

Abstract
Enhancers frequently contain multiple binding sites for the same transcription factor. These homotypic binding sites often exhibit synergy, whereby the transcriptional output from two or more binding sites is greater than the sum of the contributions of the individual binding sites alone. Although this phenomenon is frequently observed, the mechanistic basis for homotypic binding site synergy is poorly understood. Here, we identify a bona fide cardiac-specific Prkaa2 enhancer that is synergistically activated by homotypic MEF2 binding sites. We show that two MEF2 sites in the enhancer function cooperatively due to bridging of the MEF2C-bound sites by the SAP domain-containing co-activator protein myocardin, and we show that paired sites buffer the enhancer from integration site-dependent effects on transcription in vivo. Paired MEF2 sites are prevalent in cardiac enhancers, suggesting that this might be a common mechanism underlying synergy in the control of cardiac gene expression in vivo.
KEY WORDS: AMPK, MEF2, Prkaa2, Mouse, Myocardin, Transcription
Summary: In mice, bridging of paired MEF2C-bound MEF2 sites in a cardiac enhancer of the Prkaa2 gene by the transcriptional co-activator myocardin promotes cooperativity between the two homotypic elements.
INTRODUCTION
Myocyte enhancer factor 2 (MEF2) transcription factors are crucial regulators of cardiac gene expression (Black and Cripps, 2010; Potthoff and Olson, 2007). MEF2 proteins have highly conserved N-terminal MADS and MEF2 domains, which facilitate binding to an AT-rich sequence found in the promoters and enhancers of numerous cardiac genes (Black and Cripps, 2010; Black and Olson, 1998). In mice, Mef2c is the earliest Mef2 gene to be expressed in the heart, and mice lacking Mef2c die at embryonic day (E) 9.5 due to profound cardiac defects (Lin et al., 1997). Although MEF2C is widely appreciated as a regulator of cardiac gene expression during development and in adulthood (Black and Cripps, 2010; Potthoff and Olson, 2007), the mechanisms regulating MEF2C-dependent gene expression remain incompletely resolved.
The SAP domain protein myocardin is a positive-acting transcriptional co-activator that has been extensively studied as a co-factor for serum response factor (SRF) (Miano, 2015; Parmacek, 2007; Pipes et al., 2006; Wang and Olson, 2004). Alternative splicing of myocardin produces two isoforms: a short form, myocardin-856, which is expressed in smooth muscle and interacts with SRF; and a long form, myocardin-935, which is expressed in cardiac muscle and can interact with either MEF2 or SRF (Creemers et al., 2006). Interaction of myocardin-935 with MEF2 strongly potentiates the transcriptional activity of MEF2 (Creemers et al., 2006). However, the functional interaction of myocardin with MEF2 remains largely unexplored, and the role of this complex in vivo is not known.
Here, we identified a conserved, cardiac-specific enhancer of Prkaa2, the gene encoding the catalytic α2 subunit of AMP-activated protein kinase (AMPK). The Prkaa2 enhancer is dependent on MEF2C for enhancer activity in vivo, and it is cooperatively activated by MEF2C and myocardin-935. Mechanistically, we found that the Prkaa2 enhancer is cooperatively activated by the bridging of two conserved, essential MEF2 sites via myocardin dimerization and concomitant interaction with MEF2C. Moreover, the presence of paired MEF2 sites confers robustness to the Prkaa2 enhancer in vivo, buffering it from integration site-dependent effects on transcriptional output.
RESULTS AND DISCUSSION
Identification of a myocardial-specific Prkaa2 enhancer
Because of its central role in the heart as a master regulator of energy balance and homeostasis and due to its regulatory changes during heart failure, AMPK has been extensively studied, but, remarkably, the in vivo transcriptional regulation of the genes encoding AMPK subunits has not previously been investigated. The first intron of the Prkaa2 gene contains a ∼1 kb evolutionarily conserved element marked by activating histone marks in an in vitro model of cardiomyocyte differentiation (Wamstad et al., 2012) (Fig. 1A). We tested this element for enhancer activity in transgenic mouse embryos and found that it functions as a cardiac enhancer from the cardiac crescent stage, throughout embryonic development, and in adulthood in a pattern that appeared essentially identical to the endogenous pattern of Prkaa2 mRNA expression (Fig. 1B-P). Transverse sectioning of X-gal-stained embryos showed that staining was only present in cardiac progenitors at E7.75 and thereafter only in the myocardial layer of the heart at E9.5 and E11.5 (Fig. 1D,G,J,M). The Prkaa2 enhancer did not appear to be active outside of the myocardium, although we cannot rule out activity at later developmental or adult stages in other non-myocardial cell types within the heart.
