

Abstract
Naproxen (NPX) is used in the treatment of rheumatoid arthritis (RA) for alleviation of pain and inflammation. In view of the extensive albumin binding of NPX, this study investigates whether chronic inflammation and sex influence the physiologic albumin concentrations, plasma protein binding, and pharmacokinetics (PK) of NPX. The PK of NPX was evaluated in a rat model of RA [collagen-induced arthritis (CIA) in Lewis rats] and in healthy controls. These PK studies included 1) NPX in female and male CIA rats that received 10, 25, or 50 mg/kg NPX i.p.; and 2) NPX in healthy female and male rats after i.p. dosing of NPX at 50 mg/kg. Plasma albumin concentrations were quantified by enzyme-linked immunosorbent assay, and protein binding was assessed using ultrafiltration. The NPX concentrations in plasma and filtrates were determined by liquid chromatography–tandem mass spectrometry (LC-MS/MS). Plasma concentration-time data of NPX were first assessed by noncompartmental analysis (NCA). Nonlinear PK as indicated by dose-dependent NCA clearances and distribution volumes was observed. A two-compartment model with a first-order absorption process incorporating nonlinear protein binding in plasma and tissues jointly described the PK data of all groups. Saturable albumin binding accounts for the nonlinearity of NPX PK in all rats as well as part of the PK differences in arthritic rats. The CIA rats exhibited reduced albumin concentrations, reduced overall protein binding, and reduced clearances of unbound NPX, consistent with expectations during inflammation. The net effect of chronic inflammation was an elevation of the Cmax and area under the plasma concentration-time curve (AUC) of unbound drug.
Introduction
Rheumatoid arthritis (RA) is a chronic systemic inflammatory autoimmune disease. It produces joint pain, stiffness, and swelling due to synovial inflammation and effusion. There is a sex preference, with more women affected than men (van Vollenhoven, 2009). The pathogenesis of RA involves the biosynthesis and actions of many proinflammatory mediators, including cytokines and prostaglandins (PG) (McInnes and Schett, 2011). In particular, PG play a key role in the generation of cardinal signs of acute inflammation, such as pain, fever, redness, and swelling, by promoting blood flow into the inflamed tissues and increasing microvascular permeability (Funk, 2001; Ricciotti and FitzGerald, 2011), and contribute to chronic inflammation through amplifying cytokine signaling as well as inducing and recruiting inflammatory cells at affected sites (Aoki and Narumiya, 2012).
Nonsteroidal anti-inflammatory drugs (NSAIDs) are extensively used to treat inflammatory diseases, including RA, because of their effective analgesic and anti-inflammatory properties (Crofford, 2013). The primary pharmacological mechanism of NSAIDs is blocking the biosynthesis of PG from arachidonic acid through inhibition of the enzymatic activity of cyclooxygenase (COX). There are two distinct isoforms. COX-1 is a constitutive enzyme that synthesizes PG to help maintain homeostatic functions, including protection of gastric mucosa and platelet activation. In contrast, the expression of COX-2 is inducible under inflammatory conditions and serves as the main source of PG responsible for various inflammatory responses (Crofford et al., 2000; Smith et al., 2000; Ricciotti and FitzGerald, 2011). Despite the clinical effectiveness of NSAIDs, their side effects, such as gastrointestinal bleeding and potential cardiovascular toxicity, were also associated with the strong inhibition of either COX-1 or COX-2 (Lanas, 2009; Coxib and Traditional NSAID Trialists’ Collaboration et al., 2013).
Naproxen (NPX), a traditional NSAID, has been widely used in the management of RA (Davies and Anderson, 1997). It is a nonselective COX inhibitor that shows inhibitory effects for both isoforms with moderate potencies (Vane, 1971). Compared with selective COX inhibitors, NPX is better tolerated with respect to gastrointestinal complications and cardiovascular risk (Lussier et al., 1978; Watson et al., 2002). Similar to most NSAIDs, NPX is highly bound to human plasma proteins, especially albumin, with very strong binding (>99%) at therapeutic concentrations (Mortensen et al., 1979). Hypoalbuminemia frequently occurs in RA owing to increased catabolism (Wilkinson et al., 1965), and thereby, the disposition of highly albumin-bound drugs might be altered during RA. The clearance and distribution volume of NPX both increased in RA patients compared with normal subjects due to higher unbound NPX concentrations (van den Ouweland et al., 1987). Nonlinear pharmacokinetics (PK) caused by saturation of albumin binding at high total drug concentrations were reported (Stoeckel et al., 1981; Lin et al., 1985; Wong et al., 1999). Dose-dependent clearance of total drug and a plateau effect on the plasma concentration-time curve (AUC) were shown at higher NPX doses in humans (Runkel et al., 1974). Sodium naproxen is a Biopharmaceutical Classification System I compound with high solubility and permeability, and good absorption has been found in healthy and RA subjects (van den Ouweland et al., 1987; Vree et al., 1993).
The PK of NPX has been assessed in various species (Lauroba et al., 1986; Huntjens et al., 2006, 2010; Elsinghorst et al., 2011). However, the underlying mechanisms causing the nonlinear PK have either been neglected or not been fully explored. The NPX PK profiles in earlier studies were described using simple linear clearance models without consideration of the nonlinearity. In addition, the influences of chronic inflammation (RA) and sex on the protein binding and disposition of NPX have not yet been well investigated. Our studies are consonant with recent emphasis (Danska, 2014) that translational studies of sex differences should be expanded.
Collagen-induced arthritis (CIA) in Lewis rats is an animal disease model that closely resembles many features of human RA (Stuart et al., 1982; Holmdahl et al., 2001). This model has been successfully applied to assess the PK/pharmacodynamic (PD) properties of various drugs, including dexamethasone, anakinra, and abatacept (Earp et al., 2008b; Liu et al., 2011; Lon et al., 2013).
In the current study, the PK of NPX was examined in female and male normal and CIA rats after various i.p. doses of NPX. A global PK model incorporating sex and disease effects on nonlinear protein binding was successfully applied. This study was designed to support a subsequent preclinical PD study of NPX in CIA rats (Li et al., 2017).
Materials and Methods
Reagents and Chemicals.
Naproxen (purity >98.5%), sodium naproxen, liquid chromatography–mass spectrometry–grade acetonitrile, and high-performance liquid chromatography–grade formic acid were all obtained from Sigma-Aldrich (St Louis, MO). (S)-Naproxen-d3 [internal standard (IS), purity >98%] was purchased from Toronto Research Chemicals Inc. (Toronto, Canada). Milli-Q water was used (Millipore Corporation, Bedford, MA).
Animals.
