

Abstract
The therapeutic effect of glial progenitor transplantation in diseases of dysmyelination is currently attributed to the formation of new myelin. Using magnetic resonance imaging (MRI), we show that the therapeutic outcome in dysmyelinated shiverer mice is dependent on the extent of cell migration but not presence of mature and compact myelin. Human or mouse glial restricted progenitors (GRPs) were transplanted into rag2−/− shiverer mouse neonates and followed for over one year. Mouse GRPs produced mature myelin as detected with multi-parametric MRI, but showed limited migration without extended animal lifespan. In sharp contrast, human GRPs migrated extensively and significantly increased animal survival, but production of mature myelin did not occur until 46 weeks post-grafting. We conclude that human GRPs can extend the survival of transplanted shiverer mice prior to production of mature myelin, while mouse GRPs fail to extend animal survival despite early presence of mature myelin. This paradox suggests that transplanted GRPs provide therapeutic benefits through biological processes other than mature myelin capable to facilitate rapid conduction, challenging the current dogma of the role of myelin in the function of the central nervous system.
Keywords: Glial Progenitors, Transplantation, Myelin, MRI, Shiverer
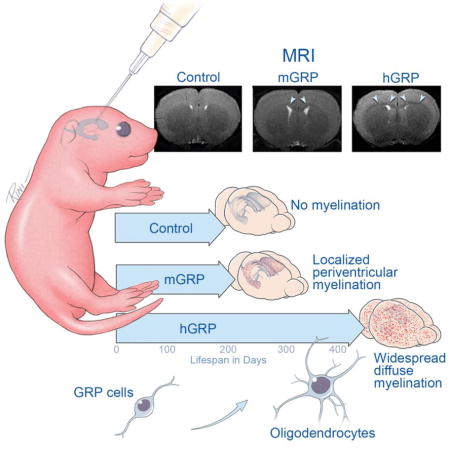
Graphical abstract
INTRODUCTION
Dysmyelinating disorders of the central nervous system (CNS), also referred to as leukodystrophies, are characterized by dysfunctional myelin sheaths. These conditions are associated with severe neurological consequences with onset typically at the early stages of development (Perlman and Mar, 2012). The pathology is related to inherited genetic mutations affecting primarily glial cells and their ability to form myelin. Accordingly, therapeutic strategies aiming at the early replacement of dysfunctional glia with exogenous counterparts to wrap axons with new, proper myelin are an attractive therapeutic option (Yandava et al., 1999). Indeed, in recent years, several types of neural progenitor cells were shown suitable for such replacement. The most promising results so far have been achieved with the use of progenitors derived from fetal CNS including CD133-expressing neural stem cells (Buchet et al., 2011; Uchida et al., 2012) or A2B5+/PSA-NCAM− glial restricted progenitors (GRPs) (Rao and Mayer-Proschel, 1997; Walczak et al., 2011; Windrem et al., 2008). Transplantation of either of these human cell types into neonatal dysmyelinated immunodeficient shiverer mice results in their extensive engraftment, migration throughout the neuraxis, and differentiation towards mature glia. Transplanted mice display extensive chimerization with the majority of glia (both oligodendrocytes and astrocytes) of human origin and murine axons are extensively myelinated by transplanted human cells. While the lifespan of dysmyelinated mice can be significantly extended by neonatal transplantation with human oligodendrocyte precursors (Windrem et al., 2008), it has not been established whether the presence of mature and compact myelin facilitating rapid conduction occurs early enough in order to elongate lifespan. Due to their close proximity to axons, oligodendrocytes are ideally suited to support neurons, e.g. by supplying motor neurons with lactate via monocarboxylate transporters (Lee et al., 2012).
While the landmark study by Windrem et al. demonstrated that global replacement of glia is feasible (Windrem et al., 2008), the 25% success rate in extending lifespan that was achieved is expected to be too low for a clinically useful therapy. Still, encouraging preclinical data led to Phase I clinical trials of fetal-derived human neural stem cells transplanted in children suffering from Pelizaeus-Merzbacher disease (Gupta et al., 2012). That study met its primary objective in that it was safe; however, at one year post-transplantation, the activity of the graft remained unclear. Magnetic resonance imaging (MRI), the primary method for non-invasive assessment of myelin, was used in this clinical study; however, interpretation of the obtained imaging results without properly validated imaging studies in animals proved to be difficult and hence the functional status of the transplanted cells remained inconclusive.
The aim of our current study was to employ MRI, a non-invasive and clinically used imaging method, for monitoring of appearance of transplant-derived mature myelin serially over time. As it is possible that the variability in therapeutic effects in the study of Windrem et al. (Windrem et al., 2008) may be related to the inter-species mismatch between donor and recipient, we also included allogeneic GRPs, in order to test the hypothesis that these cells may have a greater therapeutic effect due to faster myelination and intra-species compatibility (Muse et al., 2001). Mice transplanted with either human or mouse GRPs were followed for 400 days with T2-weighted MRI, magnetization transfer (MT) MRI, and diffusion tensor imaging (DTI), which are commonly used for the evaluation of myelination in the clinic and in several rodent models (Laule et al., 2007; Song et al., 2002). We report here that mouse GRPs (mGRPs) indeed produce mature myelin very early, but without therapeutic effect. In contrast, human GRPs (hGRPs) rescued nearly 50% of mice before the mature, compact myelin could be detected. These results indicate that biological processes other than the axonal insulation by mature and compact myelin play a significant role and should be taken into consideration for further development of cell therapy in dysmyelinated diseases.
MATERIALS AND METHODS
Cells
mGRPs were derived from mid-gestation (E13) proteolipid protein-green fluorescent protein (PLP-GFP) transgenic mice as previously described (Phillips et al., 2012) with one modification that the cells were dissected from the fetal forebrain tissue. hGRPs (Sandrock et al., 2010) (“Q cells®”) were provided by Q Therapeutics, Inc. and used for transplantation immediately upon thawing. mGRPs were cultured in serum-free Dulbeccos’s modified Eagle’sF12 medium supplemented with N2, B27, bovine serum albumin, and basic fibroblast growth factor. Before transplantation, cells were trypsinized, centrifuged and resuspended in phosphate-buffered saline at a concentration of 100,000 cells/μl.
Cell transplantation
The Johns Hopkins institutional animal care and use committee approved this study. GRPs were transplanted bilaterally into the lateral ventricles of cryoanesthetized neonatal rag2−/− shiverer mice of either sex (n=9 for mGRP transplants and n=40 for hGRP transplants) within three days of birth. A total of 200,000 cells were transplanted into each site with 31G Hamilton needle attached to gastight #1701 syringe (Hamilton Reno, NV) at the rate of 2μl/min. Control animals were subjected to the identical procedure except injection of saline. The pups were then returned to their mother until weaning at 3 weeks. Mice were checked daily for survival recordings. For histopathology, pentobarbital anesthetized mice were sacrificed and transcardially perfused with 4% paraformaldehyde (PFA). Brain and spinal cord tissue was harvested and immersed over night in 4% PFA at 4°C. Age-matched non-transplanted rag2−/− shiverers (n=9) and wild type mice (n=5) of either sex were used as controls.
MRI
In vivo MRI was performed on a horizontal 11.7 Tesla MR scanner (Bruker Biospin, Billerica, MA, USA) with a triple-axis gradient (maximum gradient strength = 74 Gauss/cm). During imaging, mice were anesthetized with isoflurane (1%) together with air and oxygen mixed at 3:1 ratio via a vaporizer and positioned in an animal holder with circulating warm water (Bruker Biospin, Billerica, MA, USA). Respiration was monitored via a pressure sensor (SAII, Stony Brook, NY, USA) and maintained at 40–60 breaths per minute. After imaging, animals recovered within 5 minutes.