Fig. 1.

Identification of a cardiac-restricted Prkaa2 enhancer. (A) Human:mouse conservation (top box), human:opossum conservation (second box), H3K27 acetylation (Wamstad et al., 2012) in cardiomyocytes (third box), and H3K27 acetylation (Wamstad et al., 2012) in cardiac precursors (fourth box) in the Prkaa2 locus. The red-boxed peak highlights the 931 bp Prkaa2 enhancer. Red-filled peaks, noncoding sequences conserved between 75% and 100%; blue-filled peaks, coding sequences conserved between 75% and 100%; white peaks, conservation between 50% and 75%. (B-P) Whole-mount (B,E,H,K,N-P) and sections (D,G,J,M) of X-gal-stained Prkaa2[931]-lacZ transgenic embryos and postnatal hearts. Prkaa2[931] enhancer activity recapitulates the expression pattern of endogenous Prkaa2 detected by whole-mount in situ hybridization (C,F,I,L) from E7.75 through E11.5. e, endocardium; hrt, heart; LV, left ventricle; m, myocardium; NT, neural tube; pcm, precardiac mesoderm; RA, right atrium, RV, right ventricle. Scale bars: 100 µm.
The location of the enhancer in the first intron of Prkaa2 and the concordance of enhancer activity with endogenous Prkaa2 expression (Fig. 1) strongly suggest that this enhancer regulates Prkaa2 expression. As an explicit test of this notion, we used CRISPR/Cas9 to delete the enhancer from the mouse genome and compared Prkaa2 expression in the presence and absence of this enhancer (Fig. S1). Mice of all genotypes (Prkaa2+/+, Prkaa2+/enhΔ, Prkaa2enhΔ/enhΔ) occurred at predicted Mendelian frequency, and no overt phenotypes were observed (data not shown). However, Prkaa2enhΔ/enhΔ mice had a 64.4% reduction in Prkaa2 expression in the heart at E9.5 compared with Prkaa2+/+ mice (Fig. S1B). This establishes that this intronic element is a bona fide Prkaa2 transcriptional enhancer. These data also suggest that additional cardiac enhancers for Prkaa2 must exist to account for the remaining 36% of cardiac gene expression in Prkaa2enhΔ/enhΔ embryos.
The Prkaa2 enhancer is a transcriptional target of MEF2C
We next generated a small series of deletion fragments within the 931 bp Prkaa2 enhancer and found that a 200 bp fragment from nucleotides 429-628 of the 931 bp intronic enhancer was necessary and sufficient to direct expression exclusively to the myocardium at E11.5 (Fig. S2). This 200 bp fragment contains two perfectly conserved MEF2 consensus sites (Fig. S2F), suggesting that this Prkaa2 enhancer might be regulated by MEF2. Indeed, activity of the enhancer was completely abolished when the Prkaa2[931]-lacZ transgene was crossed onto a Mef2c-null background (Fig. 2A,A′). Moreover, endogenous Prkaa2 expression was also significantly reduced by 77% in the hearts of Mef2c-null mice (Fig. S3). Notably, expression of endogenous Prkaa2 was not completely abolished in the absence of MEF2C function, further supporting the likely existence of additional, MEF2C-independent Prkaa2 cardiac enhancers. In electrophoretic mobility shift assays (EMSAs), MEF2C bound specifically to each of the Prkaa2 MEF2 sites (Fig. 2B).
Fig. 2.

