‐250 electrocatalysts exhibited more negative onset potentials and higher current densities than Z9‐900‐250 and Pt/carbon black. These results indicated that the Z9‐800‐250 hybrid is a promising electrocatalyst for the OER. Very recently, Aijaz et al. reported a highly active electrocatalyst for the OER comprising core‐shell Co@Co3O4 NPs embedded in CNT‐grafted N‐doped carbon‐polyhedra, which was obtained by the pyrolysis of a Co‐based MOF in H2 atmosphere and a subsequent controlled oxidative calcination.95 This electrocatalyst displayed an overpotential of 410 mV at 10 mA cm−2, comparable to RuO2, which has been demonstrated as the benchmark electrocatalyst for the OER.
Figure 1.
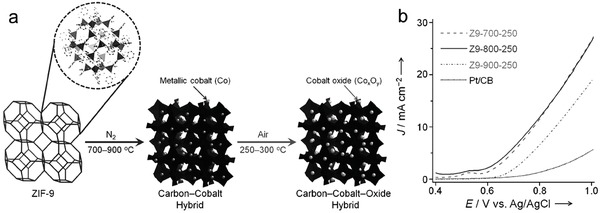
a) Schematic presentation of the formation of CoxOy‐C hybrids through the two‐step thermal treatment of ZIF‐9. b) Polarization curves of Z9‐700‐250, Z9‐800‐250, Z9‐900‐250, and Pt/CB as electrocatalysts for the OER in 0.1 M KOH. The resulting hybrids were designated as Z9‐x‐y, where x and y stand for the thermal treatment temperatures in the first and second steps, respectively. Reproduced with permission.94
Although nanoporous carbon was successfully synthesized from MOFs in 200896 and was widely used in the oxygen reduction reaction (ORR),97, 98, 99 the direct use of nanoporous carbon derived from MOFs in water splitting was demonstrated only recently by Xia et al.100 In that study, the pyrolysis synthesis of hollow nitrogen‐doped carbon nanotube frameworks (NCNTFs) derived from a Co‐based MOF (ZIF‐67) was conducted, which provided the C and N source for the growth of N‐doped CNT catalyzed by the metallic Co NPs formed in situ and served as the template for the formation of the hollow framework.100 The as‐prepared NCNTFs exhibited remarkable electrocatalytic activity and stability for the OER in an alkaline medium.
Because the catalytic activity of the electrocatalysts for the HER and OER is strongly related to the morphology/microstructure, the number of active sites, the BET surface area and the charge transfer capability, modifications of MOF‐based or MOF‐derived electrocatalysts with special morphologies/nanostructures, high surface areas, abundant active sites and excellent charge transfer capability has been exploited in the last five years. In the following section, recent progress in the design of MOFs as catalysts for electrocatalytic water splitting is summarized.
3.1.1. Morphology Control/Nanostructuring
Among the various parameters, the morphology of the electrocatalyst, which can provide more active sites and enhance the surface adsorption capability of the reactants, plays a crucial role in the activity for water splitting.38, 39, 101 In general, the morphology of MOF‐based catalysts can be tuned through the preparation method. For example, a Zn‐based MOF, MOF‐5, was prepared by an ionic liquid (IL)‐based method and displayed a distinct flower‐shaped morphology with a diameter of approximately 10 µm, very different from the regular cubic structure of MOF‐5 prepared by traditional methods. The as‐prepared MOF‐5(IL) displayed superior activity for the HER compared to cubic MOF‐5 due to the enhanced oxidation desorption reaction of the hydrogen atoms.101
In addition to the direct use of MOFs in electrocatalytic water splitting, MOFs have also been applied as precursors for the synthesis of electrocatalysts with controlled particle sizes and morphologies.102, 103, 104, 105 The morphology of the electrocatalysts derived from MOFs precursors can be tailored through the choice of the type of MOF and the temperature and atmosphere used for the subsequent calcination. For example, a CoP electrocatalyst with a concave polyhedrons (CPHs) morphology was synthesized by topological conversion using ZIF‐67 polyhedrons as the precursor.102 The morphology of the CoP CPHs is shown in Figure 2 a. CoP NPs synthesized from direct calcination of Co(NO3)2 show a different morphology (Figure 2b). For the HER, the CoP CPHs electrocatalyst showed a low overpotential of 133 mV at 10 mA cm−2, while the CoP NPs had an overpotential of 187 mV under the same conditions. In addition, the Tafel slopes of the CoP CPHs and CoP NPs are 51 and 63 mV dec−1, respectively. The electrocatalytic performance of the porous CoP CPHs is superior to most of the reported non‐noble‐metal‐based catalysts, such as CoP microspheres and FeP nanosheets, for the HER.106, 107
Figure 2.

Scanning electron microscopy (SEM) images of CoP CPHs (a) and CoP NPs (b). Reproduced with permission.102 Copyright 2015, Royal Society of Chemistry.
In addition to CoP CPHs, electrocatalysts with other controlled morphologies, such as nano‐octahedrons, spindle‐like 3D structures and porous nanocages, were also synthesized from MOFs and were investigated as electrocatalysts for the OER and HER, showing attractive activity.108, 109, 110 For example, Wu et al. introduced a MOF‐assisted strategy for the synthesis of MoCx nano‐octahedrons as electrocatalysts for the HER.108 The MoCx nano‐octahedrons (Figure 3 a) exhibited superior electrocatalytic activity and stability compared to irregular‐shaped MoCx NPs with a similar composition (Figure 3b) in both acidic and basic solutions. The overpotential of MoCx nano‐octahedrons was 87 and 92 mV at 1 mA cm−2 in acidic and basic solutions, respectively, while it was approximately 230 mV for the irregular‐shaped MoCx NPs in acidic and basic solutions.
Figure 3.

Transmission electron microscopy (TEM) images of MoCx nano‐octahedrons (a) and MoCx NPs (b). Reproduced with permission.108 Copyright 2015 Nature Publishing Group.
In another study, MOF‐derived Ni‐Co‐based metal oxides with nanocage morphology were compared with a catalyst with a nanocube morphology and the same composition and comparable surface area (≈31 m2 g−1) for water splitting.110 Ni‐Co Prussian‐blue‐analog (PBA) nanocages were derived from the Ni‐Co‐PBA nanocubes as precursors and were converted to Ni‐Co mixed oxides by calcination in air. As shown in Figure 4 a,b, the Ni‐Co mixed oxides inherited the nanocage morphology of the Ni‐Co PBA particles but had rougher surfaces than the pristine Ni‐Co mixed oxide nanocubes. As shown in Figure 4c,d, the Ni‐Co mixed oxide nanocages exhibited lower onset potential, lower Tafel slope and higher current density than the nanocubes for the OER. The enhanced OER activity of the Ni‐Co mixed oxide nanocages was assigned to the hollow and porous nanocage morphology.
Figure 4.
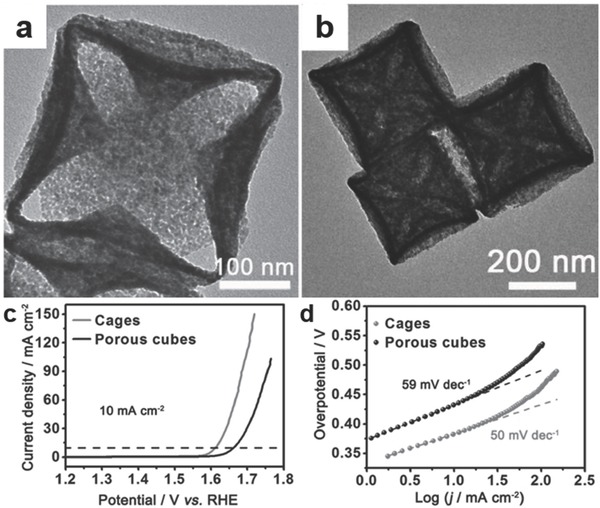
TEM images of the as‐prepared Ni‐Co oxide nanocages (a) and porous nanocubes (b); Polarization curves (c) and Tafel plots (d) of the Ni‐Co mixed oxide nanocages and porous nanocubes for the OER. Reproduced with permission.110
3.1.2. Constructing Hybrids/Composites
Because of the large variation of organic moieties in MOFs, highly nanoporous carbon may be formed by thermal carbonization of MOFs in inert atmospheres.111, 112 Such MOF‐derived nanoporous carbon materials usually exhibit exceptionally high surface areas, which can form strong coupling with metal oxides/metal to enhance the electrocatalytic activity and stability for water splitting.67, 70, 113, 114, 115, 116, 117 Thus, many investigations have attempted the synthesis of carbon‐metal/metal oxide composites from MOFs as electrocatalysts for the HER and OER. For example, hybrid porous nanowire arrays (NAs) composed of strongly interacting Co3O4 and carbon (Co3O4C‐NA) with a high specific surface area of 251 m2 g−1 and a large carbon content of 52.1 wt.% were successfully prepared by a facile carbonization of MOF grown on Cu foil,113 and displayed superior performance as the working electrode for OER without additional substrates or binders. A low onset potential of 1.47 V vs. RHE and a stable current density of 10 mA cm−2 at 1.52 V vs. RHE in 0.1 M KOH for 30 h were achieved. Co3O4 C‐NA also showed a much lower Tafel slope of 61 mV dec−1 than IrO2/C (87 mV dec−1) in 1 M KOH. For comparison, the carbon‐free counterpart (Co3O4‐NA), prepared by the calcination of Co3O4C‐NA in air, which exhibited a similar porous NA structure and a cubic spinel phase, displayed a higher onset potential of 1.50 V vs. RHE, a larger operating potential of 1.64 V vs. RHE at 10 mA cm−2 and a much higher Tafel slope of 123 mV dec−1. Co3O4C‐NA delivered superior OER activity and stability compared with the state‐of‐the‐art noble‐metal electrocatalyst.113
The strong coupling was utilized for the development of a MoS2‐based composite (MoS2/3D‐NPC) as the electrocatalyst for the HER, in which the MoS2 nanosheets grew in situ in the nanopores of 3D nanoporous carbon (3D‐NPC) derived from MOF and showed much better performance than the respective components (MoS2 NPs, 3D‐NPC) of the catalyst, as well as the physically mixed MoS2 NPs and NPC composite (MoS2+3D‐NPC).118 The MoS2/3D‐NPC composite displayed a small overpotential of 210 mV at 10 mA cm−2 for the HER, better than those of 3D‐NPC (>300 mV), MoS2 NPs (≈250 mV) and MoS2 +3D‐NPC (≈250 mV) as shown in Figure 5 a. The Tafel slope of the MoS2/3D‐NPC composite reached 51 mV dec−1, in contrast to 95 mV dec−1 for the MoS2 NPs as shown in Figure 5b. The high electrocatalytic activity of the MoS2/3D‐NPC electrocatalyst was attributed to the efficient charge transfer in the HER due to the more exposed active sites and robust interaction between the MoS2 and the MOF‐derived conductive carbon when the MoS2 nanosheets were grown in the pores of the 3D‐NPC.
Figure 5.
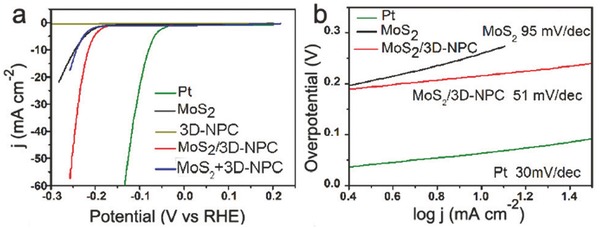
a) Polarization curves and b) Tafel plots of the MoS2/3D‐NPC composite, 3D‐NPC, MoS2 NPs, mixed MoS2+3D‐NPC and Pt. Reproduced with permission.118 Copyright 2015, Royal Society of Chemistry.
In another study, Lu et al. reported the development of a core (Au NP, ≈50–100 nm)‐shell (Zn‐Fe‐C, ≈30–60 nm) composite (Figure 6 a–c) prepared by direct pyrolysis of a Zn‐Fe‐MOF shell coated on an Au NP in an inert atmosphere.119 The core‐shell catalyst (Au@Zn‐Fe‐C) displayed a low onset potential of ‐0.08 V vs. RHE in 0.5 M H2SO4, which was much more positive than those of Au (‐0.225 V) and Zn‐Fe‐C (‐0.292 V) but slightly more negative than that of commercial Pt/C (‐0.006 V). The Tafel slope of Au@Zn‐Fe‐C was 130 mV dec−1, which was lower than the slopes of Au (167 mV dec−1) and Zn‐Fe‐C (271 mV dec−1). The onset overpotential and lower Tafel slope contributed to the more favorable HER kinetics of Au@Zn‐Fe‐C, demonstrating a synergistic effect between the Au NP core and Zn‐Fe‐C shell.