Male and female Lewis rats (5–8 weeks old) were purchased from Harlan (Indianapolis, IN), weighing approximately 110–160 g for females and 170–220 g for males, age-matched for each sex group at the time of PK studies. All rats were housed individually in the University Laboratory Animal Facility under controlled temperature (22°C) and humidity, 12-hour light/12-hour dark cycles, and free access to water and food. Rats were acclimated for 1 week before the studies. These studies were performed in accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council, 2011) and were approved by the University at Buffalo Institutional Animal Care and Use Committee.
Induction of CIA in Lewis Rats.
The induction of CIA in Lewis rats was conducted following protocols and reagents supplied by Chondrex (Redmond, WA). The detailed procedures were as previously described (Earp et al., 2008a). Paw edema and body weights were monitored from the first day of induction through day 15 for females and day 20 for males. Hind-paw swelling was used as the indicator for edema and was measured by digital calipers (VWR Scientific, Rochester, NY). The peak disease status was observed on day 16 for females and day 21 for males. Approximately 60% of the males and 80% of the females successfully developed arthritis in one or both hind paws.
Pharmacokinetics of NPX.
The NPX doses were prepared freshly as a sodium NPX solution in phosphate-buffered saline (PBS) (pH = 8) and filtered through 0.22-μm filters before use. The drug was administered i.p. in a volume of 1 ml/kg.
The female and male CIA rats were randomly divided into three subgroups per sex group and received an i.p. bolus injection with 11, 27.5, or 55 mg/kg sodium NPX PBS solution (equivalent to 10, 25, or 50 mg/kg NPX) on day 16 (females) and day 21 (males) post disease induction. Healthy male and female rats were dosed i.p. with 55 mg/kg sodium NPX in PBS (50 mg/kg NPX). Serial blood samples were collected from the saphenous vein using ethylenediaminetetraacetic acid (EDTA) as the anticoagulant at 15, 30, and 45 minutes and 1, 2, 4, 6, 9, 12, and 24 hours postdose. There were three CIA rats and four healthy rats sampled at each time point. Blood samples were immediately centrifuged at 2000 × g for 15 minutes at 4°C, and the obtained plasma samples were stored at –80°C before analysis.
Blood Collection for Albumin and Protein Binding.
Eight CIA females and eight healthy females were sacrificed on day 16 after arthritis induction by aortal exsanguinations under isoflurane anesthesia. Four CIA males and four healthy males were sacrificed on day 21 after induction. Blood samples were collected into syringes precoated with EDTA and centrifuged immediately at 2000 × g for 15 minutes at 4°C. Plasma samples were collected, and 25 µl of the plasma from each rat was separated and stored for albumin determination. The remaining fractions were pooled for each animal group and used in subsequent protein-binding studies.
Plasma Albumin Determination.
The plasma concentrations of albumin in CIA and healthy rats (both sex groups) were quantified using an anti-rat albumin enzyme-linked immunosorbent assay (ELISA) kit (Bethyl Laboratories, Montgomery, TX). Rat plasma samples were diluted (1:1,000,000) by conjugate diluent prior to assay. All other procedures followed the manufacturer’s protocol. Four rats from each animal group were used, and all samples were run in triplicate. The range of the standard curve was 1.95–125 ng/ml, and a four-parameter logistic model was applied to fit the standard curve.
Plasma Protein Binding of NPX.
Plasma protein binding of NPX was measured by ultrafiltration using Centrifree micropartition devices (Millipore Corporation) with a 30 kDa molecular mass cutoff filter. In brief, 16 µl of NPX PBS solution (0.2, 0.5, 1, 2, 5, 10, 20, and 50 mg/ml) was added at 1% of the total volume to pooled blank plasma samples from each group to yield eight plasma samples containing NPX per group (2, 5, 10, 20, 50, 100, 200, and 500 µg/ml). After incubation at 37°C for 30 minutes, aliquots (460 µl) of plasma of each concentration were transferred into three prerinsed ultrafiltration devices and centrifuged at 2000 × g for 20 minutes. The filtrates and remaining plasma samples were stored at −80°C until analysis of both free and total NPX concentrations. Preliminary studies showed that there was negligible nonspecific binding of NPX to the ultrafiltration device.
Drug Analysis.
The NPX concentrations in all samples were determined by liquid chromatography–tandem mass spectrometry (LC-MS/MS). In brief, plasma samples (15 μl) from the PK and protein-binding studies were spiked with 10 μl of IS working solutions (2.5 μg/ml) followed by precipitation with 400 μl of acetonitrile containing 0.1% formic acid. The mixtures were vortexed for 1 minute and sonicated for 20 seconds, then vortexed for another 10 seconds after sonication and centrifuged at 13,000 × g for 10 minutes. Then, 300 μl of the supernatant was transferred to a 2-ml tube containing 1.2 ml of water and vortexed for 10 seconds. Finally, 10 μl of the mixture was injected into the LC-MS/MS for analysis.
The NPX concentrations in the filtrates (free drug concentrations) from the protein-binding studies were pretreated using the same method as plasma samples, with slight modifications. In brief, 75 μl of each filtrate sample was spiked with 10 μl of IS working solutions (2.5 μg/ml) followed by precipitation with 300 μl of acetonitrile containing 0.1% formic acid. The remaining sample-preparation procedures were as described earlier for the plasma samples.
The LC-MS/MS system consisted of a Shimadzu (Kyoto, Japan) high-performance liquid chromatography module including a binary pump, a degasser, an autosampler, and a column oven, and an Applied Biosystems (Foster City, CA) PE/Sciex API3000 mass spectrometer equipped with a turbo ion spray interface. Sample separations were achieved on a Targa C18 Column (particle size 5 μm, 100 × 2.1 mm; Higgins Analytical Inc., Mountain View, CA). The mobile phase consisted of eluent A [water/acetonitrile (95:5, v/v) containing 0.1% acetic acid] and eluent B [acetonitrile/water (95:5, v/v) containing 0.1% acetic acid] and was pumped at a flow rate of 0.23 ml/min with a gradient elution. The gradient profile was as follows: 0–4 minutes, 50% B; a linear increase to 95% B from 4 to 6.5 minutes; a linear decrease to 50% B over 0.1 minute; 50% B for 4.4 minutes; and stopped at 11.00 minutes. The autosampler was maintained at 4°C during the run. The mass spectrometer was operated in the negative ionization mode for the detection of ion transitions at m/z 229.2/169.9 for NPX and 232.0/169.9 for IS. The system was controlled by Analyst software version 1.4 (Applied Biosystems Sciex) for data acquisition and analysis.
Linearity was found over a concentration range of 0.125–40 µg/ml for plasma and 0.01–30 µg/ml for filtrate samples. The coefficients of variation (CV%) for intra- and interday accuracies and precisions were all <10%. The recovery of the sample-preparation method approached 100%. Naproxen in rat plasma was previously found to be stable under various conditions (Shi et al., 2015).