The Prkaa2 cardiac enhancer requires MEF2C for activity. (A,A′) Prkaa2[931]-lacZ transgenic mice were crossed into Mef2c+/+ (wild type, A) and Mef2c−/− (A′) backgrounds, and enhancer activity was examined by X-gal staining at E8.5; hrt, heart. (B) Prkaa2 MEF2 site 1 (lanes 1-6) or Prkaa2 MEF2 site 2 (lanes 7-12) was used in EMSA with reticulocyte lysate (−, lanes 1 and 7) or with recombinant MEF2C (+, lanes 2-6 and 8-12). MEF2C efficiently bound to both of the Prkaa2 MEF2 sites (lanes 2 and 8). Binding to each site was competed by an excess of unlabeled control MEF2 site from the myogenin gene (C) or by unlabeled self probe (1) or (2), respectively, but not by mutant versions of the unlabeled competitors [mC, m(1), m(2)]. (C,D) P19CL6 cells were co-transfected with parental pTK-β-gal reporter (lanes 1, 2), wild-type Prkaa2-β-gal (lanes 3, 4), or a mutant version of the Prkaa2-β-gal reporter with both MEF2 sites disrupted (lanes 5, 6). Co-transfection of an expression plasmid for MEF2C (C) or MEF2C-VP16 (D) is indicated with a plus symbol; a minus symbol indicates that an equivalent amount of the parental expression plasmid was added. Results are reported as mean+s.e.m.; n=14 (C) or n=8 (D) independent biological replicates. Note the difference in the values on the x-axes for transactivation by MEF2C (C) versus MEF2C-VP16 (D). *P<0.05, ****P<0.0001, two-way ANOVA with Bonferroni's post-hoc test.
MEF2C significantly activated the Prkaa2 cardiac enhancer in P19CL6, a cardiac progenitor-like cell line, and this activation was dependent on the presence of intact MEF2 sites (Fig. 2C). We also examined the ability of MEF2C-VP16, a fusion of MEF2C with a potent transactivation domain from herpes simplex virus, to activate the Prkaa2 enhancer (Fig. 2D). Similar to wild-type MEF2C, MEF2C-VP16 activated the Prkaa2 reporter in a MEF2 site-dependent fashion (Fig. 2D), but activation was much more robust than by wild-type MEF2C (∼5-fold for MEF2C compared with >2000-fold for MEF2C-VP16).
Cooperative activation of the Prkaa2 enhancer by MEF2C and myocardin
One possible explanation for the dramatic difference in transactivation of the Prkaa2 enhancer by MEF2C-VP16 compared with MEF2C is that P19CL6 cells might be missing one or more MEF2C co-activator proteins required for robust activation, and fusion of the VP16 activation domain compensated for the missing co-factor. The long form of myocardin is restricted to the heart and has been shown to coactivate MEF2 (Creemers et al., 2006). Therefore, to determine if myocardin might be involved in Prkaa2 regulation in vivo, we crossed the Prkaa2[931]-lacZ transgene onto a myocardin-null (Myocd−/−) background and examined β-galactosidase activity at E9.5 (Fig. 3A). X-gal staining of Myocd−/− embryos was noticeably weaker than staining of wild-type embryos (Fig. 3A,A′). Using a quantitative luminescence assay, we found ∼80% reduction in β-galactosidase activity in Myocd-null hearts compared with wild type or Myocd heterozygotes (Fig. 3A″). These data indicate that myocardin regulates the Prkaa2 enhancer in vivo.
Fig. 3.

Myocardin-935 regulates the Prkaa2 cardiac enhancer. (A-A″) Prkaa2[931]-lacZ transgenic mice were crossed into Myocd+/+ (wild type, A), Myocd+/− and Myocd−/− (A′) genetic backgrounds and enhancer activity was examined qualitatively by X-gal staining (A,A′) or quantitatively by chemiluminescence β-galactosidase assay (A″) at E9.5. hrt, heart. Data in A″ are expressed as the mean β-galactosidase activity (+s.e.m.) with the mean activity on the Myocd+/+ background normalized to a value of 1. n=4 (Myocd+/+), n=5 (Myocd+/−) and n=3 (Myocd−/−) independent biological replicates. *P<0.05, one-way ANOVA with Bonferroni's post-hoc test. (B) P19CL6 cells were co-transfected with the parental pTK-β-gal reporter (lanes 1-4), wild-type Prkaa2-β-gal (lanes 5-8), or mutant versions of the Prkaa2-β-gal reporter containing disruptions in MEF2 site 1 (lanes 9-12), site 2 (lanes 13-16) or both MEF2 sites (lanes 17-20). Co-transfections with expression plasmids for MEF2C and myocardin-935 are indicated with a plus symbol; a minus symbol indicates that an equivalent amount of the parental expression plasmid was added. Results shown are the mean fold activation over the pTK-β-gal reporter in the presence of parental expression vectors+ s.e.m.; n=9 independent biological replicates. ***P<0.001, two-way ANOVA with Bonferroni's post-hoc test.