Figure 6.

TEM images (a, b) and energy‐dispersive X‐ray spectroscopy (EDX) mapping (c) of Au@Zn‐Fe‐C hybrids. Reproduced with permission.119 Copyright 2016 American Chemical Society.
3.1.3. Functional Modification
The charge transfer capability of the electrocatalyst plays a critical role in determining the activity for OER and HER. For MOF‐based catalysts, the addition of functional ions and/or ligands can enhance the charge transfer capability. For example, Wang et al. developed a new electrocatalyst by assembling Co ions and benzimidazolate ligands into a MOF (Co‐ZIF‐9) for the OER with high proton transfer capability.120 Co‐ZIF‐9 was effective for the electrocatalytic OER, and its turnover frequency (TOF) reached ≈1.76 × 10−3 s−1, which is similar to the active Co‐based electrocatalyst reported in the literature.121 Wang et al.'s work made an important step in water splitting chemistry by integrating the redox‐active metal centers and organic motifs into a MOF structure.
In addition to the design of MOFs, some carbon‐based functional additives were incorporated into MOF‐based catalysts to enhance the charge transfer capability.122, 123 For example, GO was used to modify the MOFs to enhance the charge transfer capability and improve the electrocatalytic activity for water splitting.123 The GO‐incorporated Cu‐MOF composite displayed good performance as a bifunctional catalyst for the HER and OER in 0.5 M H2SO4.123 The (GO 8 wt.%) Cu‐MOF exhibited the highest activity for the HER. The onset overpotential for the HER was 202 mV for Cu‐MOF and decreased from 123 to 87 mV as the GO amount increased from 2 to 8 wt.%. For the OER, the Tafel slope was 65 mV dec−1 for (GO 8 wt.%) Cu‐MOF and 89, 81 and 61 mV dec−1 for Cu‐MOF, (GO 2 wt.%) Cu‐MOF and Pt/C, respectively. The improved activity of the GO‐MOF composite for water splitting in acidic solution was attributed to the enhanced charge transfer capability and synergistic effects of GO and MOF.
Functional carbon‐based materials were also added to MOF‐based materials in the precursor stage for the preparation of highly active MOF‐derived electrocatalysts.73, 124 For example, Tang et al. synthesized an active electrocatalyst derived from a POMOFs/GO composite for the HER.73 The introduction of GO to POMOFs improved the conductivity and the HER activity of MoO2@PC‐RGO and functioned as a support for the formation of a closely connected network. This hybrid presented superior activity for the HER in acidic media due to the synergistic effects among the MoO2 NPs, PC and RGO substrates.73 MoO2@PC‐RGO initiated H2 evolution near its thermodynamic overpotential (0 mV), which was similar to that of the Pt/C catalyst, while MoO2@PC had an onset overpotential of 66 mV. The overpotentials at 10 mA cm−2 were 38 mV for Pt/C, 64 mV for MoO2@PC‐RGO and 136 mV for MoO2@PC. Tafel slopes of 30, 41 and 60 mV dec−1 were obtained for Pt/C, MoO2@PC‐RGO and MoO2@PC in 0.5 M H2SO4.
In another work, rGO was incorporated with CoP to enhance the charge transfer capability of CoP for the HER and OER in alkaline media.124 This layered composite was prepared by pyrolysis and phosphating processes with rationally designed sandwich‐type ZIF‐67 MOF/GO as a template and precursor, as shown in Figure 7 . The MOF‐derived porous CoP nanostructure guaranteed a large quantity of exposed active sites, and the close contact between CoP and rGO contributed to a continuous conductive network, which was beneficial for the electron transfer process. For the HER, CoP/rGO‐400 was more active than rGO and CoP due to the synergistic effect between the CoP and rGO in the composite. In 1 M KOH, a low Tafel slope of 38 mV dec−1 was achieved with the CoP/rGO‐400 electrocatalyst, comparable to that of Pt/C (36 mV dec−1) and much lower than CoP (60 mV dec−1). For the OER in the same alkaline solutions, CoP/rGO‐400 displayed a Tafel slope of 66 mV dec−1, which was superior to CoP, rGO and even the state‐of‐the‐art IrO2. These contributions shed light on the rational design of a series of RGO‐incorporated MOF‐based electrocatalysts for the OER and HER.
Figure 7.
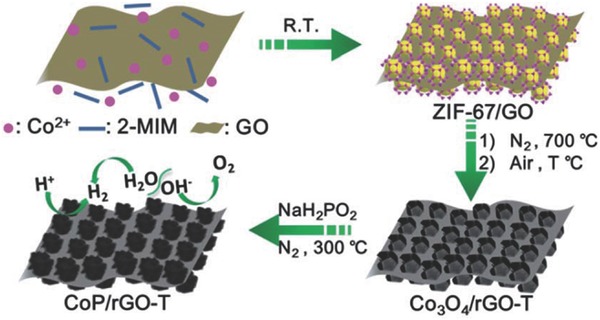
Illustration of the fabrication procedure for the CoP/rGO nanocomposite. Reproduced with permission.124 Copyright 2016, Royal Society of Chemistry.
3.1.4. Intrinsically Conductive MOFs
MOF is typically not conductive and may not be a great choice for electrocatalysis‐based applications. As such, one of the most challenging and rewarding endeavors in this field is to synthesize porous MOFs with good charge mobility and conductivity. Recently, the development of new type MOFs with enhanced intrinsic conductivity has attracted increasing attention.125, 126, 127 In 2009, Takaishi et al. reported one of the first conductive MOFs, Cu[Cu(pdt)2] (pdt = 2,3‐pyrazinedithiolate), and its electrical conductivity was 6 × 10−4 S cm−1 at 300 K.128 However, this kind of MOF collapsed upon desolvation. Thus, it is critical to develop alternative MOFs with high conductivity and structural stability for the potential electrocatalysis application.
Dincă and co‐workers have made great contributions to the development of conductive MOFs for the potential use in electrocatalysis‐based applications.129 For example, Sun et al. treated Mn2+ with 2,5‐disulfhydrylbenzene‐1,4‐dicarboxylic acid (H4DSBDC) and obtained isolated Mn2(DSBDC).130 This MOF was a thiolated analogue of Mn2(DOBDC) MOF derived from 2,5‐dihydroxybenzene‐1,4‐dicarboxylic acid (H4DOBDC). The porous Mn2(DSBDC) with one‐dimensional (1D) (–Mn–S–)∞ chains showed a high surface area (978 m2 g−1) and high charge mobility (0.01 cm2 V−1 s−1) similar to those of the most common organic semiconductors. Although Mn2(DSBDC) displayed a relatively high charge mobility, its conductivity was rather low (3.9 × 10−13 S cm−1 at 297 K). The replacement of d5 MnII by d6 FeII centers introduces high energy, loosely bound minority‐spin carriers and then enhances the conductivity.131 They found that the bulk electrical conductivity values of both Fe2(DSBDC) and Fe2(DOBDC) were ≈6 orders of magnitude higher than those of the Mn2+ analogues, Mn2(DOBDC) and Mn2(DSBDC), which was attributed to the loosely bound Fe2+β‐spin electron. This study can provide important insights for the rational design of conductive MOFs, highlighting in particular the advantages of iron for synthesizing conductive MOF‐based materials.
Besides Fe, Cd was also demonstrated to be a promising candidate to construct conductive MOFs. Park et al. found that isostructural MOFs M2(TTFTB) (M = Mn, Co, Zn and Cd; H4TTFTB = tetrathiafulvalene tetrabenzoate) exhibited a striking correlation between their single‐crystal conductivities and the shortest S···S interaction defined by neighboring tetrathiafulvalene (TTF) cores, which inversely correlated with the ionic radius of the metal ions.132 The Cd analogue, with the largest cation and shortest S···S contact, showed the highest electrical conductivity (2.86 × 10−4 S cm−1) that was 72 times higher than that of Zn2(TTFTB) (3.95 × 10−6 S cm−1). Mn2(TTFTB) and Co2(TTFTB), which display intermediate S···S distances between those observed in the Zn and Cd analogues, also showed intermediate conductivity values of 8.64 × 10−5 and 1.49 × 10−5 S cm−1, respectively, both tracking inversely with increasing S···S distance.
Highly conductive two‐dimensional (2D) MOFs made from nitrogen‐based ligands were reported in 2014 by Sheberla et al.133 They found that the reaction of NiCl2 with hexaaminotriphenylene (H6HATP) in aqueous NH3 solution led to the isolation of a new MOF, Ni3(HITP)2 (HITP = 2,3,6,7,10,11‐hexaiminotriphenylene). Ni3(HITP)2 films grown on a quartz substrate displayed a conductivity of 40 S cm−1 at room temperature, while pellets of the same material showed a bulk conductivity of 2 S cm−1. The isostructural material made from CuII, Cu3(HITP)2, also displayed a high bulk conductivity of 0.2 S cm−1 at room temperature.
The large bulk conductivity of HITP‐based materials enabled the application in chemiresistive sensing for ammonia vapor or volatile organic compounds (VOCs).134, 135 Cu3(HITP)2 functioned as reversible chemiresistive sensors, which was capable of detecting sub‐ppm levels of ammonia vapor.134 It was found that the chemiresistive response can be altered by the choice of metal node and Ni3(HITP)2 was unresponsive to ammonia, suggesting that the copper sites are critical for ammonia sensing. These studies highlight the utility of 2D conductive MOFs in the production of tunable functional materials, such as chemical sensors.
Very recently, Miner et al. investigated an intrinsically conductive Ni3(HITP)2 MOF as an active, well‐defined and tunable electrocatalyst toward the ORR in alkaline solutions.136 Ni3(HITP)2 exhibited ORR activity competitive with the most active Pt‐free electrocatalysts due to the combination of high crystallinity of MOFs, the physical durability and electrical conductivity of graphitic materials, and the diverse, well‐controlled synthetic accessibility of molecular species. In addition, the Ni3(HITP)2 MOF with high electrical conductivity is directly used as the electrode material in electrochemical double layer capacitors (EDLCs) without conductive additives or other binders.137 The performance of this MOF exceeded that of most carbon‐based materials with capacity retention greater than 90% over 10000 cycles, suggesting conductive MOFs as a new generation of active materials for supercapacitors. These studies highlight the direct use of conductive MOFs in fuel cells and supercapacitors, which can shed light to their future application in the electrocatalytic water splitting.
3.1.5. A Brief Summary
The catalytic activities of some typical MOFs and MOF‐derived electrocatalysts for the HER and/or OER are summarized in Table 1 . Taking advantage of the porous structures of MOF precursors, the MOF‐derived materials had various morphological features, such as polyhedrons, nanocubes, nanocages, nano‐octahedrons, nanosheets and nanowires, and outperformed their NP counterparts of similar compositions for catalyzing the water splitting reactions. Remarkably, in the HER under acidic conditions, porous CoP with a CPHs structure prepared from Co‐MOFs exhibited a >50 mV decrease in the overpotential required to reach 10 mA cm−2 compared to CoP NPs.102 As MOFs typically have abundant carbon and nitrogen sources, the carbonation of MOFs can lead to the formation of catalytically active metal‐based material composites with carbon materials having finely tuned porosity, graphitic degree and nitrogen content (if any). These hybrids have demonstrated good HER/OER performance, as shown in Table 1. Particularly, a strongly interacting Co3O4 and carbon hybrid grown on Cu foil directly catalyzed the OER with high efficiency, reaching 10 mA cm−2 at an overpotential as low as 290 mV in 0.1 M KOH.113 In addition to applying MOFs alone to synthesize electrocatalytic materials, GO and rGO were functionalized with MOFs to improve the charge transfer of the catalysts, as well as the catalytic performance. Despite the effectiveness of MOF‐derived materials in the HER and/or OER, most can only promote the HER in acidic media (while industrial water splitting favors an alkaline solution) and few can catalyze the overall water splitting process (both HER and OER) in the same electrolyte. Therefore, more efforts should be made to design bifunctional electrocatalysts from MOFs with the combined knowledge of MOF structures and the mechanism of the water splitting process. More importantly, current MOF‐derived catalysts can only sustain constant hydrogen or oxygen production within limited timescales, ranging from several hours to a few days. Robust MOF‐derived materials that can operate on a longer timescale are thus needed to go a step further toward the real application of water splitting.