Protein-Binding and Pharmacokinetic Data Analysis.
The binding capacity and association constants were estimated by fitting the bound versus free drug concentrations using an equation describing two classes of binding sites (Wong et al., 1999):
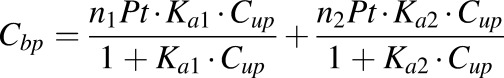 |
(1) |
where 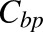 and
and 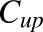 are the bound and free plasma NPX concentrations; Ka1 and Ka2 are the association constants for the first and second class of binding sites; n1 and n2 are the numbers of first and second class binding sites; and Pt is the albumin concentration in plasma.
are the bound and free plasma NPX concentrations; Ka1 and Ka2 are the association constants for the first and second class of binding sites; n1 and n2 are the numbers of first and second class binding sites; and Pt is the albumin concentration in plasma.
The relationship between bound and total drug concentrations is:
| (2) |
where 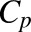 is the total plasma concentration of NPX.
is the total plasma concentration of NPX.
Based on the assessment of the plasma protein-binding data, 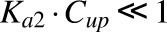 , and substitution for bound concentrations using eq. 2 yields:
, and substitution for bound concentrations using eq. 2 yields:
| (3) |
where there is one positive root for free drug concentration:
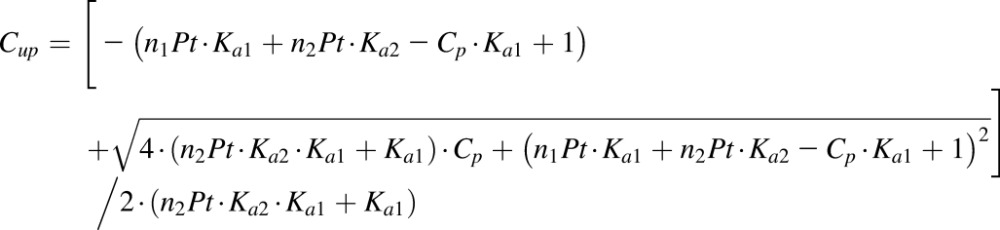 |
(4) |
The protein binding of NPX in tissues is considered to occur primarily with albumin in the interstitial fluid (ISF) (Aukland and Nicolaysen, 1981; Rodgers and Rowland, 2006). Assuming that the binding affinities and numbers of binding sites on each protein molecule in ISF are the same as in plasma, then the relationship between total (Ct) and unbound tissue concentrations (Cut) of NPX could also be described by eq. 4 with a difference in ISF and plasma protein concentrations, where Pt is multiplied by E/P, the ratio of protein concentrations in ISF and plasma.
According to the “free hormone hypothesis” (Mendel, 1989), disposition processes often operate only on free drug. Therefore, NPX plasma concentration-time profiles were characterized by compartment models based on free drug concentrations with binding calculated from total concentrations. Several models including one- and two-compartment PK models (with or without an absorption process) with binding were tested, and the final PK model selected (Fig. 1) was a two-compartment model with a first-order absorption process incorporating nonlinear protein binding. The final PK model equations and initial conditions are:
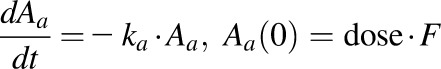 |
(5) |
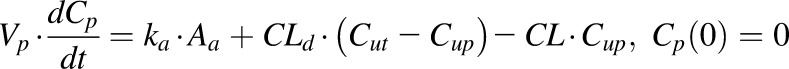 |
(6) |
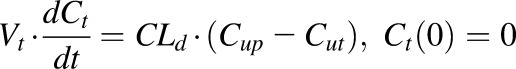 |
(7) |
where Aa indicates the amount of NPX at the absorption site; ka is the first-order absorption rate constant; CL and CLd are the plasma and distribution clearances of unbound NPX; Vp and Vt are the volumes of distribution of total NPX in central and peripheral compartments; and F is the bioavailability of the i.p. dose calculated to be about 0.9 from literature-reported i.v. data in rats (Lauroba et al., 1986). The drug was given i.p. to minimize stress in the animals and based on observations that NPX absorption is rapid, reproducible, and essentially complete by this route (Huntjens et al., 2006).
Fig. 1.

The pharmacokinetic model for naproxen incorporating protein binding in the central and peripheral compartments. Symbols are defined in the text and Tables 1 and 4.
Initial noncompartmental analysis (NCA) of the PK data was performed using the Phoenix WinNonlin 6.4 software (Certara Corporation, Princeton, NJ). All model fittings were performed using the maximum likelihood algorithm in ADAPT 5 (Biomedical Simulations Resource, Los Angeles, CA) (D’Argenio et al., 2009). The model code is provided in the Supplemental Materials. All protein-binding and PK data were naive-pooled before analysis. The protein-binding profiles were first fitted and the estimated binding parameters were fixed in the PK model. The variance model used was:
| (8) |
where Vi represents the variance of the ith data point; Yi is the ith model-predicted plasma concentration; and σ1 and σ2 are variance model parameters and were estimated together with other system parameters during model fitting. Model selection was based on the goodness-of-fit criteria, which included the Akaike information criterion, visual inspection of the fitted profiles, and coefficients of variation (CV%) of the parameter estimates.
Statistical Analysis.
All data were analyzed statistically by one-way analysis of variance and Tukey’s multiple comparison test using SPSS software version 22 (IBM SPSS Statistics, Chicago, IL), and p < 0.05 was considered to be statistically significant.
Results
Quantification of Plasma Albumin.
An anti-rat albumin ELISA was applied to determine whether sex and the presence of arthritis influence the plasma concentrations of albumin. As shown in both Fig. 2 and Table 1, the plasma albumin concentrations in CIA rats were significantly lower than those in healthy animals, consistent with the situation in humans where hypoalbuminemia is a feature of RA (Wilkinson et al., 1965). It is interesting to note that albumin concentrations also differed significantly with sex in rats, with lower values observed in males.
Fig. 2.

Plasma albumin concentrations in CIA males (CM), healthy males (HM), CIA females (CF), and healthy females (HF) determined by ELISA. a and b, ***P < 0.001, significant difference compared with CM and HF; c, ***P < 0.001, significant difference compared with HF; d, **P < 0.01, significant difference compared with CM.
TABLE 1.