Image acquisition was performed using a quadrature transmit volume coil (70 mm diameter) and a 4-channel mouse brain receive-only phased array coil (Bruker Biospin, Billerica, MA, USA). Multi-slice T2-weighted images were acquired using the rapid acquisition with refocused echoes (RARE) sequence with an echo time (TE) of 50 ms, a repetition time (TR) of 3600 ms, 4 signal averages, echo train length of 8, field of view (FOV) of 15 × 15 mm, 32 slices, and a native resolution of 0.078 × 0.078 × 0.50 mm. MT images were acquired using a RARE sequence with a chain of ten 3 ms Gaussian saturation pulses with a power of 12 μT and an offset frequency of −2 kHz from water and the following parameters: TE/TR = 7.5 ms/3000 ms, 8 signal averages, echo train length of 8, the same FOV as the T2-weighted images, and a native resolution of 0.117 × 0.117 × 0.500 mm. Images with no saturation pulse (M0) were also acquired, and MTR maps were calculated using the expression MTR = 1−Mt/M0. For DTI, a modified three-dimensional (3D) diffusion-weighted gradient and spin echo (DW-GRASE) sequence (Wu et al., 2013) was used with the following parameters: TE/TR = 27.5/600 ms, 2 signal averages, 20 imaging echoes (4 spin echoes distributed along the phase encoding direction and 16 gradient echoes distributed along the slice selection direction) after each excitation with twin navigator echoes in the end for motion and phase corrections, diffusion gradient duration = 5 ms, diffusion time = 12 ms, b = 3000 s/mm2, FOV = 16 × 16 × 16 mm, matrix size =128 × 128 × 60, and a native imaging resolution = 0.125 × 0.125 × 0.267 mm (Aggarwal et al., 2010). Two non-diffusion-weighted and six diffusion-weighted images were acquired. With respiratory gating, the total imaging time was approximately 3 hours.
Image processing
The 3D images acquired using the DW-GRASE sequence were reconstructed from raw data in MATLAB (www.mathworks.com) with navigator-based motion and phase correction(Aggarwal et al., 2010). Using the log-linear fitting method implemented in DTIStudio (http://www.mristudio.org), diffusion tensor was calculated at each pixel along with the apparent diffusion coefficient (ADC), fractional anisotropy (FA), primary eigenvector, axial diffusivity (λ||, the primary eigenvalue), and radial diffusivity (λ⊥, the average of the secondary and tertiary eigenvalues) (Jiang et al., 2006). The six diffusion-weighted images were averaged to generate the isotropic diffusion-weighted (iDW) images. Skull stripping was performed by manually outlining the boundary of the brain in the iDW images using Amira (FEI Visualization Sciences Group, http://www.vsg3d.com/amira) and removing signals from non-brain tissues. The skull-stripped mouse brain images were first rigidly aligned to the in vivo mouse brain images (referred to as the template image in the following paragraphs) in our MRI based mouse brain atlas (Wu et al., 2013) using the DiffeoMap software (Chuang et al., 2011). The rigidly aligned FA, λ||, and λ⊥ images from control, shiverer, and transplanted mice were then spatially normalized to the template image using the dual-channel (iDW+FA) large deformation diffeomorphic metric mapping (LDDMM)(Ceritoglu et al., 2009). At each voxel, the mean and standard deviation values of FA, λ||, and λ⊥ were calculated to generate group-averaged FA, λ||, and λ⊥ maps and standard deviation maps for shiverer control group. For the transplanted animals, a Z score maps with respect to the shiverer control group were computed at each pixel as
where Xtransplanted is the FA, λ||, or λ⊥ value of a transplanted animal at a voxel and X̄ref and SDref are the mean and standard error of the shiverer control group at the same voxel.
Data analysis
For spatial profile analysis and region of interest analysis, the midsection of the corpus callosum at bregma 0.38 mm was chosen, as this region was consistently myelinated in all transplanted mice. For correlating MRI parameters to the level of donor-derived myelination, we compared the MR images to histological coronal sections. We selected clusters of 8 voxels in the MR images and matched them to regions in the histological sections corresponding to the appropriate resolution (125 × 250 μm regions for comparing to FA and RD, 116 × 232 μm regions for comparing to MTR, and 156 × 312 μm regions for comparing to T2-weighted images). GFP fluorescence signal intensity was measured to reflect myelination. There was no variability in MRI parameters for the shiverer control group in the region of the brain, allowing us to exclude micro-anatomical differences in the corpus callosum as a confounding factor in our analysis. A full range of regions of interest was selected – from non-myelinated to heavily myelinated regions. Group average spatial profiles were generated by measuring MRI parameters across the cortical-corpus callosal-subcallosal region in coronal sections at bregma 0.38 mm. T2-weighted signal intensities were normalized in reference to the cortical region.
Histology and immunohistochemistry
The left hemispheres were cryopreserved and cut into 30 μm coronal sections and processed for eriochrome cyanine staining and immunofluorescence. For immunofluorescence, sections were first washed with PBS, blocked with 5% bovine serum albumin, and incubated overnight at 4 °C with primary antibodies: anti-myelin basic protein (AbD Serotec, MCA4095) and anti-human specific GFAP and anti-human specific cytoplasmic marker (Stem Cells Inc., STEM123, STEM121 respectively). Sections were rinsed with PBS and incubated with Alexa fluor secondary antibodies (Invitrogen). Images were obtained using an inverted microscope (Zeiss, Axio Observer.Z1) equipped with a motorized table. Fluorescent images were analyzed with Zen software for quantification of relative fluorescent intensity.
TEM and g-ratio analysis
The right hemispheres were used for TEM and Toluidine blue staining. Brain tissue was sectioned coronally using a microtome. Every other section was collected for immunohistochemistry to confirm successful transplantation in the right hemisphere. Sections collected for toluidine blue and electron microscopy were fixed in 4% glutaraldehyde. Sections for toluidine blue were immersed in a solution of 0.5% toluidine blue in 1 % sodium borate, washed with distilled water, dehydrated with ethanol, and dried on a hot plate at 60°C. Coronal sections of the corpus callosum were postfixed in OsO4 and embedded in Epon. Thin sections of 70 nm were stained with citrate/uranyl acetate as previously described (Farah et al., 2011). Images were acquired using a Zeiss Libra transmission electron microscope. Corpus callosum g-ratios were determined from TEM by manually measuring axon diameter and outer myelin layer diameter (axon average diameter/fiber average diameter) using Photoshop. All axons with > three lamellae were quantified (50–100 axons per animal). Calculations for the number of myelinated axons per unit area were adjusted for differences in axon diameter. To assess compaction of exogenous myelin, areas of 17 μm2 in the corpus callosum were selected. The distance between lamellae was measured by dividing the total myelin thickness by the number of myelin lamellae. An average of four measurements was calculated for each axon.
Statistical analysis
For the comparison of group average spatial profiles, one-way ANOVA was performed using IBM SPSS Statistics 22 software. G-ratio profiles were analyzed using PROC MIXED (SAS 9.2, SAS Institute Inc., Cary, NC, USA) and the least square means were used for comparison between groups. A value of p<0.01 was considered as statistically significant. Coronal profile graphs are presented as mean ± SD.