Table 1.
Electrocatalytic activity of some typical MOF‐based and MOF‐derived electrocatalysts for the HER and OER
| No. | Electrocatalyst | Target reaction | Medium | Overpotential at 10 mA cm−2 (mV) | Tafel slope (mV dec−1) | Durability (h) | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | (GO 8 wt%) Cu‐MOF composite | OER | 0.5 M H2SO4 | 110 at 2 mA cm−2 | 65 | / | 123 |
| HER | 209 at 30 mA cm−2 | 84 | / | ||||
| 2 | N‐doped carbon nanotube frameworks (NCNTFs) | OER | 1 M KOH | 370 | 93 | ≈3 | 100 |
| 3 | Zn0.30Co2.70S4 polyhedra | HER | 0.5 M H2SO4 | 80 | 47.5 | 60 | 66 |
| 4 | Co3S4 polyhedra | 380 | 85.3 | 10 | |||
| 5 | CoP CPHs | HER | 0.5 M H2SO4 | 133 | 51 | 12 | 102 |
| 6 | MoCx nano‐octahedrons | HER | 0.5 M H2SO4 | 142 | 53 | 11 | 108 |
| 1 M KOH | 151 | 59 | 11 | ||||
| 7 | Ni‐Co mixed oxide cages | OER | 1 M KOH | 380 | 50 | 10 | 110 |
| 8 | Ni‐Co mixed oxide cubes | 430 | 59 | 10 | |||
| 9 | CoNi hydroxide ultrathin nanosheets | OER | 1 M KOH | 324 | 33 | ≈3 | 104 |
| 10 | Spindle‐like Co/Fe metal oxides in N‐doped porous carbon | OER | 0.1 M KOH | 390 | 72.9 | / | 109 |
| 11 | CoPx NPs embedded in N‐doped carbon matrices | OER | 1 M KOH | 319 | 52 | 24 | 67 |
| HER | 154 | 51 | 24 | ||||
| 12 | Co3O4C‐NA | OER | 0.1 M KOH | 290 | 70 | 30 | 113 |
| 13 | MoS2/3D‐NPC | HER | 0.5 M H2SO4 | 210 | 51 | ≈3 | 118 |
| 14 | Au@Zn−Fe−C | HER | 0.5 M H2SO4 | 123 | 130 | 12 | 119 |
| 15 | Atomically isolated nickel species anchored on graphitized carbon | HER | 0.5 M H2SO4 | 34 | 41 | 25 | 117 |
| 16 | Co NPs embedded in porous N‐rich carbon | OER | 1 M KOH | 370 | 76 | 10 | 115 |
| HER | 298 | 131 | 10 | ||||
| 17 | Co@Co3O4 encapsulated in CNT‐grafted N‐doped carbon polyhedra | OER | 0.1 M KOH | 410 | 54.3 | 45 | 95 |
| 18 | Layered CoP/rGO composite | HER | 0.5 M H2SO4 | 105 | 50 | 22 | 124 |
| 1 M KOH | 150 | 38 | 22 | ||||
| OER | 1 M KOH | 340 | 66 | 22 |
3.2. Photocatalytic Water Splitting
Some MOFs have shown favorable photocatalytic activity for water splitting, dye degradation, CO2 reduction and Cr(VI) reduction.138, 139, 140, 141 For photocatalytic water splitting, the direct use of MOFs as photocatalysts for the HER was first reported in 2010 by Silva et al.138 They demonstrated two types of Zr‐based MOFs formed by terephthalate (UiO‐66) and 2‐aminoterephthalate ligands (UiO‐66‐NH2) as stable photocatalysts for the HER. However, the H2 generation rates of both photocatalysts were low (248 and 372 µmol h−1 g−1 for UiO‐66 and UiO‐66‐NH2, respectively). Thus, it is critical to incorporate a Pt co‐catalyst and to optimize the Pt amount to enhance the activity of MOF‐based photocatalysts.142 The optimum Pt amount (1.5 wt.%) achieved a high H2 evolution rate of 500 µmol h−1 g−1 with a Pt/Ti‐MOF‐NH2 photocatalyst.142 Recently, Shen et al. used an active MoS2 to replace expensive Pt in the UiO‐66‐CdS system, and superior photocatalytic activity was achieved with the MoS2/UiO‐66‐CdS photocatalyst.143 As shown in Figure 8 a, the incorporation of CdS and UiO‐66 enhanced the photocatalytic activity of both parts of the reaction, and the optimum HER activity was obtained at a UiO‐66 to CdS weight ratio of 1:1. A high H2 evolution rate of 25,770 µmol h−1 g−1 was obtained in the presence of 1 wt.% MoS2, approximately 2‐fold higher than that of 1 wt.% Pt/UiO‐66‐CdS. As shown in Figure 8b, the H2 evolution rate of MoS2/UiO66‐CdS reached a maximum when the MoS2 amount in the composite was 1.5 wt.% (32,500 µmol h−1 g−1). Shen et al.'s work demonstrated the advantage of using MoS2 as a highly active co‐catalyst to replace Pt in MOF‐based photocatalysts to enhance the HER activity.
Figure 8.
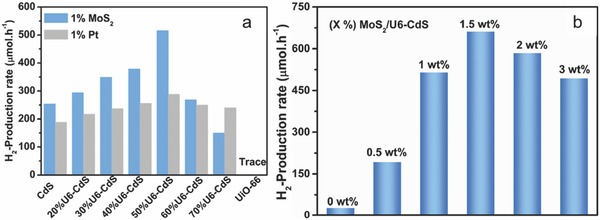
a) The H2 production rate over CdS, UiO‐66 and UiO‐66‐CdS composites loaded with 1 wt.% MoS2 or Pt. b) The H2 production rate over UiO‐66‐CdS composite (1:1, w/w) loaded with different amounts of MoS2. Reproduced with permission.143 Copyright 2015, Elsevier.
Lin and co‐workers were the first to use an Fe‐based MOF (MIL‐101) coated with amorphous TiO2 as a template to prepare Fe2O3@TiO2 composite as a photocatalyst for the HER.144 This MOF‐derived Fe2O3@TiO2 composite exhibited superior HER activity to either Fe2O3 or TiO2 alone and a mixture of the two. However, this composite had a low H2 evolution rate of ≈600 µmol h−1 g−1 after depositing Pt NPs as co‐catalysts. More recently, Pham et al. reported a hollow Fe2O3‐TiO2‐PtOx photocatalyst for the HER that was synthesized from MIL‐88B as a template.145 The hollow Fe2O3‐TiO2‐PtOx nanocomposite displayed an enhanced hydrogen evolution rate of 1100 µmol h−1 g−1 by separating the two co‐catalysts (Fe2O3 and PtOx) on two surface sides of TiO2. In 2015, Bala et al. reported a MOF‐derived Co3O4/TiO2 photocatalyst with rich p‐n heterojunctions for the HER.88 The Co3O4/TiO2 composite with the optimum Co loading amount of 2 wt.% exhibited the maximum photocatalytic activity, with a hydrogen evolution rate of 7000 µmol g−1 h−1, which was almost 7 times greater than that of the conventional Co3O4/TiO2 nanocomposite.88
As demonstrated previously, the catalytic activity of photocatalysts for the HER and OER is strongly related to the band gap energy, the visible light absorption capability, the composition and quantity of active sites/co‐catalysts and the charge transfer capability. In the following section, we summarize several important examples of MOFs as photocatalysts for water splitting under solar irradiation, including band gap engineering to improve the visible light response, active site/co‐catalyst selection, incorporation and optimization to enhance the activity and structural evolution, functional coupling and the creation of heterojunctions to enhance the charge transfer capability.
3.2.1. Engineering the Band Gap and/or Enhancing the Visible Light Adsorption Capability
Tremendous effort has been made to develop photocatalysts with high activity under visible light irradiation, and it is primarily important to tailor the band structure.34, 35, 36, 37 A hybrid organic/inorganic compound with a MOF structure, Cu3PO4(C2N3H2)2OH, which was synthesized by hydrothermal route in the presence of 1,2,4‐triazole as a structure directing agent, was investigated as a photocatalyst for the OER under visible light.146 This Cu‐based MOF had a band gap of 2.58 eV, which ensured visible‐light‐driven photocatalytic activity. When Cu3PO4(C2N3H2)2OH was used as the photocatalyst, the OER was successful under visible light irradiation, with an O2 evolution rate of ≈1875 µmol h−1 g−1. In principle, MOFs with open metal coordination sites might be able to activate small molecules, such as water, triggering chemical reactions under mild conditions. Therefore, the water oxidation reaction based on this type of porous material should be explored.
In an attempt to enhance the activity of MOF‐based photocatalysts for the HER under visible light irradiation, Toyao et al. reported a Ti‐MOF‐NH2 photocatalyst using 2‐amino‐benzenedicarboxylic acid (H2BDC‐NH2) as an organic linker (Figure 9 ) for the photocatalytic HER under visible light irradiation.142, 147 Ti‐MOF showed an absorption edge of 350 nm, while the edge of Ti‐MOF‐NH2 reached ≈500 nm. In the photocatalytic process, the organic linker absorbed visible light, and the electrons transferred from its excited state to the CB of the photoactive titanium‐oxo cluster, as shown in Figure 9. Pt/Ti‐MOF‐NH2 exhibited efficient photocatalytic activity for the HER under visible light irradiation, and a H2 evolution rate of ≈367 µmol h−1 g−1 was obtained. In contrast, Pt/Ti‐MOF displayed no photocatalytic activity for the HER. These results suggested that the band gap/visible light response played a critical role in the photocatalytic water splitting. However, the structural stability of Pt/Ti‐MOF‐NH2 should be carefully addressed in the future.
Figure 9.
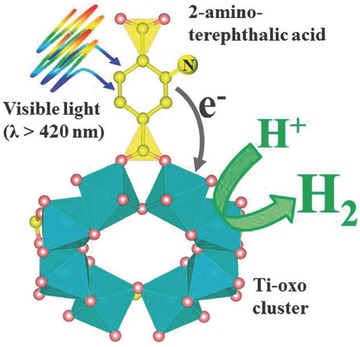
Schematic illustration of photocatalytic hydrogen production reaction over Ti‐MOF‐NH2 under visible light irradiation (λ > 420 nm). Reproduced with permission.142 Copyright 2013, Royal Society of Chemistry.
Although MOF‐based photocatalysts with organic linkers were successfully developed for water splitting under visible light, the longest wavelength available was only 500 nm due to the limited visible light response and low oxidation power of the BDC‐NH2 organic linker.142, 147 Thus, it is critical to extend the wavelength range by using other organic linkers that have wide visible light absorption and high oxidation power. Bis(4′‐(4‐carboxyphenyl)‐terpyridine) Ru(II) (Ru(tpy)2) was found to have wider absorption in the visible light region and a lower highest occupied molecular orbital (HOMO) level than BDC‐NH2. A Ru complex‐modified Ti‐based MOF (Ti‐MOF‐Ru(tpy)2) was synthesized and exhibited high photocatalytic activity for the HER under visible light, with a wavelength up to 620 nm.148
In addition to the reduction in the band gap of MOF, the visible light response can be increased by developing a dye‐sensitized system.44, 149 Since both MOFs and dyes contain a benzene ring, the strong π–π stacking and van der Waals interactions between MOFs and dyes could enhance the electron transfer in a dye‐sensitized system.150 For example, a rhodamine B (RhB)‐sensitized MOF was developed for the HER.149 RhB‐sensitized UiO‐66 exhibited high activity for HER under visible light irradiation with a H2 evolution rate of 116 µmol h−1 g−1, which was 30 times greater than that of Pt@UiO‐66.149 However, the poor stability of RhB restricted its application as an effective photosensitizer.151 Recently, Erythrosin B (ErB) dye, with superior stability to RhB, was selected to sensitize UiO‐66 MOF for photocatalytic H2 production.44 With the optimized amount of ErB, Pt‐UiO‐66‐30 (ErB/photocatalyst weight ratio of 3) displayed the highest H2 evolution rate of 460 µmol h−1 g−1, while Pt‐UiO‐66 without sensitization showed no HER activity. In addition, the photocatalytic activity of ErB‐sensitized UiO‐66 was higher than those of ErB‐sensitized P25‐TiO2 and SiO2 for the HER, demonstrating the advantage of MOFs as photocatalysts for water splitting. The enhancement in photocatalytic H2 production was attributed to the efficient transfer of the charge carriers from the photoexcited ErB to the MOFs. These results suggested that the dye‐sensitized MOFs showed great potential for the photocatalytic HER.