Parameter estimations for plasma protein binding of NPX
| Parameters | Definition | Estimates (CV%) |
|||
|---|---|---|---|---|---|
| CIA Females | Healthy Females | CIA Males | Healthy Males | ||
| Ka1 (µM−1) | Association constant for first binding site | 0.28 (3.53) | 0.25 (3.33) | 0.26 (4.00) | 0.26 (1.50) |
| Ka2 (µM−1) | Association constant for second binding site | 0.0041 (4.2) | 0.0043 (4.35) | 0.0056 (11.75) | 0.0054 (2.65) |
| n1 | Number of first-class binding sites | 1 (Fixed) | |||
| n2 | Number of second-class binding sites | 4 (Fixed) | |||
| Pt (µM) | Measured albumin concentration | 347 (Fixed) | 550 (Fixed) | 282 (Fixed) | 422 (Fixed) |
| n1Pta (µM) | Binding capacity of first binding site | 347 | 550 | 282 | 422 |
| n2Pta (µM) | Binding capacity of second binding site | 1388 | 2200 | 1128 | 1688 |
Binding capacities were calculated as the product of ni × Pt.
Protein Binding of NPX.
Protein-binding studies of NPX were carried out using plasma from the four animal groups. Plasma protein binding of NPX in rats showed concentration dependence, with higher unbound fractions at higher total plasma concentrations (Fig. 3). Higher percentage binding of NPX to rat plasma proteins (>93%) was observed at normal therapeutic concentrations in all groups with very small variation (CV% < 2.36%). However, a significant decrease in binding was found at total concentrations greater than 50 µg/ml in CIA rats and above 100 µg/ml in healthy rats (Supplemental Table S1). Rosenthal plots (Fig. 3) presenting the bound/unbound drug concentration ratios versus bound concentrations showed biphasic profiles, indicating that there are two classes of binding sites on the protein. The bound versus free concentration profiles were fitted to the binding equation with two classes of binding sites (eq. 1) (Supplemental Model Code for Protein Binding). Preliminary fittings allowing n1 and n2 to vary yielded the nearest integers of 1 and 4, which were then fixed for subsequent assessment. As shown in Fig. 4, there was very good agreement between the observed and fitted data, except that the bound concentrations in CIA males were slightly underpredicted at the highest free concentrations. The association constants listed in Table 1 indicate that NPX is bound to rat plasma albumin with high affinity, and the first class (n1) of binding sites differed from the second (n2) as Ka2 was only about 2% of Ka1. In addition, the binding capacities (nPt) varied with lower binding capacities in the CIA rats and male rats (Table 1).
Fig. 3.

Rosenthal plots of bound/unbound concentrations (Cb/Cf) versus bound concentrations (Cb) for NPX binding in rat plasma. Embedded figures are plots of free fractions (fu) versus total concentrations (Ct) for NPX binding. Closed circles depict CIA rats; open circles depict healthy rats.
Fig. 4.

Relationship of bound (Cb) versus unbound (Cf) concentrations of NPX in the four groups. Curves depict fitting of data using eq. 1.
Pharmacokinetics of NPX.
The PK of NPX was investigated in CIA rats and compared with that in healthy animals. Following i.p. doses of NPX (10, 25, and 50 mg/kg) in CIA rats and 50 mg/kg in healthy rats, the NPX PK profiles of all groups shown in Figs. 5 and 6 were biexponential, with parallel elimination phases between the dose groups. A sharper initial decline was observed in higher dose groups, indicating that distribution rates increased with dose/concentration. The primary PK parameters for total drug were initially calculated by NCA for comparison with published PK results. In CIA rats, plasma NPX concentrations increased less than dose proportionally in both sex groups, as indicated by the dose-normalized AUC, which decreased nearly 2-fold over the 5-fold range of doses. In addition, the apparent volume of distribution (V/F) and apparent clearance (CL/F) significantly increased in the highest dose groups (50 mg/kg) compared with those in the 10 mg/kg dose groups (Table 2). Together, these results indicated the occurrence of dose-dependent PK of NPX in CIA rats. The NCA results for arthritic and healthy rats are listed in Table 3. The AUC of total NPX in arthritic rats was significantly lower than that in healthy animals. Both V/F and CL/F values in CIA rats were about 30–50% higher than the corresponding values in healthy rats. Despite these differences, NCA did not indicate any significant differences in the PK parameters between female and male rats. In addition, the terminal half-life (t1/2) was similar in all dose groups in CIA rats and between arthritic and healthy rats at the same dose level (Tables 2 and 3). The apparent clearance values for NPX are considerably smaller than hepatic plasma flow (circa 1200 ml/h/kg) (Davies and Morris, 1993), indicating that this is a low-clearance compound and should have little first-pass loss after i.p. administration.
Fig. 5.

Plasma concentration versus time profiles of 10 mg/kg (solid circles and solid lines), 25 mg/kg (open squares and long dashed lines), and 50 mg/kg (solid triangles and short dashed lines) NPX in female (left panel) and male (right panel) arthritic rats. Symbols are observed total plasma concentrations of NPX. Curves in the upper panel depict model fittings jointly for all dose groups. Curves in the lower panel depict model-predicted unbound plasma concentrations of NPX.
Fig. 6.

Plasma concentration versus time profiles of 50 mg/kg NPX in female and male healthy rats. Symbols are observed total plasma concentrations of NPX. Dashed lines depict model fittings jointly for all groups, and solid lines depict model-predicted unbound plasma concentrations of NPX.
TABLE 2.
NCA pharmacokinetic parameters based on total NPX concentrations for 10, 25, and 50 mg/kg i.p. doses in CIA rats (mean ± S.D.)
| Dose (mg/kg) | CIA Females (n = 3) |
CIA Males (n = 3) |
||||||
|---|---|---|---|---|---|---|---|---|
| Dose-Normalized AUC |
CL/F |
V/F |
t1/2 |
Dose-Normalized AUC |
CL/F |
V/F |
t1/2 |
|
| µg∣h∣kg/ml/mg | ml/h/kg | ml/kg | h | µg∣h∣kg/ml/mg | ml/h/kg | ml/kg | h | |
| 10 | 33.84 ± 5.17 | 29.98 ± 4.21 | 133.8 ± 23.61 | 3.13 ± 0.65 | 27.09 ± 1.39 | 36.98 ± 1.85 | 135.1 ± 16.32 | 2.54 ± 0.31 |
| 25 | 25.44 ± 0.80 | 39.34 ± 1.22* | 157.0 ± 15.13 | 2.76 ± 0.23 | 24.26 ± 1.35 | 41.31 ± 2.38 | 137.9 ± 10.73 | 2.32 ± 0.18 |
| 50 | 18.74 ± 0.86** | 53.43 ± 2.53** | 287.1 ± 42.50** | 3.74 ± 0.64 | 16.20 ± 1.22** | 61.99 ± 4.87** | 229.3 ± 48.93* | 2.55 ± 0.36 |
P < 0.05; **P < 0.01 (significant difference compared with 10 mg/kg dose group in each sex group).
TABLE 3.