RESULTS
GRPs of mouse and human origin were cultured as monolayer for three days and immunostained for a panel of progenitor vs. mature markers (Figure 1). GRPs from both species were shown to frequently express GRP signature markers with the following phenotype distribution for mGRPs: NG2 (84.9%±4.9%); A2B5 (93.6%±3%); Olig1 (78.8%±6%); Olig2 (96.1%±2%); PDGFRa (93.4%±7%). Phenotypic characterization of hGRPs showed the following distribution: A2B5 (82.2%±10%); Olig1 (60.4%±7%); Olig2 (39.2%±11%); PDGFRa (82.8%±17%). Expression of mature oligodendrocyte marker MBP was not detected in neither human or mouse cells. The neuronal marker b-III tubulin was expressed by 3.0%±2% of mouse cells and 5.6% ±4%. Nearly half of of mGRPs (48.6%±3.3%) and 69.0%±10% of hGRPs were GFAP positive. Antigenic profile indicates that hGRPs appear as less mature compared to mGRPs. Quantitative results are shown in Figure 1. Prior to transplantation, mGRPs expressed GFP only at low basal level as GFP expression is controlled by the oligodendrocyte-specific PLP promoter. Both mGRPs and hGRPs were transplanted into neonatal shiverer mouse brains to determine the efficiency of myelination and their capacity to rescue shiverer phenotype.
Figure 1. Phenotypic characterization of mouse and human GRP cells prior to transplantation.

Mouse and human GRPs were derived from mid-gestation (day 12; week 22 respectively) whole forebrain and immunostained within three days of culture. for NG2, A2B5, Olig1, Olig2, PDGFRa, MBP, b-III Tubulin, GFAP. Scale bar=100 μm applies to all images. Graphs show quantification of cells positive for specific antigens.
Longitudinal multiparametric MRI of shiverer mice transplanted with mouse GRPs
Transplanted and control neonatal rag2−/− shiverer mice as well as wild type (WT) counterparts were subjected to MRI. MRI discriminates well between white and gray matter in the brain and offers both semi-quantitative (T2-weighted signal intensity) and quantitative (magnetization transfer ratio (MTR), radial diffusivity (λ⊥) or fractional anisotropy (FA)) measures. Imaging was performed 18, 31, 46, and 62 weeks post-grafting in the hGRP transplanted group or up to 18 weeks for the mGRP transplanted group as the latter cohort did not survive beyond that time point. (Figure 2). In vivo MRI of 18-weeks old WT mice showed excellent white–gray matter contrast on T2-weighted and DTI-derived fractional anisotropy (FA) images. For example, the corpus callosum in the WT mice presented with a hypointense T2 signal and high FA values. In age-matched shiverer mice, the T2 contrast was practically absent with the white matter appearing hyperintense compared to the cortex, and FA values were reduced compared to WT mice. Quantitative analysis of all studied MRI parameters (T2, MTR, FA, radial diffusivity (RD) revealed significant differences between WT and shiverer mice in all parameters within corpus callosum.
Figure 2. MRI readouts for hGRP vs mGRP transplanted mice.

(a) Representative T2-w and diffusion MRI (FA) images are shown for WT, non-transplanted rag2−/− shiverer, hGRP-grafted, and mGRP-grafted mice. In the corpus callosum (red arrows), wild type mice and mGRP grafted mice show hypointense white matter on the T2-w images and hyperintensity on the FA maps as compared to rag2−/− shiverer controls. T2-w images and FA maps of hGRP-grafted mice resemble rag2−/− shiverer control images with hyperintense white matter on the T2-w images and hypointensity on the FA at the 18 and 31-week time points. It is not until the 62-week time point that there is a qualitative improvement of the T2-w images and FA maps resembling WT images. (b) Group average spatial profiles of normalized T2-w signal intensities, MTR, FA, and RD were generated across the cortex, corpus callosum and subcallosal gray matter at bregma 0.38 mm for rag2−/− shiverer controls (n=9, solid black line, open marker), WT controls (n=5, solid black line), mGRP transplanted shiverers (n=9, solid green line), and hGRP transplants at 18 weeks (n=9), 31 weeks (n=5), 46 weeks (n=4), and 62 weeks (n=7) post transplantation (red solid, light red dotted, light red dashed, and light red solid lines, respectively). Shaded area indicates the region of the corpus callosum. For statistical analysis, error bars represent standard deviations within each group (* indicates p value ≤ 0.01, ** indicates p value ≤ 0.001).
In mice transplanted with mGRPs, the MRI parameters were partially improved toward WT values. At 18 weeks post-grafting, the corpus callosum showed hypointense T2 signals and increased FA values. Normalization of MRI parameters in mGRP-grafted mice was limited in area coverage and present only in the vicinity of the ventricular system. To examine myelination by mGRP cells in the corpus callosum, group average spatial profiles of normalized T2-weighted signal intensities, MTR, FA, and λ⊥ were generated across the cortex, corpus callosum, and subcallosal gray matter at bregma 0.38 mm for shiverer controls, transplanted shiverers, and WT control mice (Figure 2b). Normalized T2-weighted signal intensity profiles showed decreased signal intensity in the mGRP grafted group by as much as 44.2±8.8% towards WT values (p<0.001). MTR profiles showed an increase in the corpus callosum of transplanted shiverers by as much as 62.5±14.1% improvement towards WT values (p≤0.001). WT controls and transplanted shiverers had increased FA values in the corpus callosum (p≤0.001, p≤0.001, respectively) and decreased radial diffusivity (RD; p≤0.001, p≤0.01, respectively) as compared to non-transplanted shiverer controls. Mean fractional anisotropy (FA) values in grafted mice had as much as 40.4 ± 23.6% improvement toward mean WT values in the corpus callosum; while mean λ⊥ values in grafted mice had as much as 32.6 ± 26.6% improvement toward mean WT values in the corpus callosum (Figure 2b, green).
Longitudinal multiparametric MRI of shiverer mice transplanted with human GRPs
At the same exact time point for the mGRP-transplanted group (18 weeks post-transplantation), hGRP-grafted mice had none of MRI parameters improved. Moreover, at 31 weeks post-transplantation, MRI parameters remained unimproved with hyperintensity on T2-weighted MRI, and low MTR and FA signals. Only at 46 and 62 weeks after grafting did MR imaging detect patterns that could be expected from newly myelinated white matter including hypointensity on T2 and increased MTR values, though FA and RD showed no significant improvements even at 62 weeks (Figure 2a). Group average spatial profiles of normalized T2-weighted signal intensities, MTR, FA, and λ⊥ were generated across the corpus callosum and showed similar results. T2-weighted and MTR signals showed improvement toward WT values in the hGRP transplanted shiverers at the 46 week and 62 week time points (Figure 2b, red). Normalized T2-weighted signal intensity profiles showed decreased signal intensity in the grafted group by as much as 43.4 ± 14.6% towards WT values at the 62-week time point (p<0.001). MTR profiles showed an increase in the corpus callosum of hGRP-transplanted shiverers by as much as 53.6 ± 16.4% improvement towards WT values at the 62-week time point (p≤0.001). Surprisingly, hGRP-transplanted shiverers did not show any improvement in FA or RD values for the corpus callosum.
Time course of myelination from post-mortem histology
Immunohistochemistry for MBP in WT mice showed abundant staining throughout the brain with highest signal within white matter structures. As expected, this staining was negative in shiverer mutants as they produce an abnormal, truncated form of MBP non-reacting with the antibody. In mice transplanted with mGRPs at 18 weeks, there was extensive positive MBP staining in the corpus callosum (Figure 3a). A sagittal section of the brain shows the distribution of transplanted cells myelinating the corpus callosum, fimbria, and a small cortical area limited to the periventricular region (Figure 5a). For the hGRP-transplanted animals, sacrificed at different time points immediately following MRI (18, 31, 46 and 62 weeks post-grafting), the MBP expression was very sparse at 18 weeks. Myelination became widespread at 31 weeks and over time, its extent gradually increased, resembling the pattern observed in WT animals at 62 weeks post grafting (Figure 3a).