In addition to the dye‐sensitized system based on MOF‐based photocatalysts, MOF itself can be used as a photosensitizer for PEC water splitting.152 MIL‐125 (a Ti‐based MOF) has a more negative lowest unoccupied molecular orbital (LUMO) level than the CB edge of TiO2, making electron transfer from MIL‐125 to TiO2 feasible.153 The performance of TiO2 for PEC water oxidation was significantly enhanced by the addition of a MOF‐based photosensitizer under visible light irradiation.152 The photocurrent of a TiO2 nanowire in PEC water splitting was improved by 100% from 10 to 20 µA cm−2 at 0.75 V vs. RHE under visible light irradiation after the sensitization of aminated Ti‐based MOFs on TiO2. Considering the diversity of MOFs, a large number of MOFs are expected to function as photosensitizers for solar‐driven energy conversion. In another study, functionalized MOF‐253‐Pt was prepared by immobilizing a Pt complex in microporous MOF‐253 using a post‐synthesis method. MOF‐253‐Pt acted as both a photosensitizer and a photocatalyst for visible‐light‐driven HER.154 The photocatalytic activity of MOF‐253‐Pt was ≈5 times greater than that of the Pt complex, while MOF‐253 displayed almost no activity for the HER. Further studies should be conducted to design a robust and highly active MOF bifunctional photocatalyst/photosensitizer for visible‐light‐driven HER.
3.2.2. Active Site/Co‐Catalyst Selection, Incorporation and Optimization
Numerous efforts have been made for the integration of different molecular functional components and MOFs to enhance water splitting activity.83, 84, 85, 86, 87 The combination of MOFs with functional inorganic materials, such as metal NPs and metal oxides, has been reported as a new strategy and has received increased attention.87, 155, 156 Some researchers incorporated Pt NPs into MOFs to enhance the water splitting activity.87, 155, 156 The incorporation of Pt NPs into NH2‐MIL‐101(Cr) was effective for visible‐light‐driven H2 evolution with high activity.155 Pt NPs with an average size of ≈3.75 nm were highly dispersed on NH2‐MIL‐101(Cr) with a particle size of ≈50 nm. When the Pt amount was increased from 0.5 to 1.5 wt.%, the photocatalytic activity of Pt/NH2‐MIL‐101(Cr) was improved due to the increased active sites, and the optimum Pt loading for H2 evolution was 1.5 wt.%. A further increase in the Pt amount to 3.0 wt.% decreased the activity due to the agglomeration of Pt NPs. Similarly, Pt NPs with a particle size of 1 nm were embedded into layers of a titanium picolinate framework (TiPF) by photodeposition.156 The obtained Pt‐loaded TiPF could be used as a photocatalyst for the HER, with a hydrogen evolution rate of 1593 µmol h−1 g−1. Furthermore, TiPF, without Pt NPs, exhibited much lower activity for the HER, with an evolution rate of 228 µmol h−1 g−1, suggesting that Pt active sites played a critical role as an electron trap to improve the charge separation efficiency and increase the HER activity. However, the stability of the TiPF support must be improved in future research.
Recently, earth‐abundant Co and Ni‐based catalysts have been used to replace the precious Pt metal in water splitting.157, 158 Compared to Pt, the control and reduction of the particle size of the oxides are more difficult. Han et al. utilized an MIL‐101 MOF to immobilize Co3O4 NPs for the OER.157 With cobalt loading in the range of 1.4–4.9 wt.%, ultra‐small Co3O4 NPs (2–3 nm) were successfully immobilized and well dispersed in the cages of MIL‐101. This photocatalyst displayed enhanced photocatalytic activity for the OER, with a high TOF of 0.012 s−1 per Co atom, which was more than 9 times higher than that of the unsupported Co3O4 NPs. Furthermore, MIL‐101 also promoted the charge transfer process in the OER.
Due to their large BET surface areas, most available MOF and NPs‐modified MOF photocatalysts are highly dispersed in water, making them difficult to recycle. It is therefore critical to develop MOF‐based photocatalysts that can be easily collected for reuse. One solution is to incorporate magnetic materials into MOFs.159, 160, 161 For example, a photocatalyst composed of MIL‐53(Fe) microrods with magnetic and active Fe3O4 nanosphere decoration on the surface (MIL‐53(Fe) hybrid magnetic composites, MHMCs) was reported.161 As shown in Figure 10 , the MHMCs were composed of well‐dispersed Fe3O4 nanospheres with a particle size of ≈200 nm attached to the surface of MIL‐53(Fe) microrods, creating the heterostructures. Used as a photoanode, the MHMCs delivered a remarkable photocurrent under visible light irradiation. After the OER, the used MHMCs were easily collected through magnetic separation and were recycled for further use. This structural design approach can provide an ideal platform for the development of MOF‐based materials with high activity and magnetic recyclability for energy conversion and environmental remediation.
Figure 10.

SEM images of Fe3O4 (a and b), MIL–53(Fe) (c and d) and MHMCs (e and f). Reproduced with permission.161 Copyright 2015, Royal Society of Chemistry.
3.2.3. Structural Evolution, Coupling and Creation of Heterojunctions
The charge transfer/separation efficiency is vital to the achievement of high photocatalytic activity. Several strategies have been employed to enhance the charge transfer/separation efficiency, such as developing new MOF photocatalysts through structural evolution and coupling with carbon‐based materials to form hybrid photocatalysts.162, 163, 164, 165 For example, a highly active Bi‐based MOF photocatalyst composed of Bi3+ and H2mna (2‐mercaptonicotinic acid) was developed (denoted as Bi‐mna) for water splitting under visible light.162 According to the experimental and theoretical results, a ligand‐to‐ligand charge transfer (LLCT) process was responsible for the high photocatalytic activity, which resulted in a longer lifetime of the photogenerated charges, suppressed electron‐hole recombination and increased the photocatalytic activity. These results suggested that Bi‐based MOFs are promising candidates for efficient water splitting.
In addition to the design of new photocatalysts, functional additives have been used to enhance the charge transfer capability of MOF‐based photocatalysts.163 A new hybrid photocatalyst created by coupling UiO‐66 octahedrons and g‐C3N4 (referred to as UG‐x, where x is the weight of g‐C3N4 in the hybrids) was demonstrated.163 Although both g‐C3N4 and UiO‐66 were capable of the HER, the use of each material alone displayed limited photocatalytic activity due to the high charge recombination rate and poor sunlight absorption capability. Figure 11 a shows a schematic illustration of the coating of g‐C3N4 on UiO‐66 octahedrons by annealing. UiO‐66 had very smooth and clean surfaces as shown in Figure 11b. After annealing, g‐C3N4 nanosheets were coated on the surface of UiO‐66, making the surface uneven (Figure 11c). Increased g‐C3N4 resulted in heavy coverage of g‐C3N4 on UiO‐66 (Figure 11d). At the optimum weight of g‐C3N4 in the hybrid, the UG‐50 photocatalyst showed dramatically enhanced photocatalytic activity for the HER, with a H2 evolution rate of 1411 µmol h−1 g−1, compared with UiO‐66 and g‐C3N4 (0 and 80 µmol h−1 g−1, respectively). These results confirmed that the annealing process promoted the formation of UiO‐66/g‐C3N4 heterojunctions and enhanced the charge separation efficiency in UG‐50, which enhanced the H2 evolution rate (more than 17‐fold). These results revealed the essential role of the UiO‐66/g‐C3N4 interfaces in enhancing the photocatalytic H2 production.
Figure 11.
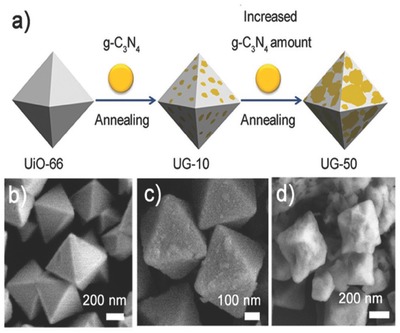
a) Schematic illustration of coating g‐C3N4 on UiO‐66 octahedrons through annealing. SEM images of b) UiO‐66, c) UG‐10 and d) UG‐50. Reproduced with permission.163
Loading CdS with RGO is a useful method to reduce the possibility of electron‐hole pair recombination since graphene has high conductivity and superior electron mobility, which can accelerate the electron transfer in photocatalytic reaction.166 Lin et al. found that the hydrogen production performance of CdS was improved by the combined action of UiO‐66 and RGO due to the increased quantity of active sites and the minimized recombination of charge carriers.164 As a support, UiO‐66 was superior to inorganic semiconductor material. The UiO‐66/CdS/1% RGO photocatalyst displayed a very high photocatalytic HER rate, which was 13.8, 2 and 1.23 times greater than commercial CdS, P25/CdS/1% RGO and UiO‐66/CdS, respectively, as shown in Figure 12 .
Figure 12.
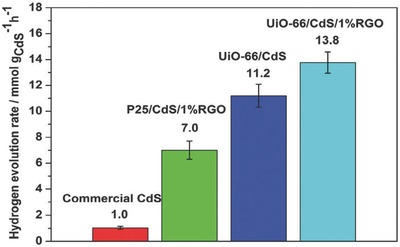
H2 evolution rates of various photocatalysts under visible light irradiation. Reproduced with permission.164 Copyright 2014, Royal Society of Chemistry.
In another study, highly porous Co3O4@carbon derived from ZIF‐67 MOFs was successfully embedded on the surface of BiVO4 nanosheets,89 in which the construction of a p‐n heterojunction between the porous Co3O4 (p‐Co3O4) and BiVO4 significantly promoted the charge transfer and reduced electron‐hole recombination. The p‐Co3O4@carbon/BiVO4 photoanode demonstrated ≈10 times higher photocurrent density than the bare BiVO4 and bulk Co3O4/BiVO4 for the OER. This system also demonstrated good stability for the OER without noticeable degradation after three recycled uses. More recently, a highly active Au@CdS/MIL‐101 heterostructure was successfully prepared.165 The MIL‐101(Cr) with large specific surface area was utilized as a support for well‐dispersed Au NPs, and CdS was selectively coated on the Au NPs.165 The good dispersion of CdS and Au NPs, as well as the strong surface plasmon resonance absorption of Au, led to enhanced electron separation and transfer. Under visible light irradiation, the Au@CdS/MIL‐101 heterostructure presented a high H2 production rate of 25,000 µmol h−1 g−1, which was 3.6 and 1.3 times greater than those of CdS and CdS/MIL‐101, respectively. These results suggested that the heterojunction in the photocatalysts played a critical role in the high photocatalytic activity, and these studies will encourage extensive research on heterostructured photocatalysts in the future.
Besides the separation efficiency of the photo‐induced charge carriers, the photocatalytic activity of the MOF‐based/derived photosensitizer/photocatalyst essentially relies on the excited state lifetime of the photo‐generated electron‐hole pair.45, 87, 167, 168, 169, 170, 171, 172, 173 For example, Lin and co‐workers synthesized a MOF based on {M[4,4′‐dcbpy]2bpy}2+ building blocks (where M = Ru or Os, bpy = 2,2′‐bipyridine and dcbpy = dicarboxy‐2,2′‐bipyridine), which can be readily excited to their long‐lived triplet metal‐to‐ligand charge transfer (3MLCT) states.171 The lifetimes of Ru2+ excited states in the Os‐doped MOF decreased progressively with increasing Os doping amount from 0.3 to 2.6 mol.%. The lifetime at 620 nm decreased from 171 ns in the pure Ru‐MOF to 29 ns in the sample with 2.6 mol.% Os doping. In the mixed‐metal samples, energy transfer was observed with an initial growth in Os emission corresponding with the decay rate of the Ru excited state. These results demonstrate rapid, efficient energy migration and long distance transfer in these isomorphous MOFs.