Comparison of NCA pharmacokinetic parameters based on total NPX concentrations for 50 mg/kg i.p. doses in CIA and healthy rats
Values are mean ± S.D.
| Groups | n | AUC |
CL/F |
V/F |
t1/2 |
|---|---|---|---|---|---|
| µg∣h/ml | ml/h/kg | ml/kg | h | ||
| CIA males | 3 | 809.8 ± 61.0* | 61.99 ± 4.87** | 229.3 ± 48.9* | 2.55 ± 0.36 |
| Healthy males | 4 | 1054.5 ± 116.5 | 47.76 ± 4.98 | 169.7 ± 18.2 | 2.46 ± 0.03 |
| CIA females | 3 | 936.0 ± 42.2* | 53.43 ± 2.53* | 287.1 ± 42.5* | 3.74 ± 0.64 |
| Healthy females | 4 | 1203.4 ± 138.2 | 41.89 ± 4.51 | 188.6 ± 24.7 | 3.13 ± 0.34 |
P < 0.05; **P < 0.01 (significant difference compared with healthy group in each sex group).
A two-compartment model with a first-order absorption process and nonlinear albumin binding (Fig. 1) was used to describe the PK of NPX (Supplemental Model Code for PK Estimation). Free NPX concentrations calculated from the total using the protein-binding parameters in Table 1 were used in modeling (eqs. 5–7) (Supplemental Model Code for PK Simulation). The model fittings applied jointly for all animals and PK groups along with the corresponding model-predicted unbound plasma concentrations of NPX are shown in Figs. 5 and 6. The final model parameter estimates are presented in Table 4. This model described the PK profiles reasonably well, except for missing the last time point, with low CV% values for all parameter estimates. Absorption of NPX from the i.p. injection site was rapid, with a ka value of about 0.8 hour−1, which is in line with reported data (Huntjens et al., 2006, 2010). The CIA rats showed lower unbound plasma clearances (CLArthritic) and larger peripheral distribution volumes of total drug (VtArthritic) compared with healthy rats, consistent with the findings in humans (van den Ouweland et al., 1987). The estimated Vt values approach the ISF volume in rats (174 ml/kg) (Shah and Betts, 2012), further indicating that NPX distributes principally to tissue interstitial space. Lower unbound distribution clearances in CIA rats (CLdArthritic) observed in our study also agree with previous findings indicating that the rheumatoid synovium is less permeable to small molecules, and inflammation might decrease the distribution of unbound NSAIDs into and out of tissues (Simkin, 1979; Wallis et al., 1985; Day et al., 1999). It is apparent that NPX is a low-extraction drug, and its unbound plasma clearance is mainly determined by the intrinsic clearance based on the well stirred model of hepatic clearance. The unbound plasma NPX concentration-time profiles for all 50 mg/kg dose groups are shown in Fig. 7. In both sex groups, arthritic rats showed higher peak unbound concentrations (13.52 vs. 8.14 µg/ml for females and 13.51 vs. 8.43 µg/ml for males) and increased unbound AUC (32.76 vs. 23.94 µg∣h/ml for females and 32.82 vs. 23.95 µg∣h/ml for males) compared with healthy rats.
TABLE 4.
Modeled pharmacokinetic parameter estimates for unbound NPX after i.p. administration
| Parameters | Definition | Estimates | CV% |
|---|---|---|---|
| ka (1/h) | Absorption rate constant | 0.814 | 7.54 |
| CLArthritic (ml/h/kg) | Unbound plasma clearance in arthritic rats | 1370 | 4.73 |
| CLHealthy (ml/h/kg) | Unbound plasma clearance in healthy rats | 1879 | 9.26 |
| Vp (ml/kg) | Central volume of distribution | 32.36a | Fixed |
| CLdArthritic (ml/h/kg) | Unbound distribution clearance in arthritic rats | 647.2 | 18.61 |
| CLdHealthy (ml/h/kg) | Unbound distribution clearance in healthy rats | 1371 | 45.23 |
| VtArthritic (ml/kg) | Peripheral distribution volume in arthritic rats | 140.7 | 9.27 |
| VtHealthy (ml/kg) | Peripheral distribution volume in healthy rats | 114.7 | 17.37 |
Physiologic parameter values obtained from Shah and Betts (2012).
Fig. 7.

Simulated unbound plasma NPX concentration versus time profiles for 50 mg/kg dose groups. Solid lines depict profiles for arthritic rats, and dashed lines reflect healthy rats.
Discussion
Protein binding has been known to play a substantial role in the PK and PD of NSAIDs (Lin et al., 1987). NPX is an acidic NSAID that is extensively bound to albumin (>99%) within the normal therapeutic range in plasma (20–200 µg/ml) (Mortensen et al., 1979). Different plasma albumin concentrations and binding kinetics of NPX with disease and sex along with altered intrinsic clearances affected the disposition of NPX.
We found lower albumin concentrations along with nonlinear NPX PK in arthritic rats producing a disproportionate increase of AUC with dose. This phenomenon occurs in humans, where the AUC of NPX tended to plateau at higher doses (500–900 mg, which is about 45–80 mg/kg in rats) (Runkel et al., 1974). The absorption of NPX is rapid and complete (Runkel et al., 1974; Segre, 1975), and NPX absorption in RA patients is similar to that in healthy subjects (van den Ouweland et al., 1987; Vree et al., 1993). Therefore, the explanation for the plateau effect in AUC was a greater clearance at high doses due to elevated free drug concentrations resulting from saturation of albumin binding (Mortensen et al., 1979; Calvo and Dominguezgil, 1983). Under inflammatory conditions, albumin is also subject to increased catabolism (Wilkinson et al., 1965). Altered albumin binding in RA results in more free drug available for elimination, leading to lower AUC and higher CL in both RA patients and CIA rats given the same dose levels. Thus, different PK behaviors were expected between healthy and arthritic rats, in accordance with findings in humans (van den Ouweland et al., 1987). Although t1/2 did not change with dose or disease, this parameter was determined by concentrations falling within the linear protein-binding range.
As seen from the ELISA results, plasma albumin concentrations differed significantly with disease and sex, with lower values in CIA rats and in male rats. A negative correlation was found between the NCA values of CL/F and plasma albumin concentrations in all groups (r = 0.94), and the values of V/F in both sex groups were also inversely related to plasma albumin concentrations. These results confirmed that chronic inflammation reduced plasma albumin and partly influenced the PK of NPX. However, different albumin concentrations between female and male rats did not produce differences in PK, suggesting that other factors (e.g., drug metabolism) are involved.