Figure 3. Histological comparison of myelination at 18, 31, and 62 weeks post transplantation.

(a) MBP immunoreactivity is consistent with the observed MRI pattern. WT mice exhibit normal robust myelin throughout the brain, shiverer controls are negative for MBP staining, and mGRP-grafted mice exhibit an intermediate amount of myelination in the corpus callosum. hGRP-grafted mice have increasing levels of immunoreactivity against MBP shows over time. (b) Eriochrome staining reveals differences in the corpus callosum among the groups (indicated by black arrows). (c) Toluidine blue staining of the mid section of the corpus callosum differences in axon size and thickness of myelination between the different cohorts. (d) Ultrastructural TEM images at 50,000× magnification. Bar in (a, b) = 1 mm, in (c) = 20 μm, in (d) = 1 μm.
Figure 5. Correlation between MRI parameters and fluorescence signal intensity from PLP-GFP cells.

(a) Sagittal section across the mid-portion of the corpus callosum at 18 weeks post transplantation reveals that PLP-GFP+ donor cells integrate into the corpus callosum, fimbria and hippocampal region of transplanted mice. This matches the normalized RD map comparing a representative transplanted mouse to the group average of shiverer controls. Scale bar in (a) = 1 mm. The color scale for the normalized RD map ranges from −5 to +5 standard deviations from the shiverer control group average. (b) Normalized T2-w signal intensity, FA, RD, and MTR values plotted against GFP signal intensity demonstrates strong correlations (R2 values equal to 0.89, 0.94, 0.88 0.80 respectively).
Eriochrome cyanin (Figure 3b), which stains hydrophobic compartments within myelin proteins and lipid molecules (Kiernan, 2007), extensively stained white matter structures of the WT controls but was negative in control shiverer mice indicating the poor myelin integrity in shiverer controls. Brain tissue of 18 weeks old mGRP-transplanted mice was positive for eriochrome in the medial corpus callosum, fimbria, and to some extent in the anterior commissure, suggesting that the hydrophobic compartments were restored in newly myelinated regions. In contrast, the hGRP-transplanted cohort showed very low eriochrome positivity at 18 and 31 weeks and its presence was limited to the medial corpus callosum. At 62 weeks, however, the staining was abundant and closely resembled the pattern observed in the WT animals.
Ultrastructural analysis of the mid section of the corpus callosum at bregma 0.38 mm with toluidine blue staining and transmission electron microscopy (TEM) (Figure 3c and d) revealed pronounced differences between WT, transplanted cohorts, and shiverer brain. Wild type mice and mGRP transplants had a significantly increased number of myelinated axons per area as compared to control shiverer mice at 18 weeks. hGRP transplanted mice had few myelinated axons at early time points. It was not until 62 weeks that the hGRP transplants had a significantly increased number of myelinated axons compared to control shiverer mice (Figure 4a). Cohorts transplanted with mGRPs had increased myelin wraps per axon. In mice transplanted with hGRPs, the myelin sheath was comparable to that in shiverer controls at the 18-week time point, but subsequent time points displayed numerous axons with structurally normal myelin, trending toward thicker myelin sheaths over time (Figure 4b). Quantitative assessment of the g-ratio interval (axonal diameter/total diameter of the myelinated fiber) revealed significant differences between WT and shiverer mice (Figure 4d). Human GRP and mGRP-transplanted cohorts had leftward shifts in their g-ratio profiles approaching WT values. The g-ratio differences between the shiverer control cohort and all transplanted mouse cohorts were found to be significant (p<0.01) and there was a progressive shift towards thicker myelin in hGRP transplants from week 18 to 62. Interestingly, we observed a change in axon size in the transplanted cohorts (Figure 4c). The increase in diameter size in mGRP-transplanted mice was comparable to the larger WT axon sizes. We quantified the number of axons containing single myelin wraps to test whether, prior to overt compact myelination, early interactions between axons and nascent myelin sheaths of newborn oligodendrocytes may promote axonal health. At 18 weeks post-transplantation, there was no significant difference between the mGRP and hGRP grafts in the number of axons containing single myelin wraps (Figure 4f). This suggests that nascent myelination was not a primary factor promoting axonal health in the hGRP grafts. Rather, hGRP grafts promoted therapeutic benefits by other means.
Figure 4. Ultrastructural quantitative analysis of GRP grafts.

(a) Quantification of myelinated axons per ROI within the white matter for all experimental groups. (b) Quantification of the myelin lamellae per axon in transplanted animals and in controls. (c) Measurements of axonal diameter within the corpus callosum for all experimental groups. (d) G-ratio profiles determined from TEM images of myelinated axons, cross-sectioned through the corpus callosum. There was no significant difference in g-ratios between WT, mGRP and hGRPs. (e) Quantification of axons with nascent myelin sheaths. (n=3 per group, *p value ≤ 0.01, ** p value ≤ 0.001).
Correlative analysis between myelination detected by MRI and histology
Myelination by engrafted mGRP cells was not uniform across the white matter with various regions being either highly or poorly myelinated, and this provided an opportunity to examine the sensitivity of the MRI markers to myelination by donor cells. In mGRP donor cells, GFP fluorescence is expressed under the PLP promoter, which is constitutively active in mature oligodendrocytes. GFP expression can therefore be used as an indirect marker for donor-derived myelin. The spatial patterns of myelination by mGRP cells were visualized using Z-maps based on three-dimensional DTI data, in which changes in FA and λ⊥ standardized to values measured in shiverer controls were highlighted. In a representative transplanted mouse brain, a large part of the corpus callosum and fimbria regions had reduced λ⊥ values that were more than five times the standard deviation below the mean shiverer control values (dark blue regions in Figure 5a). The spatial pattern observed on MRI corresponded well with donor-derived GFP+ cells and immunoreactivity for MBP. The GFP fluorescence signal intensity was compared to MRI parameters in the same corresponding regions of the anterior corpus callosum in transplanted mice. Scatter plots of normalized T2-weighted signal, MTR, FA, and RD (λ⊥) values versus GFP signal intensity (Figure 5b) showed significant linear correlations (p < 0.0001, R2 > 0.8). As GFP signal intensity increased, indicating myelination, FA and MTR increased while normalized T2-weighted signals and λ⊥ decreased. Additionally, the correlation between MRI parameters and immunoreactivity against MBP was in good agreement (Figure 6). In the case of hGRP grafts, a similar correlation comparison was not possible as on histology the entire white matter was uniformly and completely myelinated when the myelin-related signal was detected on MRI.
Figure 6. Correlation between MRI parameters and MBP immunostaining.

(a) Fractional anisotropy, (b) radial diffusivity, (c) MTR, and (d) normalized T2-w signal intensity values were plotted against MBP fluorescence signal intensity (R2 values equal to 0.37, 0.18, 0.28, and 0.15 respectively).
Engraftment, extent of migration, and therapeutic effects of transplanted GRPs
Engraftment of mGRPs was visualized by GFP expression, which is driven by the PLP promoter resulting in a strong expression in mature oligodendrocytes. Undifferentiated cells expressed low levels but were still detectable (Figure 7d, inset). Transplanted mGRPs migrated out of the ventricles, the site of transplantation, and engrafted in the brain parenchyma. We previously reported on the dynamics of such migration at early timepoints (Walczak et al., 2011). Here, we show extensive migratory behavior over the period of 18 weeks. There was a marked migration of transplanted cells primarily localizing within the periventricular corpus callosum, stria terminalis, and fimbria. Occasionally, transplanted cells also migrated into areas of the cerebral cortex (Figure 7a).
Figure 7. Species-specific differences in migration potential and therapeutic effect.