Morris and co‐workers have made outstanding contributions to the study of the lifetimes of the photo‐induced electron and holes in MOF‐based photocatalyst.174, 175, 176, 177 The effect of the doping amount of ruthenium(II) tris(5,5′‐dicarboxy‐2,2′‐bipyridine), Rudcbpy, on the lifetime of the emissive 3MLCT state of the Rudcbpy‐doped UiO‐67 MOF (Zr6(μ3‐O)4(μ3‐OH)4(bpdc)6 (bpdc = biphenyldicarboxylic acid)) was systematically investigated by Morris and co‐workers.174 They found that the lifetime of the emissive 3MLCT state corresponding to Rudcbpy centers incorporated into the MOF backbone was found to be sensitive to the Rudcbpy doping amount in the material. The excited state properties of the Rudcbpy‐doped UiO‐67 at low doping concentrations resembled that of Rudcbpy in dimethylformamide (DMF) displaying a long‐lived (≈1.4 µs) 3MLCT state. Increasing the Rudcbpy doping concentration in UiO‐67 was accompanied by a significant decrease in the emission lifetime, which was proposed to be due to the homogeneous energy transfer between the Rudcbpy centers.174, 175 Single Rudcbpy preferentially occupied the larger octahedral cages of UiO‐67 by incorporation into the backbone of the cage and experienced a DMF‐like solvation environment. At higher doping concentrations of Rudcbpy, in addition to incorporation of Rudcbpy into the backbone of the octahedral cavities, populations of encapsulated Rudcbpy were also found in separate octahedral UiO‐67 cavities. The decreased lifetime of the slow phase with increased doping concentration was attributed to the intermolecular energy transfer between neighboring Rudcbpy molecules incorporated into the backbone of the octahedral cages.
In another study, Morris and co‐workers found that this Rudcbpy‐doped UiO‐67 can be grown onto conductive fluorine‐doped tin oxide (FTO) coated glass substrates without changing its excited state properties or dynamics.175 The Rudcbpy dopant within the UiO‐67 films interacted with each other and underwent self‐quenching via a resonance energy transfer mechanism.175 The average distance between Rudcbpy was decreased in the film relative to similarly doped powders. This is attributed to an electrostatic effect upon formation of the framework due to increased charge at the bpdc self‐assembled monolayer on the surface of the substrate. These Rudcbpy‐doped UiO‐67 films can also be grown onto TiO2 as a sensitizing material for photovoltaic applications such as sensitized solar cells, which can broad the potential applications of MOFs in various solar energy‐based research fields.177
In addition, it was found that the coupling of nanosized carbon nitride nanosheets (CNNS) with UiO‐66 via a facile electrostatic self‐assembly method increased the lifetime of the photo‐generated electron‐hole pair of CNNS photocatalyst.178 It was demonstrated that the electrons from the photo‐excited CNNS can transfer to UiO‐66, which substantially suppressed the electron‐hole pair recombination in the CNNS, and also provided long‐lived electrons for the reduction of CO2 molecules that were adsorbed in UiO‐66. The calculated lifetime values were 481.4 ns for CNNS and 846.3 ns for UiO‐66/CNNS composite. As a result, the UiO‐66/CNNS photocatalyst exhibited a much higher photocatalytic activity than that of CNNS. This work highlights the synergistic incorporation of MOFs to C3N4‐based photocatalysts to increase the lifetime and improve the separation efficiency of the photo‐generated electron‐hole pair.
3.2.4. A Brief Summary
The catalytic activities of some typical MOF‐based and MOF‐derived photocatalysts for the HER and OER are listed in Table 2 . In addition to the main active components, the activity of the photocatalysts for the HER and OER is heavily dependent on the sacrificial reagent and the co‐catalyst/active sites. Compared to MOF‐based electrocatalysts, in which the MOFs were more frequently used as precursors, MOFs have been widely used as photocatalysts directly, although they were often coupled with active sites/co‐catalysts to enhance their activity. The amine‐functionalized Ti‐MOF displayed superior photocatalytic activity for the HER due to the improved visible light response. The addition of metal/metal oxide improved the photocatalytic activity. However, the addition of an excessive amount of metal/metal oxide reduced the photoactivity for the HER and OER. For Pt, a loading amount of 1.5 wt.% in the Ti‐MOF‐NH2 photocatalyst resulted in the highest H2 evolution rate. Similarly, for CoOx NP‐modified MOF, the optimum CoOx amount of 3.9 wt.% was demonstrated for the OER, suggesting that the amount and the particle size of the co‐catalyst should be controlled and optimized to achieve high photocatalytic activity for the HER and OER. In addition to metals and metal oxides, functional materials, such as CdS, RGO, MoS2 and g‐C3N4, have been added to MOF to improve the photocatalytic activity for the HER. As shown in Table 2, MoS2 increased the photoactivity for the HER and was superior to Pt as a co‐catalyst.130 The H2 production rate of UiO‐66/CdS increased 25‐fold when 1.5 wt.% MoS2 was incorporated. The construction of dye‐sensitized systems can contribute to high photocatalytic activity; the selection of the dye, its amount and the incorporation method should be carefully investigated. In addition to the direct use of MOFs as photocatalysts, nanostructures and composites derived from MOFs were investigated as efficient photocatalysts for the HER and OER. Porous Co3O4 nanocages derived from PBA Co3[Co(CN)6]2 displayed high activity for the OER.103 However, the recycled performance of many MOF‐based photocatalysts must be enhanced. Additionally, in most of the studies summarized in this review, the quantum yield of the MOF‐based photocatalysts for water splitting was not presented, and the values that were presented are rather low and must be improved in the future.
Table 2.
Photocatalytic activity of some typical MOF‐based and MOF‐derived photocatalysts for the HER and OER
| No. | Photocatalyst | Target reaction | Sacrificial reagent | Production rate (µmol h−1 g−1) | Quantum yields (%) | Recycled times | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | Bi‐based MOF | OER | AgNO3 | ≈180 | / | / | 162 |
| 2 | Ti‐MOF | HER | Triethanolamine (TEOA) | 0 | / | / | 142 |
| 3 | Ti‐MOF‐NH2 | ≈170 | / | / | |||
| 4 | 0.5 wt.% Pt/Ti‐MOF‐NH2 | ≈330 | / | / | |||
| 5 | 1.5 wt.% Pt/Ti‐MOF‐NH2 | ≈500 | ≈1.3 at 420 nm | 3 | |||
| 6 | 2 wt.% Pt/Ti‐MOF‐NH2 | ≈460 | / | / | |||
| 7 | 2.6 wt.% CoOx NPs‐MIL‐101 | OER | [Ru(bpy)3]2+–Na2S2O8 | ≈11,000 | / | / | 157 |
| 8 | 3.9 wt.% CoOx NPs‐MIL‐101 | ≈15,000 | / | 2 | |||
| 9 | 4.9 wt.% CoOx NPs‐MIL‐101 | ≈13,000 | / | / | |||
| 10 | Small‐sized Ni NPs anchored in MOF‐5 | HER | TEOA | 3,022 | 7.8 at 520 nm | 4 | 158 |
| 11 | MnTD⊂MIL‐101 | OER | Ceric ammonium nitrate | 2,250 | / | / | 84 |
| 12 | MIL‐101 | 125 | / | / | |||
| 13 | UiO‐66 | HER | Na2S, Na2SO3 | 0 | / | / | 164 |
| 14 | UiO‐66/CdS | 1,700 | / | 6 | |||
| 15 | UiO‐66/CdS/1%RGO | 2,100 | / | / | |||
| 16 | MoS2/UiO‐66/CdS | HER | Lactic acid | 32,500 | 23.6 at 420 nm | 4 | 143 |
| 17 | UiO‐66/CdS | 1,250 | / | / | |||
| 18 | g‐C3N4 | HER | L‐ascorbic acid | 80 | / | / | 163 |
| 19 | UiO‐66 | 0 | / | / | |||
| 20 | g‐C3N4/UiO‐66 (1:1, w/w) | 1,141 | / | / | |||
| 21 | Ti‐MOF‐Ru(tpy)2 | HER | TEOA | ≈200 | 0.2 at 500 nm | 3 | 148 |
| 22 | Pt complex immobilized MOF‐253 | HER | CH3CN | ≈58,000 | 1.63 at 440 nm | 2 | 154 |
| 23 | Pt@UiO‐66 | HER | TEOA | 3.9 | / | / | 149 |
| 24 | 2.54 mg g−1 of RhB/Pt@UiO‐66 | 5.6 | / | / | |||
| 25 | 11.92 mg g−1 of RhB/Pt@UiO‐66 | ≈100 | / | 3 | |||
| 26 | ErB dye‐sensitized Pt/UiO‐66 octahedrons | HER | Methanol | 460 | 0.25 at 420 nm | 3 | 44 |
| 27 | Hollow Fe2O3‐TiO2‐PtOx | HER | Lactic acid | 1,100 | / | 5 | 145 |
| 28 | Porous Co3O4 nanocages | OER | [Ru(bpy)3]2+–Na2S2O8 | ≈7,900 | / | / | 103 |
| 29 | Porous MnxCo3−xO4 nanocages | ≈4,900 | / | / | |||
| 30 | Porous FexCo3−xO4 nanocages | 4,000 | / | / | |||
| 31 | Co3O4/TiO2 p–n heterojunction | HER | Methanol | 7,000 | / | / | 88 |
4. Challenges and Perspectives
In this review, the recent research progress of MOF‐based and MOF‐derived catalysts for electrocatalytic and photocatalytic water splitting reactions has been summarized. The applications of MOFs as direct catalysts and as precursors of catalysts for water splitting were included, and both the OER and HER were considered. MOFs have demonstrated promising applications in the field of electrocatalytic and photocatalytic water splitting for H2 production, and several impressive results are available. However, the field of MOF‐mediated water splitting is in its infancy, and remarkable enhancements are needed to make MOF‐mediated water splitting systems fully competitive. For example, the poor stability of MOF‐based catalysts in water is a major concern, which is attributed to the higher affinities of their ions for water molecules than the carboxylate ligands. The leaching of metallic and organic components from the MOFs into the reaction medium during the water splitting reactions is another major concern. The reaction mechanisms of MOF‐based catalysts for water splitting are still not well understood and require further investigation. The recycling of MOF‐based catalysts is also a major challenge due to the ultrafine size of most MOF‐based materials. The main challenges and perspectives for MOF‐based catalysts in electrocatalytic and photocatalytic water splitting reactions are discussed in the following sections.
4.1. MOF‐Based Catalysts for Electrocatalytic Water Splitting
There are three strategies to design MOF‐based electrocatalysts for electrocatalytic water splitting: morphology control/nanostructuring, constructing hybrids/composites and functional modifications. The morphology of the catalyst plays an important role in achieving high electrocatalytic activity. Electrocatalysts with special morphologies, such as CPHs and nano‐octahedrons, may show superior electrocatalytic activity than their related NPs. By tailoring the composition of the MOFs and the calcination temperature and atmosphere, various morphologies can be obtained with different catalytic properties for the water splitting reactions. For example, a MOF with a flower‐shaped morphology was developed with high electrocatalytic activity.101 However, due to the diversity of MOFs, it is hard to identify the most efficient morphology for the electrocatalytic OER/HER. A more systematic experimental investigation or theoretical study of the morphology‐activity relationship of MOFs is required.
In addition to the morphology, the interaction or synergistic effect between components in the electrocatalysts has a strong influence on the electrocatalytic activity of MOF‐derived materials and requires more attention in the development of MOF‐based and MOF‐derived electrocatalysts for the water splitting reactions. For MOF‐derived metal and metal oxide‐based catalysts, the leaching of metals during the operation is a major concern and could cause a serious decrease in the electrocatalytic activity and stability for water splitting in acidic or alkaline electrolytes. An effective solution to the deterioration is to encapsulate metal NPs into carbon nanotubes or carbon shells. The use of MOFs as precursors for the synthesis of core‐shell structured hybrid electrocatalysts may provide a new research direction for the development of MOF‐based catalysts for efficient water splitting. Despite the various MOF‐derived nanostructures, precise control of the morphology of these materials is absent because of the limited knowledge of the transformation process. A clearer understanding of this issue is urgently needed and would be helpful for the design and construction of fine nanostructures from MOFs with high surface areas, regular pores and tunable compositions for the electrochemical water splitting reactions.
Imidazolate linkers are often used as functional modifiers to improve the proton transfer process in the electrocatalytic water splitting reactions, which may be applied as a general method to design MOFs with improved charge transfer capability. In addition, functional carbon‐based materials, such as GO and rGO, can be used as modifiers for MOF‐based electrocatalysts to improve the charge transfer process. For example, CoP/rGO can be used as a bifunctional catalyst on both the anode and cathode for water splitting in alkaline solutions, with better activity (135 mV dec−1) than the integrated Pt/C and IrO2 catalyst couple (201 mV dec−1).124 However, the mechanism must be clarified. In addition, other carbon‐based materials, such as g‐C3N4, could be used to improve the charge transfer efficiency in the electrocatalytic OER and HER.