We next examined cooperative activation of the Prkaa2 enhancer by MEF2C and myocardin-935 in P19CL6 cells (Fig. 3B). MEF2C weakly, but significantly, activated the wild-type Prkaa2-β-gal reporter on its own (Fig. 3B, lanes 5 and 6; Fig. S4). Activation of the wild-type reporter was very potently augmented by cotransfection of a myocardin-935 expression plasmid (Fig. 3B, lanes 6 and 8; Fig. S4). The weak activation of the wild-type reporter by myocardin alone (Fig. 3B, lane 7) is likely to be due to low levels of MEF2 in P19CL6 cells, since mutation of the MEF2 sites in the Prkaa2 enhancer abolished the myocardin-dependent activation of the reporter (Fig. 3B, lane 19). Mutation of either MEF2 site dramatically reduced, but did not abolish, cooperative activation by myocardin and MEF2C (Fig. 3B, lanes 9-16). Mutation of both MEF2 sites completely abolished activation of the reporter by MEF2C and myocardin (Fig. 3B, lanes 17-20). Importantly, no synergy was observed between MEF2C and the short form of myocardin (myocardin-856) on the Prkaa2 enhancer under conditions in which myocardin-935 potently augmented MEF2C-dependent transactivation of the enhancer (Fig. S4).
Bridging of two MEF2 sites by myocardin dimerization
MEF2C and myocardin activated the Prkaa2 reporters with a single intact MEF2 site by ∼10-fold but activated the wild-type reporter with two intact MEF2 sites by more than 60-fold (Fig. 3B), suggesting that the two sites function cooperatively. Previous work has shown that myocardin homodimerizes through a conserved leucine zipper (LZ) domain and that homodimerization facilitates stronger activation of SRF-dependent reporter genes containing two or more SRF binding sites (Wang et al., 2003). Based on these observations, we hypothesized that myocardin dimerization might facilitate interaction between the two Prkaa2 MEF2 sites, and we tested this notion using an in vitro pull-down experiment (Fig. 4A,B). Addition of MEF2C or myocardin-935 alone resulted in minimal pull-down of MEF2 site 1 by MEF2 site 2 (Fig. 4B, lane 1). By contrast, inclusion of both MEF2C and myocardin-935 resulted in robust and highly significant pull-down of MEF2 site 1 by MEF2 site 2 (Fig. 4B, lane 4). Mutation of the myocardin LZ motif in a manner predicted to disrupt homodimerization (Wang et al., 2003) completely abolished the complex and resulted in only baseline pull-down of MEF2 site 1 (Fig. 4B, lane 5). These experiments demonstrate that myocardin-935 dimerization can facilitate interaction between two MEF2C-bound MEF2 sites.
Fig. 4.

Bridging of MEF2C-bound MEF2 sites by myocardin-935. (A) Schematic of the in vitro pull-down assay. (B) qPCR detection of co-precipitated MEF2 site 1 after incubation with biotinylated MEF2 site 2 in the presence of reticulocyte lysate control (lane 1), MEF2C alone (lane 2), myocardin-935 (wt) alone (lane 3), MEF2C plus myocardin-935 (lane 4), or MEF2C plus a leucine zipper mutant form of myocardin-935 (LZ mut) (lane 5). Results are presented as mean fold enrichment over the reticulocyte lysate control+s.d. ****P<0.0001, two-way ANOVA with Bonferroni's post-hoc test. (C-H) The MEF2 sites in the Prkaa2 cardiac enhancer act synergistically in vivo. The wild-type Prkaa2[931]-lacZ transgenic reporter (C) and versions containing mutations in MEF2 site 1 (D), site 2 (E) or both MEF2 sites (F) were used to generate multiple independent transgenic lines or F0 embryos, and representative E11.5 embryos are shown. hrt, heart; LV, left ventricle. (G,H) The presence of both MEF2 sites is required for Prkaa2 enhancer activity in the adult heart. (I) Quantitation of β-galactosidase activity in E11.5 hearts from each of the Prkaa2[931]-lacZ transgenic reporter constructs shown in C-F. Each point on the graph represents a single embryonic heart from an independently generated transgenic embryo. Data are expressed as mean RLU/µg of excised heart tissue+s.e.m. Note the log scale on the y-axis. *P<0.05, **P<0.01, two-way ANOVA with Bonferroni's post-hoc test.