(a) GFP expression of transplanted mGRPs shows engraftment of donor cells in the brain parenchyma primarily localizing within the periventricular regions (18 weeks post-grafting). (b) hGRP-grafted mice stained for human nuclear antigen, STEM 121 reveals widespread migration of transplanted cells from the lateral ventricles into the brain parenchyma with a clear propensity to localize within white matter structures (18 weeks post-grafting). (c) Despite donor-derived myelination, transplanted mice (n=5) did not have significantly extended survival as compared to the non-transplanted shiverer controls (n=10). Shiverer mice grafted with hGRP (n=21) had extended survival with the majority of the group surviving over 200 days longer than the average life span of the non-transplanted shiverer controls. (d) Engraftment of mGRPs 18 weeks post-grafting has been assessed based on expression of GFP, which is driven by PLP promoter resulting in strong expression in mature oligodendrocytes and basal expression also in undifferentiated cells (inset: PLP-GFP=green; MBP=red). (e) Immunohistochemistry of the spinal cord of hGRP-transplanted shiverer 18 weeks after transplantation shows distribution of transplanted hGRPs immunostained for human specific STEM121 marker (green) and donor-derived myelin (MBP, red) in the transplanted shiverer mice. Higher magnification sections (f, g) of the spinal cord and highest magnification (h) to appreciate single human cells expressing MBP. Bar in (a)=1000μm, in (f)=100 μm.
Engraftment of hGRPs was visualized by human-specific markers (human nuclear antigen, STEM 121), detecting large quantities of transplanted cells within most of the entire brain. There was apparent and widespread migration of transplanted cells from the site of injection (lateral ventricle) towards the parenchyma with a clear propensity to localize within white matter structures including the corpus callosum or fimbria (Figure 7b), anterior commissure, optic tract and nerve, hippocampal region, and olfactory bulbs (not shown). Cells were detected in abundance also in structures more remote from ventricular system including the cortex and even the lumbar spinal cord (Figure 7e–g).
Despite evidence of early but spatially limited myelination in MRI and histology, mice transplanted with mGRPs did not have extended survival as compared to non-transplanted shiverer controls. All transplanted mice died at the age of 193 ± 5 days (Figure 7c, green), while non-transplanted shiverer controls had an average lifespan of 180 ± 20 days. In contrast, mice grafted with hGRPs had delayed myelination as detected by MRI and histology; however, lifespan was radically extended with 48% of mice surviving over 400 days (Figure 7c, red).
Temporal characterization of cell fate in hGRP-grafted shiverer mice
Engrafted hGRPs were identified by double immunostaining for human-specific nuclear antigen (HuNA) and Olig2 or GFAP at various time points after transplantation (Figure 8a). Quantification of the number of human cells, as identified by HuNA, revealed that the amount of cells slightly increased over time, with a maximum around 31 weeks (Figure 8b). The majority of human cells expressed Olig2 at earlier time points (70% ± 5 at 18 weeks, 83% ± 5 at 31 weeks). Only 25% ± 12 of all human cells expressed Olig2 at 62 weeks (Figure 8c).
Figure 8. Temporal characterization of cell fate in hGRP-grafted shiverer mice.

(a) Engrafted human cells 18, 31, and 62 weeks after transplant into neonatal shiverer mice were stained for HuNA and counterstained with Olig2 or hGFAP (50 μm and 20 μm scale bar, respectively). (b) Quantification of the number of human cells, identified by HuNa, at successive time points after transplantation in the corpus callosum. (c) Quantification of human cell fate 18, 31, and 62 weeks after transplantation in the corpus callosum (Olig2 and hGFAP). (d) Quantification of hGRP-derived mature astrocytes in the corpus callosum.
A minority of hGRPs expressed GFAP. The percent of human cells expressing GFAP remained steady at 18, 31, and 62 weeks (23% ± 7, 15% ± 4, and 12% ± 2 respectively, Figure 8c). However, the population of these human astrocytes shifted towards more mature astrocytes over time (Figure 8d), as assessed by their morphology. Quantification of MBP-expressing oligodendrocytes was not feasible due to the extensive network of processes and sparse signal in oligodendrocyte cell bodies, but based upon increasing brain area with MBP positivity from week 31 to 62 and loss of human progenitor cell content it can be concluded that grafted cells matured over that interval towards myelinating oligodendrocytes.
DISCUSSION
Therapeutic effects are independent from production of mature and compact myelin by transplanted GRPs
To the best of our knowledge, this is a first report demonstrating a side-by-side assessment of myelination by GRPs from xenogeneic (human-to-mouse) vs. allogeneic sources in dysmyelinated mice using in vivo MRI for longitudinal monitoring of myelination, with direct correlation with post-mortem tissue analysis. We made intriguing and provocative observations that may have important implications for clinical application of cell therapy in patients suffering from dysmyelination. We have shown that xenografting of hGRP results in improved survival of shiverer mice which is consistent with a report by Windrem et al. (Windrem et al., 2008), where about 25% of grafted mice survived long-term. The timing of myelination by hGRPs is also consistent with that report where extensive myelination was only observed beyond 35 weeks (Windrem et al., 2008). This delayed myelination puts into question the role of mature and compact myelin in prolonging the life of grafted dysmyelinated mice. Paradoxically, mGRP-grafted mice died shortly after the 18-week time point despite exhibiting a more pronounced, but locally limited presence of mature and compact myelin.
Post-transplantation week 18 as critical time interval for enhancement of survival
Our first point of analysis (18 weeks post-transplantation) is a critical time point as it is near the maximum normal lifespan for shiverer mice, with death usually occurring at week 20. For cell therapy to be effective, the therapeutic benefits should therefore start prior to week 20. Our imaging and histological analysis revealed that mGRPs quickly differentiated into mature myelinating oligodendrocytes by 18 weeks as detected by MRI and eriochrome staining, albeit the engrafted area was limited to the periventricular region. In contrast, hGRP grafts had no detectable myelin on MRI, and histology detected engraftment of human cells throughout the brain, but only sparse myelination. While myelination was delayed in the hGRP-transplanted mice, we observed an increase in axon diameter size in xenografted animals at all time points as compared to the mGRP allografts and shiverer mice. Other reports have shown that oligodendrocyte elimination results in reduced axon diameters (Colello et al., 1994). Overall the lifespan of hGRP-transplanted mice was dramatically prolonged despite the lack of mature and compact myelin at the 18-week interval (Figure 3) suggesting that the therapeutic benefit of hGRPs is due to properties beyond merely axon insulation and facilitation of rapid conduction.
Extensive cell migration and global engraftment are required for improved animal survival
Biodistribution of mGRPs at 18 weeks corresponded well with myelin as detected by MRI and MBP immunohistochemistry, while in the case of hGRPs the widely distributed cells were not producing mature and compact myelin. These observations are consistent with other reports showing only a small portion of the corpus callosum (Nair et al., 2005) or spinal cord (Mothe and Tator, 2008) myelinated by rodent progenitors in shiverer mice. The observed extensive migratory potential of hGRPs in shiverer mice is consistent with other reports including transplantation of hGRPs (Walczak et al., 2011; Windrem et al., 2008) or primary human neural stem cells (Uchida et al., 2012). Similarly, an extensive migration and a very slow process of differentiation was observed for human cells transplanted into the spinal cord of adult mice. A substantial differentiation towards oligodendrocytes could be observed only after 33–49 weeks (Buchet et al., 2011). This slow process of differentiation may be a factor that contributes towards better migratory properties of human cells compared to mouse cells. Human glial progenitors were shown to have competitive advantage displacing endogenous mouse glia even when transplanted into naïve, non-myelin deficient recipients. (Windrem et al., 2014).