4.2. MOF‐Based Catalysts for Photocatalytic Water Splitting
As a photocatalyst for water splitting, the efficiency of a MOF‐based material is strongly affected by its sunlight absorption capability (band gap), the number of active sites and the charge separation/transfer efficiency. This review provided a summary of recent developments involving the use of MOFs as photocatalysts, with particular emphasis on several key strategies (band gap engineering, photosensitization, active site/co‐catalyst selection and optimization and the coupling of MOFs with other functional materials) to develop MOF‐based and MOF‐derived photocatalysts with high performance.
The band gap energy of MOFs can be tailored by changing the metal‐oxo clusters and bridging organic linkers. Several organic dyes have been used to sensitize MOF‐based photocatalysts, leading to significantly enhanced photoactivity under visible light irradiation. However, the stability of these organic‐linker‐modified MOFs and the organic dyes should be carefully considered in future research. MOFs themselves can also be used as photosensitizers for PEC water splitting and can significantly enhance the photoactivity of TiO2 under visible light.139 In the future, the match of the band structure of MOFs and TiO2 should be clarified by theoretical calculations, such as DFT, which may provide useful design guidelines for MOF‐based photosensitizers.
The incorporation of active sites, such as Pt NPs, into MOFs was found to dramatically enhance the photocatalytic activity.142, 143 However, the limited reserves and high cost of Pt hinder its large‐scare application. Recently, highly efficient, small‐sized Ni particles embedded in MOF‐5 as co‐catalysts/active sites with low overpotential for photocatalytic HER under visible light was demonstrated.145 A low overpotential of −0.37 V was obtained by this photocatalyst, which was comparable to that of Pt@MOF‐5. The development of non‐precious metal NPs in MOFs to enhance the photocatalytic activity requires more attention in the future. It is critical to develop MOF‐based photocatalysts that can be easily collected and reused. One solution is to incorporate magnetic materials into the MOFs. After water splitting, the magnetic photocatalyst can be easily collected by magnetic separation and recycled for further use. Magnetic MOF‐based composites should be next‐generation photocatalysts for water splitting.
The charge transfer/separation efficiency plays a vital role in the photocatalytic water splitting reactions. Various methods have been used to improve the charge transfer/separation efficiency. For example, electrons can be transferred from photoexcited organic linkers to metal‐clusters in Zr‐based MOFs (LLCT process).149 In the H2 generation process via the LLCT mechanism, the CB edge position of a titanium‐oxo cluster is more suitable for charge transfer since the CB potential of a titanium‐oxo cluster is more positive than that of its zirconium counterpart. Thus, Ti‐based MOFs are expected to be highly active photocatalysts for the HER. In addition to the design of new photocatalysts, functional additives, such as g‐C3N4, have been used to enhance the charge transfer/separation efficiency of MOF‐based photocatalysts. For example, g‐C3N4/UiO‐66 hybrid photocatalysts showed enhanced activity in the visible‐light‐driven HER, which was attributed to the efficient interfacial charge transfer from photoexcited g‐C3N4 to UiO‐66.150 These results demonstrated the potential use of MOFs and g‐C3N4 to construct active photocatalysts for water splitting. Based on the extensive studies of the application of MOFs in photocatalytic water splitting, the application of MOFs in solar cells will be a research focus in the future, although it is out of the scope of this article. It will be exciting to watch the rapid development of such new materials in the years to come.
Acknowledgements
The authors would like to thank the Australia Research Council for supporting the project under contract DP150104365 and DP160104835.
Biographies
Wei Wang obtained his Ph.D. degree in Chemical Engineering at Nanjing Tech University, China, in June, 2013. He is now a postdoctoral fellow in Curtin University, Australia, since December, 2013. His research interests include anode catalytic materials and coke formation mechanism for solid oxide fuel cells (SOFCs) operating on hydrocarbons, fuel selection and application for SOFCs, photocatalysts for degradation of organic substances, photoanodes and counter electrodes for dye‐sensitized solar cells.

Xiaomin Xu received his Bachelor (2013) and Master (2016) degrees in Chemical Engineering from Nanjing Tech University, China. He is now pursuing a Ph.D. degree in Chemical Engineering at Curtin University, Australia. His research interests include the structure, synthesis and characterization of perovskite materials and their applications in the electrocatalytic water splitting reactions.

Wang W., Xu X., Zhou W., Shao Z., Adv. Sci. 2017, 4, 1600371.
References
- 1. Deluga G. A., Salge J. R., Schmidt L. D., Verykios X. E., Science 2004, 303, 993. [DOI] [PubMed] [Google Scholar]
- 2. Ni M., Leung D. Y. C., Leung M. K. H., Int. J. Hydrogen Energy 2007, 32, 3238. [Google Scholar]
- 3. Hafizi A., Rahimpour M. R., Hassanajili S., Appl. Energy 2016, 165, 685. [Google Scholar]
- 4. Zhang J., Wang T., Liu P., Liu S., Dong R., Zhuang X., Chen M., Feng X., Energy Environ. Sci. 2016, 9, 2789. [Google Scholar]
- 5. Xu X., Chen Y., Zhou W., Zhu Z., Su C., Liu M., Shao Z., Adv. Mater. 2016, 28, 6442. [DOI] [PubMed] [Google Scholar]
- 6. Yin X. L., He G. Y., Sun B., Jiang W. J., Xue D. J., Xia A. D., Wan L. J., Hu J. S., Nano Energy 2016, 28, 319. [Google Scholar]
- 7. Lu Q., Yu Y., Ma Q., Chen B., Zhang H., Adv. Mater. 2016, 28, 1917. [DOI] [PubMed] [Google Scholar]
- 8. Zhu Y., Zhou W., Sunarso J., Zhong Y., Shao Z., Adv. Funct. Mater. 2016, 26, 5862. [Google Scholar]
- 9. Zhu L., Ran R., Tadé M., Wang W., Shao Z., Asia‐Pac. J. Chem. Eng. 2016, 11, 338. [Google Scholar]
- 10. Pi Y., Zhang N., Guo S., Guo J., Huang X., Nano Lett. 2016, 16, 4424. [DOI] [PubMed] [Google Scholar]
- 11. Audichon T., Napporn T. W., Canaff C., Morais C., Comminges C., Kokoh K. B., J. Phys. Chem. C 2016, 120, 2562. [Google Scholar]
- 12. Subbaraman R., Tripkovic D., Strmcnik D., Chang K. C., Uchimura M., Paulikas A. P., Stamenkovic V., Markovic N. M., Science 2011, 334, 1256. [DOI] [PubMed] [Google Scholar]
- 13. Kavian R., Choi S. I., Park J., Liu T., Peng H. C., Lu N., Wang J., Kim M. J., Xia Y., Lee S. W., J. Mater. Chem. A 2016, 4, 12392. [Google Scholar]
- 14. Yu L., Xia B. Y., Wang X., Lou X. W., Adv. Mater. 2016, 28, 92. [DOI] [PubMed] [Google Scholar]
- 15. Xu X., Pan Y., Zhou W., Chen Y., Zhang Z., Shao Z., Electrochim. Acta 2016, 219, 553. [Google Scholar]
- 16. Peng Z., Jia D., Al‐Enizi A. M., Elzatahry A. A., Zheng G., Adv. Energy Mater. 2015, 5, 1402031. [Google Scholar]
- 17. Friebel D., Louie M. W., Bajdich M., Sanwald K. E., Cai Y., Wise A. M., Cheng M. J., Sokaras D., Weng T. C., Alonso‐Mori R., Davis R. C., Bargar J. R., Nørskov J. K., Nilsson A., Bell A. T., J. Am. Chem. Soc. 2015, 137, 1305. [DOI] [PubMed] [Google Scholar]
- 18. Xu X., Su C., Zhou W., Zhu Y., Chen Y., Shao Z., Adv. Sci. 2016, 3, 1500187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wu H., Geng J., Ge H., Guo Z., Wang Y., Zheng G., Adv. Energy Mater. DOI: 10.1002/aenm.201600794. [Google Scholar]
- 20. Li J. C., Hou P. X., Zhao S. Y., Liu C., Tang D. M., Cheng M., Zhang F., Cheng H. M., Energy Environ. Sci. 2016, 9, 3079. [Google Scholar]
- 21. Zheng Y., Jiao Y., Zhu Y., Li L. H., Han Y., Chen Y., Du A., Jaroniec M., Qiao S. Z., Nat. Commun. 2014, 5, 3783. [DOI] [PubMed] [Google Scholar]
- 22. Liu Y., Yu G., Li G. D., Sun Y., Asefa T., Chen W., Zou X., Angew. Chem. Int. Ed. 2015, 54, 10752. [DOI] [PubMed] [Google Scholar]
- 23. Zou X., Huang X., Goswami A., Silva R., Sathe B. R., Mikmeková E., Asefa T., Angew. Chem. Int. Ed. 2014, 53, 4372. [DOI] [PubMed] [Google Scholar]
- 24. Zheng Y., Jiao Y., Li L. H., Xing T., Chen Y., Jaroniec M., Qiao S. Z., ACS Nano 2014, 8, 5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ma T. Y., Dai S., Jaroniec M., Qiao S. Z., Angew. Chem. Int. Ed. 2014, 53, 7281. [DOI] [PubMed] [Google Scholar]
- 26. Chen H., Yu G., Li G. D., Xie T., Sun Y., Liu J., Li H., Huang X., Wang D., Asefa T., Chen W., Zou X., Angew. Chem. Int. Ed. 2016, 55, 11442. [DOI] [PubMed] [Google Scholar]
- 27. Bhunia M. K., Melissen S., Parida M. R., Sarawade P., Basset J. M., Anjum D. H., Mohammed O. F., Sautet P., Bahers T. L., Takanabe K., Chem. Mater. 2015, 27, 8237. [Google Scholar]
- 28. Ma F., Wu Y., Shao Y., Zhong Y., Lv J., Hao X., Nano Energy 2016, 27, 466. [Google Scholar]
- 29. Lin Z., Xiao J., Li L., Liu P., Wang C., Yang G., Adv. Energy Mater. 2016, 6, 1501865. [Google Scholar]
- 30. Huang Z. F., Song J., Pan L., Wang Z., Zhang X., Zou J. J., Mi W., Zhang X., Wang L., Nano Energy 2015, 12, 646. [Google Scholar]
- 31. Wu H. B., Hng H. H., Lou X. W. D., Adv. Mater. 2012, 24, 2567. [DOI] [PubMed] [Google Scholar]