The two MEF2 sites in the Prkaa2 cardiac enhancer exhibit cooperative activity in vivo
To determine whether the Prkaa2 MEF2 sites are required for enhancer activity in vivo, we generated transgenic mouse embryos with the MEF2 sites mutated singly and in combination (Fig. 4C-H). The wild-type (wt) Prkaa2 enhancer directed robust expression in E11.5 and adult hearts (Fig. 4C,G). Mutation of both MEF2 sites resulted in complete loss of detectable X-gal staining in every transgenic founder examined at E11.5 (Fig. 4F) and in adult hearts (Fig. 4H). By contrast, mutation of either of the MEF2 sites alone reduced (but did not completely abolish) enhancer activity (Fig. 4D,E). These observations are consistent with the transactivation data (Fig. 3B), where we observed that mutation of a single site profoundly reduced, but did not completely abolish, transactivation whereas mutation of both MEF2 sites completely abolished transactivation by MEF2C and myocardin-935.
To determine if the Prkaa2 MEF2 sites function cooperatively in vivo, we generated numerous independent F0 transgenic founder embryos with each of the transgene constructs, and quantified β-galactosidase activity in E11.5 hearts (Fig. 4I). The wild-type enhancer showed consistently strong activity (n=10 independent transgenic lines) with only a few outliers and a mean activity of 792,489 RLU/µg of cardiac tissue. Mutation (m) of either MEF2 site resulted in a profound and significant diminution of activity (mMEF2 site 1, n=12 independent transgenic lines, 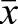 =194,591; mMEF2 site 2, n=18 independent transgenic lines,
=194,591; mMEF2 site 2, n=18 independent transgenic lines, 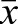 =96,627). Mutation of both MEF2 sites [mMEF2(1+2)] completely abolished enhancer activity in every transgenic founder examined (n=6 independent transgenic lines,
=96,627). Mutation of both MEF2 sites [mMEF2(1+2)] completely abolished enhancer activity in every transgenic founder examined (n=6 independent transgenic lines, 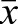 =1700). These data demonstrate a cooperative (greater than additive) relationship between the two Prkaa2 MEF2 sites in vivo. Interestingly, nearly all Prkaa2-lacZ transgenic lines with two intact MEF2 sites exhibited strong activity that ranged by less than a single order of magnitude, whereas transgenic embryos made from Prkaa2-lacZ constructs with only a single intact MEF2 site showed far greater variability ranging over nearly three orders of magnitude (Fig. 4I); this suggests that the presence of two functional MEF2 sites buffers the enhancer from silencing due to positional effects (Elgin and Reuter, 2013).
=1700). These data demonstrate a cooperative (greater than additive) relationship between the two Prkaa2 MEF2 sites in vivo. Interestingly, nearly all Prkaa2-lacZ transgenic lines with two intact MEF2 sites exhibited strong activity that ranged by less than a single order of magnitude, whereas transgenic embryos made from Prkaa2-lacZ constructs with only a single intact MEF2 site showed far greater variability ranging over nearly three orders of magnitude (Fig. 4I); this suggests that the presence of two functional MEF2 sites buffers the enhancer from silencing due to positional effects (Elgin and Reuter, 2013).
Paired MEF2 sites are prevalent in predicted cardiac enhancers
We analyzed predicted enhancers from mouse embryonic stem cells (ESCs), ESC-derived cardiac cells (cardiomyocytes and cardiac progenitors) and liver cells (Creyghton et al., 2010; Wamstad et al., 2012) and found that paired MEF2 sites occur in ∼30% of predicted cardiac enhancers. Interestingly, paired MEF2 sites occur 1.7 times more frequently in predicted cardiac enhancers than in liver enhancers (95% CI 1.5-1.9) and 2.0 times more frequently than in ESCs (95% CI 1.7-2.3) (Tables S1 and S2). The significant enrichment of paired MEF2 sites in predicted cardiac enhancers compared with liver and ESC enhancers suggests that natural selection has favored this arrangement in cardiac enhancers, possibly due to an advantage in gene expression conferred by myocardin bridging of the sites. Future studies will determine how the sequence and spacing of paired MEF2 sites in the Prkaa2 enhancer and other cis-regulatory elements determines the timing and robustness of cardiac gene activation.