This study was enacted to further explore and comprehensively integrate and model the role of different plasma albumin concentrations in the binding of NPX, to examine whether nonlinear protein binding accounts for the nonlinearities in NPX PK, and to assess disease and sex effects. The bound fractions of NPX in our study are slightly higher than other data in rats (Huntjens et al., 2006). In our study, protein binding of NPX was constant at low drug concentrations and became saturated at higher concentrations. Such saturation of binding was observed at lower NPX concentrations in CIA rats, which can be explained by their lower albumin concentrations. In CIA rats, even with very small decreases in percentage bound, the unbound fractions of NPX can vary up to about 6-fold over the therapeutic concentration range, which is comparable to the situation in humans (Borgå and Borgå, 1997). Our companion study assesses whether the pharmacological effects of NPX can be directly related to the unbound drug concentrations in CIA rats (Li et al., 2017).
Initial modeling tests showed that the estimated number of binding sites (n1 and n2) was close to 1 and 4 in all groups except CIA males, where the estimated n2 value was 6.6. Nonetheless, uniform values of n (n1 = 1 and n2 = 4) were chosen for all groups in the final model, and only the association constants (Ka1 and Ka2) were estimated. The binding affinity of NPX to one class of binding site on albumin was much higher than the other. Attempted modeling of all protein-binding data using single values for Ka1 and Ka2 produced less satisfactory overall fittings. The lower binding capacity observed in CIA rats and in male rats was primarily due to their lower albumin concentrations.
From the perspective of mechanism, albumin- and drug concentration–dependent protein binding was incorporated into a classic type of two-compartment model to account for the nonlinearity of NPX PK. This modeling approach is based on the literature (Fleishaker and McNamara, 1985) in that free drug was used to govern the distribution process of total drug. However, their model only applies when protein binding and clearance are linear. Our model is more comprehensive in accounting for nonlinear binding in plasma and tissues, operating both distribution and elimination processes using unbound drug, and featuring a global analysis of all experimental data jointly. These advantages offset our ability to capture all data perfectly, especially the last time point, which differs from the fitting trend for unknown reasons.
Protein concentrations in tissue interstitial space are known to increase due to the increase of microvascular permeability with inflammation (Aukland and Johnsen, 1974; Simkin, 1979; Bell et al., 1983); therefore, different E/P values were assigned to CIA and healthy rats. The average E/P based on the literature is about 0.5 for healthy rats (Rodgers et al., 2005; Rodgers and Rowland, 2006). Assuming that increased albumin concentrations in CIA ISF are mainly attributed to the transfer of albumin from plasma to ISF, the E/P value used for CIA rats was about 0.9 based on the reduced fraction of plasma albumin concentrations. This is in accordance with findings in humans that E/P is about 0.32 in healthy subjects and increases to about 0.73 in RA patients (Fleishaker and McNamara, 1985; Day et al., 1995). Since no significant alteration of the absorption of NPX occurs with RA (van den Ouweland et al., 1987; Vree et al., 1993), the absorption rate constant (ka) was shared for all groups. The central distribution volume of total drug (Vp) was difficult to estimate, perhaps owing to the i.p. doses, and thus it was fixed to rat plasma volume to improve model stability. In the final model, different values of unbound plasma clearances (CL), unbound distribution clearances (CLd), and peripheral distribution volumes of total drug (Vt) were assigned for CIA and healthy rats. The presence of proinflammatory mediators during inflammation is commonly associated with reduced cytochrome P450 expression and activity, leading to impaired drug metabolism (Slaviero et al., 2003; Renton, 2005). Therefore, the lower value of CLArthritic was expected, as NPX undergoes significant hepatic metabolism that is primarily dependent on cytochromes CYP2C9 and CYP1A2 (Miners et al., 1996). The effects of chronic inflammation on the tissue disposition of NPX still remain unclear. However, it might be anticipated that the diffusion of unbound NPX into and out of tissues should decrease due to decreased perfusion with inflammation (Wallis et al., 1985). The lower unbound distribution clearance observed in CIA rats is consistent with this. The larger peripheral distribution volume of total NPX in CIA rats corresponded well with the NCA results. As the ISF protein concentrations increase with inflammation, the bound fraction of NPX will increase in ISF and thereby enhance the penetration of total drug into tissue, as previously reported (Fleishaker and McNamara, 1985). The estimated Vt values of all groups were smaller than ISF volume, which might be due to collagen occupying part of the ISF space. The PK parameters based on free drug were independent of dose, confirming that saturation of protein binding was the major determinant of the dose-dependent PK in CIA rats. Despite the consistency of findings regarding the influences of RA on the disposition of total and unbound NPX, our results showed no significant sex effects. It was reported that females exhibited higher free NPX concentrations than male patients with osteoarthritis (Hundal et al., 1991).
The CIA rat model exhibits many histopathological features similar to human RA, such as synovial proliferation, pannus formation, and cartilage destruction (Stuart et al., 1982). Therefore, the results of this study appear to mimic and explain the nonlinear PK of NPX in humans, as well as the effects of chronic inflammation and sex on the protein binding and disposition of NPX in RA patients.
In conclusion, plasma albumin concentrations in rats differed significantly with sex and arthritis, which leads to markedly different PK behaviors of NPX. Reduced albumin in plasma and altered protein binding of NPX was shown to be the primary cause for the dose-dependent PK of NPX in normal and arthritic rats, with the latter also exhibiting impaired clearance of free drug. The PK profiles of NPX in all groups were well described by the global PK model incorporating these diverse factors. This is the most complete exploration of the nonlinear PK of NPX with comparisons based on sex and presence of disease. This study serves as the prelude for our subsequent pharmacodynamics study of NPX in CIA rats (Li et al., 2017).
Acknowledgments
The authors thank Donna Ruszaj for technical assistance with LC-MS/MS assay development.
Abbreviations
- AUC
area under the plasma concentration-time curve
- CIA
collagen-induced arthritis
- COX
cyclooxygenase
- CV%
coefficient of variation
- ELISA
enzyme-linked immunosorbent assay
- GI
gastrointestinal
- IS
internal standard
- ISF
interstitial fluid
- LC-MS/MS
liquid chromatography–tandem mass spectrometry
- NCA
noncompartmental analysis
- NPX
naproxen
- NSAID
nonsteroidal anti-inflammatory drug
- PBS
phosphate-buffered saline
- PD
pharmacodynamics
- PG
prostaglandin
- PK
pharmacokinetics
- RA
rheumatoid arthritis
Authorship Contributions
Participated in research design: Li, DuBois, Almon, Jusko.
Conducted experiments: Li, DuBois.
Performed data analysis: Li, Jusko.
Wrote or contributed to the writing of the manuscript: Li, DuBois, Almon, Jusko.
Footnotes
X.L. received financial support from the China Scholarship Council to pursue research at the State University of New York at Buffalo. This work was funded by the National Institute of General Medical Sciences [Grant R01-GM24211].
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.