- 32. Joo J. B., Lee I., Dahl M., Moon G. D., Zaera F., Yin Y., Adv. Funct. Mater. 2013, 23, 4246. [Google Scholar]
- 33. Pan J. H., Wang X. Z., Huang Q., Shen C., Koh Z. Y., Wang Q., Engel A., Bahnemann D. W., Adv. Funct. Mater. 2014, 24, 95. [Google Scholar]
- 34. Shi L., Li P., Zhou W., Wang T., Chang K., Zhang H., Kako T., Liu G., Ye J., Nano Energy 2016, 28, 158. [Google Scholar]
- 35. Zou J. P., Wang L. C., Luo J., Nie Y. C., Xing Q. J., Luo X. B., Du H. M., Luo S. L., Suib S. L., Appl. Catal. B: Environ. 2016, 193, 103. [Google Scholar]
- 36. Sprick R. S., Bonillo B., Clowes R., Guiglion P., Brownbill N. J., Slater B. J., Blanc F., Zwijnenburg M. A., Adams D. J., Cooper A. I., Angew. Chem. Int. Ed. 2016, 55, 1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chang K., Li M., Wang T., Ouyang S., Li P., Liu L., Ye J., Adv. Energy Mater. 2015, 5, 1402279. [Google Scholar]
- 38. Wu Y., Li G. D., Liu Y., Yang L., Lian X., Asefa T., Zou X., Adv. Funct. Mater. 2016, 26, 4839. [Google Scholar]
- 39. Ma T. Y., Cao J. L., Jaroniec M., Qiao S. Z., Angew. Chem. Int. Ed. 2016, 55, 1138. [DOI] [PubMed] [Google Scholar]
- 40. Yin X. L., Liu J., Jiang W. J., Zhang X., Hu J. S., Wan L. J., Chem. Commun. 2015, 51, 13842. [DOI] [PubMed] [Google Scholar]
- 41. Fu J., Skrabalak S. E., J. Mater. Chem. A 2016, 4, 8451. [Google Scholar]
- 42. Ren Y., Chia G. H., Gao Z., Nano Today 2013, 8, 577. [Google Scholar]
- 43. Xia W., Mahmood A., Zou R., Xu Q., Energy Environ. Sci. 2015, 8, 1837. [Google Scholar]
- 44. Yuan Y. P., Yin L. S., Cao S. W., Xu G. S., Li C. H., Xue C., Appl. Catal. B: Environ. 2015, 168–169, 572. [Google Scholar]
- 45. Meyer K., Ranocchiari M., van Bokhoven J. A., Energy Environ. Sci. 2015, 8, 1923. [Google Scholar]
- 46. Wang C. C., Li J. R., Lv X. L., Zhang Y. Q., Guo G., Energy Environ. Sci. 2014, 7, 2831. [Google Scholar]
- 47. Wang C. C., Zhang Y. Q., Li J., Wang P., J. Mol. Struct. 2015, 1083, 127. [Google Scholar]
- 48. Khaletskaya K., Pougin A., Medishetty R., Rösler C., Wiktor C., Strunk J., Fischer R. A., Chem. Mater. 2015, 27, 7248. [Google Scholar]
- 49. Gao Y., Li S., Li Y., Yao L., Zhang H., Appl. Catal. B: Environ. 2017, 202, 165. [Google Scholar]
- 50. Yu X. Y., Feng Y., Guan B., Lou X. W. D., Paik U., Energy Environ. Sci. 2016, 9, 1246. [Google Scholar]
- 51. Yang Y., Lun Z., Xia G., Zheng F., He M., Chen Q., Energy Environ. Sci. 2015, 8, 3563. [Google Scholar]
- 52. Hao J., Yang W., Zhang Z., Tang J., Nanoscale 2015, 7, 11055. [DOI] [PubMed] [Google Scholar]
- 53. Zheng Y., Jiao Y., Jaroniec M., Qiao S. Z., Angew. Chem. Int. Ed. 2015, 54, 52. [DOI] [PubMed] [Google Scholar]
- 54. Nørskov J. K., Bligaard T., Logadottir A., Kitchin J. R., Chen J. G., Pandelov S., Stimming U., J. Electrochem. Soc. 2005, 152, J23. [Google Scholar]
- 55. Hong W. T., Risch M., Stoerzinger K. A., Grimaud A., Suntivich J., Shao‐Horn Y., Energy Environ. Sci. 2015, 8, 1404. [Google Scholar]
- 56. Chen D., Chen C., Baiyee Z. M., Shao Z., Ciucci F., Chem. Rev. 2015, 115, 9869. [DOI] [PubMed] [Google Scholar]
- 57. Rossmeisl J., Dimitrievski K., Siegbahn P., Nørskov J. K., J. Phys. Chem. C 2007, 111, 18821. [Google Scholar]
- 58. Rossmeisl J., Logadottir A., Nørskov J. K., Chem. Phys. 2005, 319, 178. [Google Scholar]
- 59. Rossmeisl J., Qu Z. W., Zhu H., Kroes G. J., Nørskov J. K., J. Electroanal. Chem. 2007, 607, 83. [Google Scholar]
- 60. Trasatti S., Electrochim. Acta 1984, 29, 1503. [Google Scholar]
- 61. Man I. C., Su H. Y., Calle‐Vallejo F., Hansen H. A., Martínez J. I., Inoglu N. G., Kitchin J., Jaramillo T. F., Nørskov J. K., Rossmeisl J., ChemCatChem 2011, 3, 1159. [Google Scholar]
- 62. Zhu Y., Zhou W., Chen Y., Yu J., Liu M., Shao Z., Adv. Mater. 2015, 27, 7150. [DOI] [PubMed] [Google Scholar]
- 63. Zhu Y., Zhou W., Chen Z. G., Chen Y., Su C., Tadé M. O., Shao Z., Angew. Chem. Int. Ed. 2015, 54, 3897. [DOI] [PubMed] [Google Scholar]
- 64. Zhu Y., Zhou W., Yu J., Chen Y., Liu M., Shao Z., Chem. Mater. 2016, 28, 1691. [Google Scholar]
- 65. Su C., Wang W., Chen Y., Yang G., Xu X., Tadé M. O., Shao Z., ACS Appl. Mater. Interfaces 2015, 7, 17663. [DOI] [PubMed] [Google Scholar]
- 66. Huang Z. F., Song J., Li K., Tahir M., Wang Y. T., Pan L., Wang L., Zhang X., Zou J. J., J. Am. Chem. Soc. 2016, 138, 1359. [DOI] [PubMed] [Google Scholar]
- 67. You B., Jiang N., Sheng M., Gul S., Yano J., Sun Y., Chem. Mater. 2015, 27, 7636. [Google Scholar]
- 68. Shi Z., Wang Y., Lin H., Zhang H., Shen M., Xie S., Zhang Y., Gao Q., Tang Y., J. Mater. Chem. A 2016, 4, 6006. [Google Scholar]
- 69. Li X., Fang Y., Lin X., Tian M., An X., Fu Y., Li R., Jin J., Ma J., J. Mater. Chem. A 2015, 3, 17392. [Google Scholar]
- 70. Li J. S., Li S. L., Tang Y. J., Han M., Dai Z. H., Bao J. C., Lan Y. Q., Chem. Commun. 2015, 51, 2710. [DOI] [PubMed] [Google Scholar]
- 71. Zhang H., Ma Z., Duan J., Liu H., Liu G., Wang T., Chang K., Li M., Shi L., Meng X., Wu K., Ye J., ACS Nano 2016, 10, 684. [DOI] [PubMed] [Google Scholar]
- 72. Hou Y., Huang T., Wen Z., Mao S., Cui S., Chen J., Adv. Energy Mater. 2014, 4, 1400337. [Google Scholar]
- 73. Tang Y. J., Gao M. R., Liu C. H., Li S. L., Jiang H. L., Lan Y. Q., Han M., Yu S. H., Angew. Chem. Int. Ed. 2015, 54, 12928. [DOI] [PubMed] [Google Scholar]
- 74. Yerga R. M. N., Galván M. C. Á., del Valle F., de la Mano J. A. V., Fierro J. L. G., ChemSusChem 2009, 2, 471. [DOI] [PubMed] [Google Scholar]
- 75. Xing J., Fang W. Q., Zhao H. J., Yang H. G., Chem. Asian J. 2012, 7, 642. [DOI] [PubMed] [Google Scholar]
- 76. Maeda K., Domen K., J. Phys. Chem. C 2007, 111, 7851. [Google Scholar]
- 77. Kitano M., Hara M., J. Mater. Chem. 2010, 20, 627. [Google Scholar]
- 78. Wang W., Tadé M. O., Shao Z., Chem. Soc. Rev. 2015, 44, 5371. [DOI] [PubMed] [Google Scholar]
- 79. Li P., Ouyang S., Xi G., Kako T., Ye J., J. Phys. Chem. C 2012, 116, 7621. [Google Scholar]
- 80. Iwase A., Kato H., Kudo A., ChemSusChem 2009, 2, 873. [DOI] [PubMed] [Google Scholar]
- 81. Li Y. H., Peng C., Yang S., Wang H. F., Yang H. G., J. Catal. 2015, 330, 120. [Google Scholar]
- 82. Otal E. H., Kim M. L., Calvo M. E., Karvonen L., Fabregas I. O., Sierra C. A., Hinestroza J. P., Chem. Commun. 2016, 52, 6665. [DOI] [PubMed] [Google Scholar]
- 83. Nepal B., Das S., Angew. Chem. Int. Ed. 2013, 52, 7224. [DOI] [PubMed] [Google Scholar]
- 84. Hansen R. E., Das S., Energy Environ. Sci. 2014, 7, 317. [Google Scholar]
- 85. Sasan K., Lin Q., Mao C., Feng P., Chem. Commun. 2014, 50, 10390. [DOI] [PubMed] [Google Scholar]
- 86. Pullen S., Fei H., Orthaber A., Cohen S. M., Ott S., J. Am. Chem. Soc. 2013, 135, 16997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wang C., deKrafft K. E., Lin W., J. Am. Chem. Soc. 2012, 134, 7211. [DOI] [PubMed] [Google Scholar]
- 88. Bala S., Mondal I., Goswami A., Pal U., Mondal R., J. Mater. Chem. A 2015, 3, 20288. [Google Scholar]
- 89. Hou C. C., Li T. T., Chen Y., Fu W. F., ChemPlusChem 2015, 80, 1465. [DOI] [PubMed] [Google Scholar]
- 90. Sun X., Yu Q., Zhang F., Wei J., Yang P., Catal. Sci. Technol. 2016, 6, 3840. [Google Scholar]
- 91. Nohra B., Moll H. E., Albelo L. M. R., Mialane P., Marrot J., Mellot‐Draznieks C., O'Keeffe M., Biboum R. N., Lemaire J., Keita B., Nadjo L., Dolbecq A., J. Am. Chem. Soc. 2011, 133, 13363. [DOI] [PubMed] [Google Scholar]
- 92. Qin J. S., Du D. Y., Guan W., Bo X. J., Li Y. F., Guo L. P., Su Z. M., Wang Y. Y., Lan Y. Q., Zhou H. C., J. Am. Chem. Soc. 2015, 137, 7169. [DOI] [PubMed] [Google Scholar]
- 93. Dai X., Liu M., Li Z., Jin A., Ma Y., Huang X., Sun H., Wang H., Zhang X., J. Phys. Chem. C 2016, 120, 12539. [Google Scholar]
- 94. Chaikittisilp W., Torad N. L., Li C., Imura M., Suzuki N., Ishihara S., Ariga K., Yamauchi Y., Chem. Eur. J. 2014, 20, 4217. [DOI] [PubMed] [Google Scholar]
- 95. Aijaz A., Masa J., Rösler C., Xia W., Weide P., Botz A. J. R., Fischer R. A., Schuhmann W., Muhler M., Angew. Chem. Int. Ed. 2016, 55, 4087. [DOI] [PubMed] [Google Scholar]
- 96. Liu B., Shioyama H., Akita T., Xu Q., J. Am. Chem. Soc. 2008, 130, 5390. [DOI] [PubMed] [Google Scholar]
- 97. Zhang P., Sun F., Xiang Z., Shen Z., Yun J., Cao D., Energy Environ. Sci. 2014, 7, 442. [Google Scholar]
- 98. Wei W., Liang H., Parvez K., Zhuang X., Feng X., Müllen K., Angew. Chem. Int. Ed. 2014, 53, 1570. [DOI] [PubMed] [Google Scholar]
- 99. Ge L., Yang Y., Wang L., Zhou W., Marco R. D., Chen Z., Zou J., Zhu Z., Carbon 2015, 82, 417. [Google Scholar]
- 100. Xia B. Y., Yan Y., Li N., Wu H. B., Lou X. W. D., Wang X., Nat. Energy 2016, 1, 15006. [Google Scholar]
- 101. Yang H. M., Song X. L., Yang T. L., Liang Z. H., Fan C. M., Hao X. G., RSC Adv. 2014, 4, 15720. [Google Scholar]
- 102. Xu M., Han L., Han Y., Yu Y., Zhai J., Dong S., J. Mater. Chem. A 2015, 3, 21471. [Google Scholar]
- 103. Wei J., Feng Y., Liu Y., Ding Y., J. Mater. Chem. A 2015, 3, 22300. [Google Scholar]
- 104. He K., Cao Z., Liu R., Miao Y., Ma H., Ding Y., Nano Res. 2016, 9, 1856. [Google Scholar]
- 105. Feng Y., Yu X. Y., Paik U., Chem. Commun. 2016, 52, 1633. [DOI] [PubMed] [Google Scholar]