MATERIALS AND METHODS
Plasmids, cloning and mutagenesis
A 931 bp fragment of the mouse Prkaa2 gene located between exons 1 and 2 was amplified by PCR using primers Prkaa2-F (5′-ACCCTGTAAAGAGGGAAAACCAAAAC-3′) and Prkaa2-R (5′-GCCAAAGCCTCGTGGTTCCTGCCAGC-3′) and then cloned into plasmid hsp68-lacZ (Kothary et al., 1989) to generate reporter plasmid Prkaa2[931]-lacZ for use in transgenic analyses and into plasmid pTK-β-gal (Robinson et al., 2014) to create plasmid Prkaa2-β-gal for use in transfection analyses. Plasmids pCDNA1-MEF2C, pCDNA1-MEF2C-VP16 and pCDNA3-Myocardin-935 have been described previously (Black et al., 1996; Black et al., 1995; Creemers et al., 2006). Details of deletion and site-specific mutations are provided in the supplementary Materials and Methods.
Generation and analysis of transgenic and enhancer knockout mice
Generation of transgenic mice was performed as described previously (De Val et al., 2004). Mef2c (MGI:1857491) and Myocd (MGI:2137495) knockout mice have been described previously (Li et al., 2003; Lin et al., 1997). The Prkaa2enhΔ allele was generated by CRISPR-mediated genome editing (Wang et al., 2013). Additional details of CRISPR-mediated genome editing and mouse genotyping are provided in the supplementary Materials and Methods. Adult hearts were collected from female ICR mice at 12 weeks of age; embryos were collected from 6- to 52-week-old ICR mice. No inclusion or exclusion criteria were defined, and no animals were excluded from analyses. All experiments using animals complied with federal and institutional guidelines and were reviewed and approved by the UCSF IACUC.
X-gal staining and in situ hybridization
X-gal staining was performed as described previously (Anderson et al., 2004). Whole-mount and section in situ hybridization with digoxigenin-labeled antisense probes was performed as described (Rojas et al., 2005). Detailed protocol information is provided in the supplementary Materials and Methods.
Cell culture, transfections and luminescent β-galactosidase assays
P19CL6 cells (obtained from Richard Kitsis; Peng et al., 2002) were authenticated by differentiation assay and confirmed to be free of contamination during experiments, were maintained in Minimum Essential Medium (MEM) α supplemented with 10% fetal bovine serum, and were transfected using Fugene 6 (Roche) according to the manufacturer's recommendations. Cells were harvested 48 h post-transfection, and cellular extracts were prepared and assayed for β-galactosidase activity using the Luminescent β-galactosidase Detection Kit (Clontech) as previously described (Dodou et al., 2003). For details, see the supplementary Materials and Methods.
EMSA and myocardin bridging assay
EMSAs were performed as previously described (Dodou et al., 2003). The sense strand sequences of the Prkaa2 oligonucleotides used for EMSA (with the MEF2 site underlined and mutant sequences indicated in bold) were: MEF2(1), 5′-GGGCACCATGCTAAAAATAAAATGGTTT-3′; MEF2(2), 5′-GGGAAAGTTTCTATTATTAGCAGAGATA-3′; mMEF2(1), 5′-GGGCACCATGCTAAACCCAAAATGGTTT-3′; and mMEF2(2), 5′-GGGAAAGTTTCTATTCCCAGCAGAGATA-3′. Control and mutant control MEF2 sites from the myogenin promoter have been described (Yee and Rigby, 1993). Methods for the myocardin bridging assay are described in the supplementary Materials and Methods.
Bioinformatics and MEF2 site prediction
Cardiac enhancers (Wamstad et al., 2012) were analyzed using the matchPattern function in the Biostrings package (Pages et al., 2014) within Bioconductor (Gentleman et al., 2004) and R (R Core Team, 2015) to identify MEF2 sites, as defined by the consensus sequence YTAWWWWTAR. For further details, see the supplementary Materials and Methods.