Correlation of GRP engraftment between MRI and histology
An intriguing observation was that there was a strong correlation between histological and imaging measures of myelination in mGRP grafts, but not for hGRPs, indicating that differences in the structure of new myelin must exist. For the hGRP group, there was a significant discrepancy between myelination detected by anti-MBP immunohistochemistry, by MRI, and by eriochrome staining. The MRI readouts of myelin were substantially delayed, and for DTI it never normalized during the 400 days of this study. DTI has been shown to detect myelination by transplanted neural stem cells (Uchida et al., 2012); however, the imaging in that study was only done ex vivo after perfusion fixation and hence may not be accurate.
Species-specific differences in GRP function
Species-specific differences in the timing and extent of myelination were observed despite close similarities between human and mouse progenitors in terms of the tissue of origin or developmental stage (derived from second trimester fetuses). Species-specific features of glial progenitors are expressed by developmentally imprinted differences in the timescale for myelination between rodents and humans (McMorris and McKinnon, 1996). Additionally, rodent OPCs express far more CXCL1 receptors which mediate mitogenic signaling pathways (de Castro and Bribian, 2005; Filipovic et al., 2003) than human OPCs, suggesting that human and rodent OPCs respond differently to certain factors, potentially resulting in the different timescale for migration and myelination. Furthermore, the species-specific differences of GRP migration could be due to their innate developmental programming, with murine progenitors required to migrate in a brain that is about a thousandth of the volume of a human brain. Another possible explanation for the therapeutic effect only obtained using hGRPs is the larger size and longer processes of human glia as compared to those for mouse cells (Oberheim et al., 2009), suggesting that neuroprotective molecules could be produced more efficiently and in larger quantities by human glia. In this context it would be further warranted to explore the quantity and quality of neurotrophic factors or exosomal release by oligodendrocytes of different species.
Overall, the difference in the migratory potential have important implications for cell transplantation-based therapies in patients, as it could be that human GRPs may need additional engineering to enhance their species-innate programmed migration in order to achieve similar therapeutic effects in patients as obtained in mice.
We hypothesize that the capability for extensive migration and engraftment is the most fundamental feature for cells to be successful candidates for treatment of dysmyelinating disorders, and that presence of mature and compact myelin itself is only of secondary importance. It suggests that oligodendrocytes in shiverer mice are not only incapable of the production of proper myelin, but also incapable of providing proper trophic support with the later perhaps being of greater consequences to the degenerative process observed in dysmyelination.
Role of myelin pathology in dysmyelination
Numerous studies in shiverer and other hypomyelinated mutants suggested that there is a direct correlation between the lack of myelin and decreased lifespan (Duncan et al., 1995). In support of the critical role of myelin proteins is the observation that correction of the defect within the myelin basic protein gene results in rescue of the shiverer phenotype (Readhead et al., 1987) and expression of the MBP antisense cDNA can induce the shiverer phenotype (Katsuki et al., 1988). While hypomyelination is always detrimental, studies of mutant animals with different degrees of hypomyelination demonstrate the lack of a relationship between the extent of hypomyelination and premature death (Duncan et al., 1995). Notably, it has been shown that mutant mice completely lacking myelin can survive at least 100 days (Wolf et al., 1999). This demonstrates that the supportive role of oligodendrocytes is complex and likely extends beyond myelination. Indeed, it has recently been shown that oligodendrocytes play an important nutritional role for motor neurons. Oligodendrocytes were shown to produce one of the main lactate transporters (MCT-1) and dysfunction of oligodendrocytes with reduced level of MCT-1 leads to degeneration of neurons such as in ALS (Lee et al., 2012).
In addition, it has been suggested that myelinating oligodendrocytes release metabolite-loaded exosomes in response to neuronal signals, which exert a neuroprotective effect on neurons under stress (Fruhbeis et al., 2013). This suggests that the abnormalities observed in dysmyelinated mice may be at least in part due to disrupted trophic support of neurons by dysfunctional oligodendrocytes. Lack of trophic support, in concert with increased energy demand for the propagation of action potentials in hypomyelinated axons, places metabolic strain on neurons that could contribute to their gradual degeneration. Previous studies on human spinal cord morphology have suggested that the axonal diameter can be used as a marker for axonal pathology, as smaller axons are more susceptible to injury and age-dependent changes (Armstrong et al., 2008; Ganter et al., 1999; Terao et al., 1994). Interestingly, in our study, hGRP-grafted mice exhibited an increase in axon cross-sectional area over time, compared to WT axon sizes, while shiverer controls and mGRP-grafted mice had smaller axon cross-sectional areas. The increased axonal size in the hGRP-grafted mice may suggest that host axons were regenerated beyond WT quality axons, owing to some therapeutic property intrinsic to hGRP cells, though likely independent of myelination status.
MRI as a non-invasive method for imaging graft-derived myelin
MRI is an ideal tool for non-invasive monitoring of cell-based therapies given its superior soft tissue contrast and clinical use (Walczak and Bulte, 2007). The digitized data format allows easy processing and comparisons between different subjects or the same subject over time to examine the spatiotemporal progression of myelin. To realize the full diagnostic potential of MRI, we employed imaging techniques that are commonly available on clinical scanners. They cover different aspects of the state of myelination, including the chemical composition or micro-structural organization (Laule et al., 2007; Wu et al., 2011) and were included in our study to detect myelination at different maturation stages.
Our study shows that the synthesis of hydrophobic compartment-rich myelin may be a key factor for sensitive detection of myelination using MRI as evidenced by the good correlation between the MRI and eriochrome staining results. However, MRI of myelination alone should be used with reservations for a proper evaluation of the therapeutic effects of GRPs in patients with dysmyelination, as we show here that the outcome can be independent of myelination. It has to be emphasized that the detection of myelin in mouse brain using MRI is rather limited to major white matter structures such as the corpus callosum or internal capsule. Dispersed myelin fibers within the cortex or even fasciculated tracts in the striatum are beyond sensitivity limits and practically undetectable. Moreover, we found that the therapeutic benefit is highly dependent on the extent of cell distribution, suggesting that MRI cell tracking using either fluorine or iron oxide MR contrast agents may be equally important (Janowski et al., 2012; Srivastava and Bulte, 2014). Cell delivery technique such as MRI-guided intraarterial injection may further improve cell biodistribution (Gorelik et al., 2012; Walczak et al., 2016). Nevertheless, imaging of myelin may be more relevant to evaluating the therapeutic effects at later time points. While extensive cell migration is critical for rescuing initial animal survival, the long-term sustainability and normalization of neurological deficits will certainly rely on extensive myelination.
This pre-clinical study represents a step toward validation of clinically applicable MRI monitoring of glial cell replacement therapy for dysmyelinating conditions. The process of graft-derived myelination as measured by anti-myelin immunohistochemistry is slow, and maturation of that myelin for MRI detection is even slower, requiring a time span of well over one year. Importantly, MRI diffusion parameters that were previously suggested to be highly specific for myelination (Song et al., 2002) did not improve here for the hGRP-transplanted cohorts, despite a strong evidence for extensive myelination as detected by immunohistochemical and T2-w or MT MRI methods. However, the T2-w and MT MRI parameters for myelination corresponded well with the presence of hydrophobic regions as visualized by eriochrome staining. This indicates that there are confounding factors influencing conventional diffusion MRI parameters, making them not as sensitive to myelin as T2-w and MT MRI parameters in this study.