References
- Aoki T, Narumiya S. (2012) Prostaglandins and chronic inflammation. Trends Pharmacol Sci 33:304–311. [DOI] [PubMed] [Google Scholar]
- Aukland K, Johnsen HM. (1974) Protein concentration and colloid osmotic pressure of rat skeletal muscle interstitial fluid. Acta Physiol Scand 91:354–364. [DOI] [PubMed] [Google Scholar]
- Aukland K, Nicolaysen G. (1981) Interstitial fluid volume: local regulatory mechanisms. Physiol Rev 61:556–643. [DOI] [PubMed] [Google Scholar]
- Bell DR, Mullins RJ, Powers MR. (1983) Extravascular distribution of albumin and IgG during high-permeability edema in skin. Am J Physiol 244:H599–H606. [DOI] [PubMed] [Google Scholar]
- Borgå O, Borgå B. (1997) Serum protein binding of nonsteroidal antiinflammatory drugs: a comparative study. J Pharmacokinet Biopharm 25:63–77. [DOI] [PubMed] [Google Scholar]
- Calvo MV, Dominguezgil A. (1983) Binding of Naproxen to Human-Albumin - Interaction with Palmitic Acid. Int J Pharm 16:215–223. [Google Scholar]
- Coxib and Traditional NSAID Trialists’ Collaboration, Bhala N, Emberson J, Merhi A, Abramson S, Arber N, Baron JA, Bombardier C, Cannon C, Farkouh ME, FitzGerald GA et al. (2013) Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: meta-analyses of individual participant data from randomised trials. Lancet 382:769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crofford LJ. (2013) Use of NSAIDs in treating patients with arthritis. Arthritis Res Ther 15 (Suppl 3):S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crofford LJ, Lipsky PE, Brooks P, Abramson SB, Simon LS, van de Putte LB. (2000) Basic biology and clinical application of specific cyclooxygenase-2 inhibitors. Arthritis Rheum 43:4–13. [DOI] [PubMed] [Google Scholar]
- Danska JS. (2014) Sex matters for mechanism. Sci Transl Med 6:258fs40. [DOI] [PubMed] [Google Scholar]
- D'Argenio D, Schumitzky A, Wang X. (2009) Adapt 5 User's Guide:Pharmacokinetic/Pharmacodynamic Systems Analysis Software, BMSR, University of Southern California. [Google Scholar]
- Davies B, Morris T. (1993) Physiological parameters in laboratory animals and humans. Pharm Res 10:1093–1095. [DOI] [PubMed] [Google Scholar]
- Davies NM, Anderson KE. (1997) Clinical pharmacokinetics of naproxen. Clin Pharmacokinet 32:268–293. [DOI] [PubMed] [Google Scholar]
- Day RO, Francis H, Vial J, Geisslinger G, Williams KM. (1995) Naproxen concentrations in plasma and synovial fluid and effects on prostanoid concentrations. J Rheumatol 22:2295–2303. [PubMed] [Google Scholar]
- Day RO, McLachlan AJ, Graham GG, Williams KM. (1999) Pharmacokinetics of nonsteroidal anti-inflammatory drugs in synovial fluid. Clin Pharmacokinet 36:191–210. [DOI] [PubMed] [Google Scholar]
- Earp JC, Dubois DC, Molano DS, Pyszczynski NA, Keller CE, Almon RR, Jusko WJ. (2008a) Modeling corticosteroid effects in a rat model of rheumatoid arthritis I: mechanistic disease progression model for the time course of collagen-induced arthritis in Lewis rats. J Pharmacol Exp Ther 326:532–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earp JC, Pyszczynski NA, Molano DS, Jusko WJ. (2008b) Pharmacokinetics of dexamethasone in a rat model of rheumatoid arthritis. Biopharm Drug Dispos 29:366–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsinghorst PW, Kinzig M, Rodamer M, Holzgrabe U, Sörgel F. (2011) An LC-MS/MS procedure for the quantification of naproxen in human plasma: development, validation, comparison with other methods, and application to a pharmacokinetic study. J Chromatogr B Analyt Technol Biomed Life Sci 879:1686–1696. [DOI] [PubMed] [Google Scholar]
- Fleishaker JC, McNamara PJ. (1985) Performance of a diffusional clearance model for beta-lactam antimicrobial agents as influenced by extravascular protein binding and interstitial fluid kinetics. Antimicrob Agents Chemother 28:369–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk CD. (2001) Prostaglandins and leukotrienes: advances in eicosanoid biology. Science 294:1871–1875. [DOI] [PubMed] [Google Scholar]
- Holmdahl R, Lorentzen JC, Lu S, Olofsson P, Wester L, Holmberg J, Pettersson U. (2001) Arthritis induced in rats with nonimmunogenic adjuvants as models for rheumatoid arthritis. Immunol Rev 184:184–202. [DOI] [PubMed] [Google Scholar]
- Hundal O, Rugstad HE, Husby G. (1991) Naproxen free plasma concentrations and unbound fractions in patients with osteoarthritis: relation to age, sex, efficacy, and adverse events. Ther Drug Monit 13:478–484. [DOI] [PubMed] [Google Scholar]
- Huntjens DR, Spalding DJ, Danhof M, Della Pasqua OE. (2006) Correlation between in vitro and in vivo concentration-effect relationships of naproxen in rats and healthy volunteers. Br J Pharmacol 148:396–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntjens DR, Spalding DJ, Danhof M, Della Pasqua OE. (2010) Impact of chronic inflammation on the pharmacokinetic-pharmacodynamic relationship of naproxen. Eur J Pain 14:227 e221–210. [DOI] [PubMed] [Google Scholar]
- Lanas A. (2009) Nonsteroidal antiinflammatory drugs and cyclooxygenase inhibition in the gastrointestinal tract: a trip from peptic ulcer to colon cancer. Am J Med Sci 338:96–106. [DOI] [PubMed] [Google Scholar]
- Lauroba J, Doménech J, Moreno J, Plá-Delfina JM. (1986) Relationships between biophasic disposition and pharmacokinetic behavior in nonsteroid antiinflammatory drugs. Arzneimittelforschung 36:710–714. [PubMed] [Google Scholar]
- Li X, DuBois DC, Almon RR, Jusko WJ (2017) Modeling sex differences in pharmacokinetics, pharmacodynamics, and disease progression effects of naproxen in rats with collagen-induced arthritis. Drug Metab Dispos. 45:484–491. [DOI] [PMC free article] [PubMed]
- Lin JH, Cocchetto DM, Duggan DE. (1987) Protein binding as a primary determinant of the clinical pharmacokinetic properties of non-steroidal anti-inflammatory drugs. Clin Pharmacokinet 12:402–432. [DOI] [PubMed] [Google Scholar]