Acknowledgements
We thank M. Parmacek, R. Kitsis and E. Olson for providing mice and reagents.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
C.M.A., J.H., T.B.G., B.C., T.S. and A.B.H. performed experiments and analyzed data. S.-M.X., D.E.D. and L.A.P. generated transgenic and enhancer knockout mice. R.T. and K.S.P. performed bioinformatics analyses. B.G.B. helped identify the Prkaa2 enhancer and made intellectual contributions. B.L.B. conceived and directed the project, analyzed data and wrote the paper. All authors discussed and commented on the manuscript.
Funding
J.H. was supported by an American Heart Association fellowship. T.B.G. was supported by National Institutes of Health (NIH) grant T32 HL007544. This work was supported by NIH grants HL089707 to B.G.B. and B.L.B. and HL064658 to B.L.B. Work at Lawrence Berkeley National Laboratory was supported by NIH grants R24HL123879, U01DE024427, R01HG003988, U54HG006997 and UM1HL098166 and was conducted and performed under DOE Contract DE-AC02-05CH11231. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.138487.supplemental
References
- Anderson J. P., Dodou E., Heidt A. B., De Val S. J., Jaehnig E. J., Greene S. B., Olson E. N. and Black B. L. (2004). HRC is a direct transcriptional target of MEF2 during cardiac, skeletal, and arterial smooth muscle development in vivo. Mol. Cell. Biol. 24, 3757-3768. 10.1128/MCB.24.9.3757-3768.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black B. L. and Cripps R. M. (2010). Myocyte enhancer factor 2 transcription factors in heart development and disease. In Heart Development and Regeneration (ed. Rosenthal N. and Harvey R. P.), pp. 673-699. Oxford: Academic Press. [Google Scholar]
- Black B. L. and Olson E. N. (1998). Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu. Rev. Cell Dev. Biol. 14, 167-196. 10.1146/annurev.cellbio.14.1.167 [DOI] [PubMed] [Google Scholar]
- Black B. L., Martin J. F. and Olson E. N. (1995). The mouse MRF4 promoter is trans-activated directly and indirectly by muscle-specific transcription factors. J. Biol. Chem. 270, 2889-2892. 10.1074/jbc.270.13.7055 [DOI] [PubMed] [Google Scholar]
- Black B. L., Ligon K. L., Zhang Y. and Olson E. N. (1996). Cooperative transcriptional activation by the neurogenic basic helix-loop-helix protein MASH1 and members of the myocyte enhancer factor-2 (MEF2) family. J. Biol. Chem. 271, 26659-26663. 10.1074/jbc.271.37.22499 [DOI] [PubMed] [Google Scholar]
- Creemers E. E., Sutherland L. B., Oh J., Barbosa A. C. and Olson E. N. (2006). Coactivation of MEF2 by the SAP domain proteins myocardin and MASTR. Mol. Cell 23, 83-96. 10.1016/j.molcel.2006.05.026 [DOI] [PubMed] [Google Scholar]
- Creyghton M. P., Cheng A. W., Welstead G. G., Kooistra T., Carey B. W., Steine E. J., Hanna J., Lodato M. A., Frampton G. M., Sharp P. A. et al. (2010). Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 107, 21931-21936. 10.1073/pnas.1016071107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Val S., Anderson J. P., Heidt A. B., Khiem D., Xu S.-M. and Black B. L. (2004). Mef2c is activated directly by Ets transcription factors through an evolutionarily conserved endothelial cell-specific enhancer. Dev. Biol. 275, 424-434. 10.1016/j.ydbio.2004.08.016 [DOI] [PubMed] [Google Scholar]
- Dodou E., Xu S.-M. and Black B. L. (2003). mef2c is activated directly by myogenic basic helix-loop-helix proteins during skeletal muscle development in vivo. Mech. Dev. 120, 1021-1032. 10.1016/S0925-4773(03)00178-3 [DOI] [PubMed] [Google Scholar]
- Elgin S. C. R. and Reuter G. (2013). Position-effect variegation, heterochromatin formation, and gene silencing in Drosophila. Cold Spring Harb. Perspect. Biol. 5, a017780 10.1101/cshperspect.a017780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman R. C., Carey V. J., Bates D. M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J. et al. (2004). Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5, R80 10.1186/gb-2004-5-10-r80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothary R., Clapoff S., Darling S., Perry M. D., Moran L. A. and Rossant J. (1989). Inducible expression of an hsp68-lacZ hybrid gene in transgenic mice. Development 105, 707-714. [DOI] [PubMed] [Google Scholar]
- Li S., Wang D.-Z., Wang Z., Richardson J. A. and Olson E. N. (2003). The serum response factor coactivator myocardin is required for vascular smooth muscle development. Proc. Natl. Acad. Sci. USA 100, 9366-9370. 10.1073/pnas.1233635100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q., Schwarz J., Bucana C. and Olson E. N. (1997). Control of mouse cardiac morphogenesis and myogenesis by transcription factor MEF2C. Science 276, 1404-1407. 10.1126/science.276.5317.1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miano J. M. (2015). Myocardin in biology and disease. J. Biomed. Res. 29, 3-19. 10.7555/JBR.29.20140151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pages H., Aboyoun P., Gentleman R. and DebRoy S. (2014). Biostrings: string objects representing biological sequences, and matching algorithms R package version 2.36.4.