Conclusions
We have demonstrated that transplantation of human GRPs is a viable therapeutic strategy for treatment of dysmyelination, with robust therapeutic effects seen in half of the treated dysmyelinated shiver mice. The therapeutic effect precedes the maturation of myelin as detected by MRI, suggesting that the therapeutic mechanisms of graft activity is not centered around the role of oligodendrocytes as axonal insulators. We demonstrated species-specific effects of GRP transplantation with regards to the timing of myelination, distribution across the entire neuroaxis, histological axonal morphology, and animal survival. We found that the ability of extensive migration and engraftment is a fundamental feature for GRP cells to be successful candidates for use in dysmyelinating disorders. Further studies in large animals using allo- and xenografting paradigms are needed to address whether the migratory capabilities of hGRPs will be sufficient for potential therapeutic effects in humans. Since the rescue of animal lifespan appears to rely heavily on cell distribution and not necessarily presence of mature and compact myelin, future efforts toward MRI-based monitoring of GRP therapy may benefit from including MRI cell tracking studies.
Highlights.
Mouse GRP allografts fail to extend lifespan of shiverers despite early myelination
Human GRP xenografts result in robust therapeutic effect significantly extending survival for over 50% of animals.
Distribution of xenografts is extensive but myelination is delayed with substantial presence of mature myelin occurring after 18 weeks indicating that therapeutic effects via mechanisms independent of mature, compact myelin
T2-weighted and magnetization transfer MRI facilitate accurate detection of graft-derived myelin while sensitivity of diffusion measures is limited in the context of dysmyelinating disease.
Acknowledgments
The authors acknowledge Dr. Melina Jones of Johns Hopkins University for her advice with histological analysis, Dr. David Rini for preparation of Graphical Abstract, Carol Cooke and Irina Shats for their technical support, and Q Therapeutics Inc. for providing hGRPs. This work was supported by NMSS RG 4994-A-3, MSCRFII-0193, MSCRFII0052, MSCREFE-0714, R01 NS076573, RO1 NS045062, R01NS091110 S10RR0289551 and Strategmed 1/233209/12/NCBIR/2015.
Footnotes
Conflict of Interest: Dr. James Campanelli is a senior research director at Q Therapeutics Inc. that provided cells for this study; however, he did not take part in acquisition or processing of the presented data.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aggarwal M, Mori S, Shimogori T, Blackshaw S, Zhang J. Three-dimensional diffusion tensor microimaging for anatomical characterization of the mouse brain. Magn Reson Med. 2010;64:249–261. doi: 10.1002/mrm.22426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong RA, Syed AB, Smith CU. Density and cross-sectional areas of axons in the olfactory tract in control subjects and Alzheimer’s disease: an image analysis study. Neurol Sci. 2008;29:23–27. doi: 10.1007/s10072-008-0854-0. [DOI] [PubMed] [Google Scholar]
- Buchet D, Garcia C, Deboux C, Nait-Oumesmar B, Baron-Van Evercooren A. Human neural progenitors from different foetal forebrain regions remyelinate the adult mouse spinal cord. Brain. 2011;134:1168–1183. doi: 10.1093/brain/awr030. [DOI] [PubMed] [Google Scholar]
- Ceritoglu C, Oishi K, Li X, Chou MC, Younes L, Albert M, Lyketsos C, van Zijl PC, Miller MI, Mori S. Multi-contrast large deformation diffeomorphic metric mapping for diffusion tensor imaging. Neuroimage. 2009;47:618–627. doi: 10.1016/j.neuroimage.2009.04.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang N, Mori S, Yamamoto A, Jiang H, Ye X, Xu X, Richards LJ, Nathans J, Miller MI, Toga AW, Sidman RL, Zhang J. An MRI-based atlas and database of the developing mouse brain. Neuroimage. 2011;54:80–89. doi: 10.1016/j.neuroimage.2010.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colello RJ, Pott U, Schwab ME. The role of oligodendrocytes and myelin on axon maturation in the developing rat retinofugal pathway. J Neurosci. 1994;14:2594–2605. doi: 10.1523/JNEUROSCI.14-05-02594.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Castro F, Bribian A. The molecular orchestra of the migration of oligodendrocyte precursors during development. Brain Res Brain Res Rev. 2005;49:227–241. doi: 10.1016/j.brainresrev.2004.12.034. [DOI] [PubMed] [Google Scholar]
- Duncan ID, Nadon NL, Hoffman RL, Lunn KF, Csiza C, Wells MR. Oligodendrocyte survival and function in the long-lived strain of the myelin deficient rat. J Neurocytol. 1995;24:745–762. doi: 10.1007/BF01191211. [DOI] [PubMed] [Google Scholar]
- Farah MH, Pan BH, Hoffman PN, Ferraris D, Tsukamoto T, Nguyen T, Wong PC, Price DL, Slusher BS, Griffin JW. Reduced BACE1 activity enhances clearance of myelin debris and regeneration of axons in the injured peripheral nervous system. J Neurosci. 2011;31:5744–5754. doi: 10.1523/JNEUROSCI.6810-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipovic R, Jakovcevski I, Zecevic N. GRO-alpha and CXCR2 in the human fetal brain and multiple sclerosis lesions. Developmental neuroscience. 2003;25:279–290. doi: 10.1159/000072275. [DOI] [PubMed] [Google Scholar]
- Fruhbeis C, Frohlich D, Kuo WP, Amphornrat J, Thilemann S, Saab AS, Kirchhoff F, Mobius W, Goebbels S, Nave KA, Schneider A, Simons M, Klugmann M, Trotter J, Kramer-Albers EM. Neurotransmitter-triggered transfer of exosomes mediates oligodendrocyte-neuron communication. PLoS Biol. 2013;11:e1001604. doi: 10.1371/journal.pbio.1001604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganter P, Prince C, Esiri MM. Spinal cord axonal loss in multiple sclerosis: a postmortem study. Neuropathol Appl Neurobiol. 1999;25:459–467. doi: 10.1046/j.1365-2990.1999.00205.x. [DOI] [PubMed] [Google Scholar]
- Gorelik M, Orukari I, Wang J, Galpoththawela S, Kim H, Levy M, Gilad AA, Bar-Shir A, Kerr DA, Levchenko A, Bulte JW, Walczak P. Use of MR cell tracking to evaluate targeting of glial precursor cells to inflammatory tissue by exploiting the very late antigen-4 docking receptor. Radiology. 2012;265:175–185. doi: 10.1148/radiol.12112212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta N, Henry RG, Strober J, Kang SM, Lim DA, Bucci M, Caverzasi E, Gaetano L, Mandelli ML, Ryan T, Perry R, Farrell J, Jeremy RJ, Ulman M, Huhn SL, Barkovich AJ, Rowitch DH. Neural stem cell engraftment and myelination in the human brain. Sci Transl Med. 2012;4:155ra137. doi: 10.1126/scitranslmed.3004373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janowski M, Bulte JW, Walczak P. Personalized nanomedicine advancements for stem cell tracking. Advanced drug delivery reviews. 2012;64:1488–1507. doi: 10.1016/j.addr.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, van Zijl PC, Kim J, Pearlson GD, Mori S. DtiStudio: resource program for diffusion tensor computation and fiber bundle tracking. Comput Methods Programs Biomed. 2006;81:106–116. doi: 10.1016/j.cmpb.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Katsuki M, Sato M, Kimura M, Yokoyama M, Kobayashi K, Nomura T. Conversion of normal behavior to shiverer by myelin basic protein antisense cDNA in transgenic mice. Science. 1988;241:593–595. doi: 10.1126/science.2456614. [DOI] [PubMed] [Google Scholar]