- Lin JH, Hooke KF, Yeh KC, Duggan DE. (1985) Dose-dependent pharmacokinetics of diflunisal in rats: dual effects of protein binding and metabolism. J Pharmacol Exp Ther 235:402–406. [PubMed] [Google Scholar]
- Liu D, Lon HK, Dubois DC, Almon RR, Jusko WJ. (2011) Population pharmacokinetic-pharmacodynamic-disease progression model for effects of anakinra in Lewis rats with collagen-induced arthritis. J Pharmacokinet Pharmacodyn 38:769–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lon HK, Liu D, DuBois DC, Almon RR, Jusko WJ. (2013) Modeling pharmacokinetics/pharmacodynamics of abatacept and disease progression in collagen-induced arthritic rats: a population approach. J Pharmacokinet Pharmacodyn 40:701–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lussier A, Arsenault A, Varady J. (1978) Gastrointestinal microbleeding after aspirin and naproxen. Clin Pharmacol Ther 23:402–407. [DOI] [PubMed] [Google Scholar]
- McInnes IB, Schett G. (2011) The pathogenesis of rheumatoid arthritis. N Engl J Med 365:2205–2219. [DOI] [PubMed] [Google Scholar]
- Mendel CM. (1989) The free hormone hypothesis: a physiologically based mathematical model. Endocr Rev 10:232–274. [DOI] [PubMed] [Google Scholar]
- Miners JO, Coulter S, Tukey RH, Veronese ME, Birkett DJ. (1996) Cytochromes P450, 1A2, and 2C9 are responsible for the human hepatic O-demethylation of R- and S-naproxen. Biochem Pharmacol 51:1003–1008. [DOI] [PubMed] [Google Scholar]
- Mortensen A, Jensen EB, Petersen PB, Husted S, Andreasen F. (1979) The determination of naproxen by spectrofluorometry and its binding to serum proteins. Acta Pharmacol Toxicol (Copenh) 44:277–283. [DOI] [PubMed] [Google Scholar]
- National Research Council (NRC) (2011) Guide for the Care and Use of Laboratory Animals. 8th ed National Academies Press, Washington, DC. [Google Scholar]
- Renton KW. (2005) Regulation of drug metabolism and disposition during inflammation and infection. Expert Opin Drug Metab Toxicol 1:629–640. [DOI] [PubMed] [Google Scholar]
- Ricciotti E, FitzGerald GA. (2011) Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol 31:986–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers T, Leahy D, Rowland M. (2005) Physiologically based pharmacokinetic modeling 1: predicting the tissue distribution of moderate-to-strong bases. J Pharm Sci 94:1259–1276. [DOI] [PubMed] [Google Scholar]
- Rodgers T, Rowland M. (2006) Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J Pharm Sci 95:1238–1257. [DOI] [PubMed] [Google Scholar]
- Runkel R, Forchielli E, Sevelius H, Chaplin M, Segre E. (1974) Nonlinear plasma level response to high doses of naproxen. Clin Pharmacol Ther 15:261–266. [DOI] [PubMed] [Google Scholar]
- Segre EJ. (1975) Naproxen metabolism in man. J Clin Pharmacol 15:316–323. [DOI] [PubMed] [Google Scholar]
- Shah DK, Betts AM. (2012) Towards a platform PBPK model to characterize the plasma and tissue disposition of monoclonal antibodies in preclinical species and human. J Pharmacokinet Pharmacodyn 39:67–86. [DOI] [PubMed] [Google Scholar]
- Shi X, Shang W, Wang S, Xue N, Hao Y, Wang Y, Sun M, Du Y, Cao D, Zhang K, et al. (2015) Simultaneous quantification of naproxcinod and its active metabolite naproxen in rat plasma using LC-MS/MS: application to a pharmacokinetic study. J Chromatogr B Analyt Technol Biomed Life Sci 978-979:157–162. [DOI] [PubMed] [Google Scholar]
- Simkin PA. (1979) Synovial permeability in rheumatoid arthritis. Arthritis Rheum 22:689–696. [DOI] [PubMed] [Google Scholar]
- Slaviero KA, Clarke SJ, Rivory LP. (2003) Inflammatory response: an unrecognised source of variability in the pharmacokinetics and pharmacodynamics of cancer chemotherapy. Lancet Oncol 4:224–232. [DOI] [PubMed] [Google Scholar]
- Smith WL, DeWitt DL, Garavito RM. (2000) Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem 69:145–182. [DOI] [PubMed] [Google Scholar]
- Stoeckel K, McNamara PJ, Brandt R, Plozza-Nottebrock H, Ziegler WH. (1981) Effects of concentration-dependent plasma protein binding on ceftriaxone kinetics. Clin Pharmacol Ther 29:650–657. [DOI] [PubMed] [Google Scholar]
- Stuart JM, Cremer MA, Townes AS, Kang AH. (1982) Type II collagen-induced arthritis in rats. Passive transfer with serum and evidence that IgG anticollagen antibodies can cause arthritis. J Exp Med 155:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Ouweland FA, Franssen MJ, van de Putte LB, Tan Y, van Ginneken CA, Gribnau FW. (1987) Naproxen pharmacokinetics in patients with rheumatoid arthritis during active polyarticular inflammation. Br J Clin Pharmacol 23:189–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Vollenhoven RF. (2009) Sex differences in rheumatoid arthritis: more than meets the eye.... BMC Med 7:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vane JR. (1971) Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol 231:232–235. [DOI] [PubMed] [Google Scholar]
- Vree TB, van den Biggelaar-Martea M, Verwey-van Wissen CP, Vree JB, Guelen PJ. (1993) Pharmacokinetics of naproxen, its metabolite O-desmethylnaproxen, and their acyl glucuronides in humans. Biopharm Drug Dispos 14:491–502. [DOI] [PubMed] [Google Scholar]
- Wallis WJ, Simkin PA, Nelp WB. (1985) Low synovial clearance of iodide provides evidence of hypoperfusion in chronic rheumatoid synovitis. Arthritis Rheum 28:1096–1104. [DOI] [PubMed] [Google Scholar]
- Watson DJ, Rhodes T, Cai B, Guess HA. (2002) Lower risk of thromboembolic cardiovascular events with naproxen among patients with rheumatoid arthritis. Arch Intern Med 162:1105–1110. [DOI] [PubMed] [Google Scholar]
- Wilkinson P, Jeremy R, Brooks FP, Hollander JL. (1965) The mechanism of hypoalbuminemia in rheumatoid arthritis. Ann Intern Med 63:109–114. [DOI] [PubMed] [Google Scholar]
- Wong BK, Bruhin PJ, Lin JH. (1999) Dose-dependent plasma clearance of MK-826, a carbapenem antibiotic, arising from concentration-dependent plasma protein binding in rats and monkeys. J Pharm Sci 88:277–280. [DOI] [PubMed] [Google Scholar]