- Parmacek M. S. (2007). Myocardin-related transcription factors: critical coactivators regulating cardiovascular development and adaptation. Circ. Res. 100, 633-644. 10.1161/01.RES.0000259563.61091.e8 [DOI] [PubMed] [Google Scholar]
- Peng C.-F., Wei Y., Levsky J. M., McDonald T. V., Childs G. and Kitsis R. N. (2002). Microarray analysis of global changes in gene expression during cardiac myocyte differentiation. Physiol. Genomics 9, 145-155. 10.1152/physiolgenomics.00027.2002 [DOI] [PubMed] [Google Scholar]
- Pipes G. C. T., Creemers E. E. and Olson E. N. (2006). The myocardin family of transcriptional coactivators: versatile regulators of cell growth, migration, and myogenesis. Genes Dev. 20, 1545-1556. 10.1101/gad.1428006 [DOI] [PubMed] [Google Scholar]
- Potthoff M. J. and Olson E. N. (2007). MEF2: a central regulator of diverse developmental programs. Development 134, 4131-4140. 10.1242/dev.008367 [DOI] [PubMed] [Google Scholar]
- R Core Team (2015). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org.
- Robinson A. S., Materna S. C., Barnes R. M., De Val S., Xu S. M. and Black B. L. (2014). An arterial-specific enhancer of the human endothelin converting enzyme 1 (ECE1) gene is synergistically activated by Sox17, FoxC2, and Etv2. Dev. Biol. 395, 379-389. 10.1016/j.ydbio.2014.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas A., De Val S., Heidt A. B., Xu S. M., Bristow J. and Black B. L. (2005). Gata4 expression in lateral mesoderm is downstream of BMP4 and is activated directly by Forkhead and GATA transcription factors through a distal enhancer element. Development 132, 3405-3417. 10.1242/dev.01913 [DOI] [PubMed] [Google Scholar]
- Wamstad J. A., Alexander J. M., Truty R. M., Shrikumar A., Li F., Eilertson K. E., Ding H., Wylie J. N., Pico A. R., Capra J. A. et al. (2012). Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell 151, 206-220. 10.1016/j.cell.2012.07.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D.-Z. and Olson E. N. (2004). Control of smooth muscle development by the myocardin family of transcriptional coactivators. Curr. Opin. Genet. Dev. 14, 558-566. 10.1016/j.gde.2004.08.003 [DOI] [PubMed] [Google Scholar]
- Wang Z., Wang D.-Z., Pipes G. C. T. and Olson E. N. (2003). Myocardin is a master regulator of smooth muscle gene expression. Proc. Natl. Acad. Sci. USA 100, 7129-7134. 10.1073/pnas.1232341100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Yang H., Shivalila C. S., Dawlaty M. M., Cheng A. W., Zhang F. and Jaenisch R. (2013). One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153, 910-918. 10.1016/j.cell.2013.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee S. P. and Rigby P. W. (1993). The regulation of myogenin gene expression during the embryonic development of the mouse. Genes Dev. 7, 1277-1289. 10.1101/gad.7.7a.1277 [DOI] [PubMed] [Google Scholar]