- Kiernan JA. Histochemistry of staining methods for normal and degenerating myelin in the central and peripheral nervous systems. J Histotechnol. 2007;30:87–106. [Google Scholar]
- Laule C, Vavasour IM, Kolind SH, Li DK, Traboulsee TL, Moore GR, MacKay AL. Magnetic resonance imaging of myelin. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics. 2007;4:460–484. doi: 10.1016/j.nurt.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN, Liu Y, Tsingalia A, Jin L, Zhang PW, Pellerin L, Magistretti PJ, Rothstein JD. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature. 2012;487:443–448. doi: 10.1038/nature11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMorris FA, McKinnon RD. Regulation of oligodendrocyte development and CNS myelination by growth factors: prospects for therapy of demyelinating disease. Brain Pathol. 1996;6:313–329. doi: 10.1111/j.1750-3639.1996.tb00858.x. [DOI] [PubMed] [Google Scholar]
- Mothe AJ, Tator CH. Transplanted neural stem/progenitor cells generate myelinating oligodendrocytes and Schwann cells in spinal cord demyelination and dysmyelination. Exp Neurol. 2008;213:176–190. doi: 10.1016/j.expneurol.2008.05.024. [DOI] [PubMed] [Google Scholar]
- Muse ED, Jurevics H, Toews AD, Matsushima GK, Morell P. Parameters related to lipid metabolism as markers of myelination in mouse brain. Journal of neurochemistry. 2001;76:77–86. doi: 10.1046/j.1471-4159.2001.00015.x. [DOI] [PubMed] [Google Scholar]
- Nair G, Tanahashi Y, Low HP, Billings-Gagliardi S, Schwartz WJ, Duong TQ. Myelination and long diffusion times alter diffusion-tensor-imaging contrast in myelin-deficient shiverer mice. Neuroimage. 2005;28:165–174. doi: 10.1016/j.neuroimage.2005.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberheim NA, Takano T, Han X, He W, Lin JH, Wang F, Xu Q, Wyatt JD, Pilcher W, Ojemann JG, Ransom BR, Goldman SA, Nedergaard M. Uniquely hominid features of adult human astrocytes. J Neurosci. 2009;29:3276–3287. doi: 10.1523/JNEUROSCI.4707-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlman SJ, Mar S. Leukodystrophies. Advances in experimental medicine and biology. 2012;724:154–171. doi: 10.1007/978-1-4614-0653-2_13. [DOI] [PubMed] [Google Scholar]
- Phillips AW, Falahati S, DeSilva R, Shats I, Marx J, Arauz E, Kerr DA, Rothstein JD, Johnston MV, Fatemi A. Derivation of glial restricted precursors from E13 mice. Journal of visualized experiments : JoVE. 2012 doi: 10.3791/3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao MS, Mayer-Proschel M. Glial-restricted precursors are derived from multipotent neuroepithelial stem cells. Developmental biology. 1997;188:48–63. doi: 10.1006/dbio.1997.8597. [DOI] [PubMed] [Google Scholar]
- Readhead C, Popko B, Takahashi N, Shine HD, Saavedra RA, Sidman RL, Hood L. Expression of a myelin basic protein gene in transgenic shiverer mice: correction of the dysmyelinating phenotype. Cell. 1987;48:703–712. doi: 10.1016/0092-8674(87)90248-0. [DOI] [PubMed] [Google Scholar]
- Sandrock RW, Wheatley W, Levinthal C, Lawson J, Hashimoto B, Rao M, Campanelli JT. Isolation, characterization and preclinical development of human glial-restricted progenitor cells for treatment of neurological disorders. Regenerative medicine. 2010;5:381–394. doi: 10.2217/rme.10.24. [DOI] [PubMed] [Google Scholar]
- Song SK, Sun SW, Ramsbottom MJ, Chang C, Russell J, Cross AH. Dysmyelination revealed through MRI as increased radial (but unchanged axial) diffusion of water. NeuroImage. 2002;17:1429–1436. doi: 10.1006/nimg.2002.1267. [DOI] [PubMed] [Google Scholar]
- Srivastava AK, Bulte JWM. Seeing Stem Cells at Work In Vivo. Stem Cell Rev Rep. 2014;10:127–144. doi: 10.1007/s12015-013-9468-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terao S, Sobue G, Hashizume Y, Shimada N, Mitsuma T. Age-related changes of the myelinated fibers in the human corticospinal tract: a quantitative analysis. Acta Neuropathol. 1994;88:137–142. doi: 10.1007/BF00294506. [DOI] [PubMed] [Google Scholar]
- Uchida N, Chen K, Dohse M, Hansen KD, Dean J, Buser JR, Riddle A, Beardsley DJ, Wan Y, Gong X, Nguyen T, Cummings BJ, Anderson AJ, Tamaki SJ, Tsukamoto A, Weissman IL, Matsumoto SG, Sherman LS, Kroenke CD, Back SA. Human Neural Stem Cells Induce Functional Myelination in Mice with Severe Dysmyelination. Sci Transl Med. 2012:4. doi: 10.1126/scitranslmed.3004371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walczak P, All AH, Rumpal N, Gorelik M, Kim H, Maybhate A, Agrawal G, Campanelli JT, Gilad AA, Kerr DA, Bulte JW. Human glial-restricted progenitors survive, proliferate, and preserve electrophysiological function in rats with focal inflammatory spinal cord demyelination. Glia. 2011;59:499–510. doi: 10.1002/glia.21119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walczak P, Bulte JW. The role of noninvasive cellular imaging in developing cell-based therapies for neurodegenerative disorders. Neurodegener Dis. 2007;4:306–313. doi: 10.1159/000101887. [DOI] [PubMed] [Google Scholar]
- Walczak P, Wojtkiewicz J, Nowakowski A, Habich A, Holak P, Xu J, Adamiak Z, Chehade M, Pearl MS, Gailloud P, Lukomska B, Maksymowicz W, Bulte JW, Janowski M. Real-time MRI for precise and predictable intra-arterial stem cell delivery to the central nervous system. J Cereb Blood Flow Metab. 2016 doi: 10.1177/0271678X16665853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windrem MS, Schanz SJ, Guo M, Tian GF, Washco V, Stanwood N, Rasband M, Roy NS, Nedergaard M, Havton LA, Wang S, Goldman SA. Neonatal chimerization with human glial progenitor cells can both remyelinate and rescue the otherwise lethally hypomyelinated shiverer mouse. Cell Stem Cell. 2008;2:553–565. doi: 10.1016/j.stem.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windrem MS, Schanz SJ, Morrow C, Munir J, Chandler-Militello D, Wang S, Goldman SA. A competitive advantage by neonatally engrafted human glial progenitors yields mice whose brains are chimeric for human glia. J Neurosci. 2014;34:16153–16161. doi: 10.1523/JNEUROSCI.1510-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf MK, Nunnari JN, Billings-Gagliardi S. Quaking*shiverer double-mutant mice survive for at least 100 days with no CNS myelin. Developmental neuroscience. 1999;21:483–490. doi: 10.1159/000017415. [DOI] [PubMed] [Google Scholar]
- Wu D, Xu J, McMahon MT, van Zijl PC, Mori S, Northington FJ, Zhang J. In vivo high-resolution diffusion tensor imaging of the mouse brain. Neuroimage. 2013;83:18–26. doi: 10.1016/j.neuroimage.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YC, Field AS, Duncan ID, Samsonov AA, Kondo Y, Tudorascu D, Alexander AL. High b-value and diffusion tensor imaging in a canine model of dysmyelination and brain maturation. Neuroimage. 2011;58:829–837. doi: 10.1016/j.neuroimage.2011.06.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yandava BD, Billinghurst LL, Snyder EY. “Global” cell replacement is feasible via neural stem cell transplantation: evidence from the dysmyelinated shiverer mouse brain. Proc Natl Acad Sci U S A. 1999;96:7029–7034. doi: 10.1073/pnas.96.12.7029. [DOI] [PMC free article] [PubMed] [Google Scholar]