- 106. Liu Q., Tian J., Cui W., Jiang P., Cheng N., Asiri A. M., Sun X., Angew. Chem. Int. Ed. 2014, 53, 6710. [DOI] [PubMed] [Google Scholar]
- 107. Xu Y., Wu R., Zhang J., Shi Y., Zhang B., Chem. Commun. 2013, 49, 6656. [DOI] [PubMed] [Google Scholar]
- 108. Wu H. B., Xia B. Y., Yu L., Yu X. Y., Lou X. W. D., Nat. Commun. 2015, 6, 6512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Han Y., Zhai J., Zhang L., Dong S., Nanoscale 2016, 8, 1033. [DOI] [PubMed] [Google Scholar]
- 110. Han L., Yu X. Y., Lou X. W. D., Adv. Mater. 2016, 28, 4601. [DOI] [PubMed] [Google Scholar]
- 111. Liu B., Shioyama H., Jiang H., Zhang X., Xu Q., Carbon 2010, 48, 456. [Google Scholar]
- 112. Liu S., Zhang H., Zhao Q., Zhang X., Liu R., Ge X., Wang G., Zhao H., Cai W., Carbon 2016, 106, 74. [Google Scholar]
- 113. Ma T. Y., Dai S., Jaroniec M., Qiao S. Z., J. Am. Chem. Soc. 2014, 136, 13925. [DOI] [PubMed] [Google Scholar]
- 114. Gadipelli S., Zhao T., Shevlin S. A., Guo Z., Energy Environ. Sci. 2016, 9, 1661. [Google Scholar]
- 115. Li X., Niu Z., Jiang J., Ai L., J. Mater. Chem. A 2016, 4, 3204. [Google Scholar]
- 116. Hou Y., Li J., Wen Z., Cui S., Yuan C., Chen J., Nano Energy 2015, 12, 1. [Google Scholar]
- 117. Fan L., Liu P. F., Yan X., Gu L., Yang Z. Z., Yang H. G., Qiu S., Yao X., Nat. Commun. 2016, 7, 10667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Liu Y., Zhou X., Ding T., Wang C., Yang Q., Nanoscale 2015, 7, 18004. [DOI] [PubMed] [Google Scholar]
- 119. Lu J., Zhou W., Wang L., Jia J., Ke Y., Yang L., Zhou K., Liu X., Tang Z., Li L., Chen S., ACS Catal. 2016, 6, 1045. [Google Scholar]
- 120. Wang S., Hou Y., Lin S., Wang X., Nanoscale 2014, 6, 9930. [DOI] [PubMed] [Google Scholar]
- 121. Esswein A. J., Surendranath Y., Reece S. Y., Nocera D. G., Energy Environ. Sci. 2011, 4, 499. [Google Scholar]
- 122. Jahan M., Bao Q., Loh K. P., J. Am. Chem. Soc. 2012, 134, 6707. [DOI] [PubMed] [Google Scholar]
- 123. Jahan M., Liu Z., Loh K. P., Adv. Funct. Mater. 2013, 23, 5363. [Google Scholar]
- 124. Jiao L., Zhou Y. X., Jiang H. L., Chem. Sci. 2016, 7, 1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Narayan T. C., Miyakai T., Seki S., Dincă M., J. Am. Chem. Soc. 2012, 134, 12932. [DOI] [PubMed] [Google Scholar]
- 126. Jeon I., Sun L., Negru B., Van Duyne R. P., Dincă M., Harris T. D., J. Am. Chem. Soc. 2016, 138, 6583. [DOI] [PubMed] [Google Scholar]
- 127. Lu X.‐F., Liao P.‐Q., Wang J.‐W., Wu J.‐X., Chen X.‐W., He C.‐T., Zhang J.‐P., Li G.‐R., Chen X.‐M., J. Am. Chem. Soc. 2016, 138, 8336. [DOI] [PubMed] [Google Scholar]
- 128. Takaishi S., Hosoda M., Kajiwara T., Miyasaka H., Yamashita M., Nakanishi Y., Kitagawa Y., Yamaguchi K., Kobayashi A., Kitagawa H., Inorg. Chem. 2009, 48, 9048. [DOI] [PubMed] [Google Scholar]
- 129. Sun L., Campbell M. G., Dincă M., Angew. Chem. Int. Ed. 2016, 55, 3566. [DOI] [PubMed] [Google Scholar]
- 130. Sun L., Miyakai T., Seki S., Dincă M., J. Am. Chem. Soc. 2013, 135, 8185. [DOI] [PubMed] [Google Scholar]
- 131. Sun L., Hendon C. H., Minier M. A., Walsh A., Dincă M., J. Am. Chem. Soc. 2015, 137, 6164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Park S. S., Hontz E. R., Sun L., Hendon C. H., Walsh A., Van Voorhis T., Dincă M., J. Am. Chem. Soc. 2015, 137, 1774. [DOI] [PubMed] [Google Scholar]
- 133. Sheberla D., Sun L., Blood‐Forsythe M. A., Er S., Wade C. R., Brozek C. K., Aspuru‐Guzik A., Dincă M., J. Am. Chem. Soc. 2014, 136, 8859. [DOI] [PubMed] [Google Scholar]
- 134. Campbell M. G., Sheberla D., Liu S. F., Swager T. M., Dincă M., Angew. Chem. Int. Ed. 2015, 54, 4349. [DOI] [PubMed] [Google Scholar]
- 135. Campbell M. G., Liu S. F., Swager T. M., Dincă M., J. Am. Chem. Soc. 2015, 137, 13780. [DOI] [PubMed] [Google Scholar]
- 136. Miner E. M., Fukushima T., Sheberla D., Sun L., Surendranath Y., Dincă M., Nat. Commun. 2016, 7, 10942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Sheberla D., Bachman J. C., Elias J. S., Sun C.‐J., Shao‐Horn Y., Dincă M., Nat. Mater. doi: 10.1038/NMAT4766. [DOI] [PubMed] [Google Scholar]
- 138. Gomes Silva C., Luz I., Llabrés i Xamena F. X., Corma A., García H., Chem. Eur. J. 2010, 16, 11133. [DOI] [PubMed] [Google Scholar]
- 139. Du J. J., Yuan Y. P., Sun J. X., Peng F. M., Jiang X., Qiu L. G., Xie A. J., Shen Y. H., Zhu J. F., J. Hazard. Mater. 2011, 190, 945. [DOI] [PubMed] [Google Scholar]
- 140. Fu Y., Sun D., Chen Y., Huang R., Ding Z., Fu X., Li Z., Angew. Chem. Int. Ed. 2012, 51, 3364. [DOI] [PubMed] [Google Scholar]
- 141. Shi L., Wang T., Zhang H., Chang K., Meng X., Liu H., Ye J., Adv. Sci. 2015, 2, 1500006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Toyao T., Saito M., Horiuchi Y., Mochizuki K., Iwata M., Higashimura H., Matsuoka M., Catal. Sci. Technol. 2013, 3, 2092. [Google Scholar]
- 143. Shen L., Luo M., Liu Y., Liang R., Jing F., Wu L., Appl. Catal. B: Environ. 2015, 166–167, 445. [Google Scholar]
- 144. deKrafft K. E., Wang C., Lin W., Adv. Mater. 2012, 24, 2014. [DOI] [PubMed] [Google Scholar]
- 145. Pham M. H., Dinh C. T., Vuong G. T., Ta N. D., Do T. O., Phys. Chem. Chem. Phys. 2014, 16, 5937. [DOI] [PubMed] [Google Scholar]
- 146. Bagtache R., Rekhila G., Abdmeziem K., Trari M., Mater. Sci. Semicond. Process. 2014, 23, 144. [Google Scholar]
- 147. Horiuchi Y., Toyao T., Saito M., Mochizuki K., Iwata M., Higashimura H., Anpo M., Matsuoka M., J. Phys. Chem. C 2012, 116, 20848. [Google Scholar]
- 148. Toyao T., Saito M., Dohshi S., Mochizuki K., Iwata M., Higashimura H., Horiuchi Y., Matsuoka M., Chem. Commun. 2014, 50, 6779. [DOI] [PubMed] [Google Scholar]
- 149. He J., Wang J., Chen Y., Zhang J., Duan D., Wang Y., Yan Z., Chem. Commun. 2014, 50, 7063. [DOI] [PubMed] [Google Scholar]
- 150. Wang Y., Hong J., Zhang W., Xu R., Catal. Sci. Technol. 2013, 3, 1703. [Google Scholar]
- 151. Tributsch H., Coord. Chem. Rev. 2004, 248, 1511. [Google Scholar]
- 152. Zhang L., Cui P., Yang H., Chen J., Xiao F., Guo Y., Liu Y., Zhang W., Huo F., Liu B., Adv. Sci. 2016, 3, 1500243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Butler K. T., Hendon C. H., Walsh A., J. Am. Chem. Soc. 2014, 136, 2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Zhou T., Du Y., Borgna A., Hong J., Wang Y., Han J., Zhang W., Xu R., Energy Environ. Sci. 2013, 6, 3229. [Google Scholar]
- 155. Wen M., Mori K., Kamegawa T., Yamashita H., Chem. Commun. 2014, 50, 11645. [DOI] [PubMed] [Google Scholar]
- 156. Truong Q. D., Hoa H. T., Le T. S., Catal. Commun. 2015, 59, 55. [Google Scholar]
- 157. Han J., Wang D., Du Y., Xi S., Hong J., Yin S., Chen Z., Zhou T., Xu R., J. Mater. Chem. A 2015, 3, 20607. [Google Scholar]
- 158. Zhen W., Ma J., Lu G., Appl. Catal. B: Environ. 2016, 190, 12. [Google Scholar]
- 159. Zhang C. F., Qiu L. G., Ke F., Zhu Y. J., Yuan Y. P., Xu G. S., Jiang X., J. Mater. Chem. A 2013, 1, 14329. [Google Scholar]
- 160. Zhang Y. F., Qiu L. G., Yuan Y. P., Zhu Y. J., Jiang X., Xiao J. D., Appl. Catal. B: Environ. 2014, 144, 863. [Google Scholar]
- 161. Zhang C., Ai L., Jiang J., J. Mater. Chem. A 2015, 3, 3074. [Google Scholar]
- 162. Wang G., Sun Q., Liu Y., Huang B., Dai Y., Zhang X., Qin X., Chem. Eur. J. 2015, 21, 2364. [DOI] [PubMed] [Google Scholar]
- 163. Wang R., Gu L., Zhou J., Liu X., Teng F., Li C., Shen Y., Yuan Y., Adv. Mater. Interfaces 2015, 2, 1500037. [Google Scholar]
- 164. Lin R., Shen L., Ren Z., Wu W., Tan Y., Fu H., Zhang J., Wu L., Chem. Commun. 2014, 50, 8533. [DOI] [PubMed] [Google Scholar]
- 165. Wang Y., Zhang Y., Jiang Z., Jiang G., Zhao Z., Wu Q., Liu Y., Xu Q., Duan A., Xu C., Appl. Catal. B: Environ. 2016, 185, 307. [Google Scholar]
- 166. Li Q., Guo B., Yu J., Ran J., Zhang B., Yan H., Gong J. R., J. Am. Chem. Soc. 2011, 133, 10878. [DOI] [PubMed] [Google Scholar]
- 167. Wang J.‐L., Wang C., Lin W., ACS Catal. 2012, 2, 2630. [Google Scholar]
- 168. Zhang Z.‐M., Zhang T., Wang C., Lin Z., Long L.‐S., Lin W., J. Am. Chem. Soc. 2015, 137, 3197. [DOI] [PubMed] [Google Scholar]
- 169. Xu H.‐Q., Hu J., Wang D., Li Z., Zhang Q., Luo Y., Yu S.‐H., Jiang H.‐L., J. Am. Chem. Soc. 2015, 137, 13440. [DOI] [PubMed] [Google Scholar]
- 170. Hou C.‐C., Li T.‐T., Cao S., Chen Y., Fu W.‐F., J. Mater. Chem. A 2015, 3, 10386. [Google Scholar]
- 171. Kent C. A., Mehl B. P., Ma L., Papanikolas J. M., Meyer T. J., Lin W., J. Am. Chem. Soc. 2010, 132, 12767. [DOI] [PubMed] [Google Scholar]
- 172. Kent C. A., Liu D., Ma L., Papanikolas J. M., Meyer T. J., Lin W., J. Am. Chem. Soc. 2011, 133, 12940. [DOI] [PubMed] [Google Scholar]
- 173. Kent C. A., Liu D., Meyer T. J., Lin W., J. Am. Chem. Soc. 2012, 134, 3991. [DOI] [PubMed] [Google Scholar]
- 174. Maza W. A., Morris A. J., J. Phys. Chem. C 2014, 118, 8803. [Google Scholar]
- 175. Maza W. A., Ahrenholtz S. R., Epley C. C., Day C. S., Morris A. J., J. Phys. Chem. C 2014, 118, 14200. [Google Scholar]
- 176. Maza W. A., Padilla R., Morris A. J., J. Am. Chem. Soc. 2015, 137, 8161. [DOI] [PubMed] [Google Scholar]
- 177. Maza W. A., Haring A. J., Ahrenholtz S. R., Epley C. C., Lin S. Y., Morris A. J., Chem. Sci. 2016, 7, 719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Shi L., Wang T., Zhang H., Chang K., Ye J., Adv. Funct. Mater. 2015, 25, 5360. [Google Scholar]