
Figure 1.
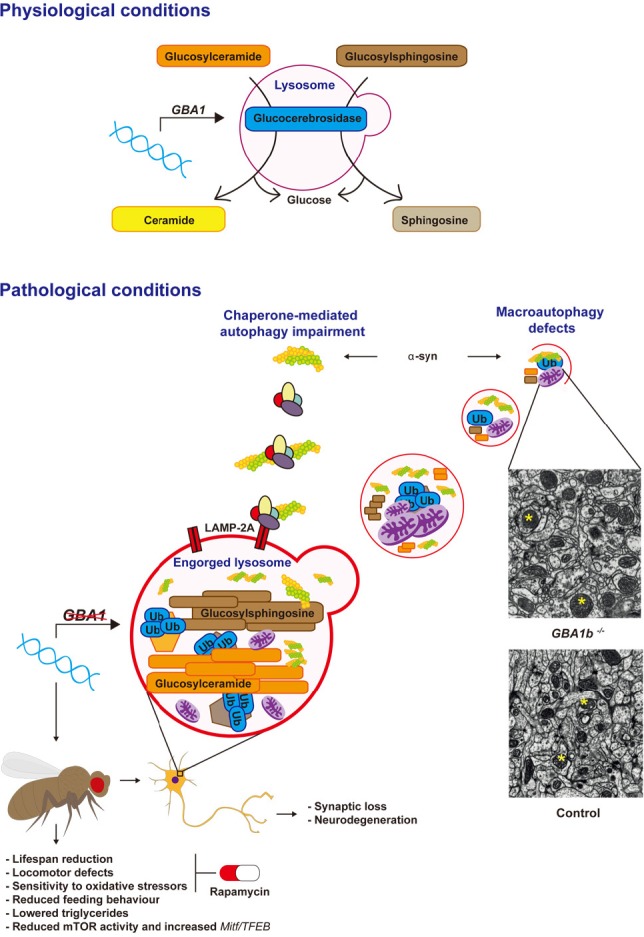
Glucocerebrosidase (GCase) deficiency results in autophagy-lysosomal system (ALS) dysfunction.
Under normal physiological conditions GCase hydrolyses glycolipid substrates (glucosylceramide and glycosylsphingosine) within the lysosome. Normal lysosomal function is required for the autophagic clearance of defective cellular organelles and mis-folded proteins. Mutations in the GBA1 gene result in GCase loss-of-function and accumulation of its substrates. This leads to lysosomal dysfunction and subsequent defects in both macroautophagy and chaperone-mediated autophagy (CMA), with the consequent accumulation of α-synuclein (α-syn). ALS dysfunction in a fly model of neuronal GCase deficiency was associated with the accumulation of abnormal giant mitochondria (representative giant mitochondria in GBA1 knockout (GBA1b−/−) and healthy mitochondria in wild type control fly brains are shown with yellow asterisks, shown under identical magnification), accumulation of autophagy substrates (p62 and polyubiquitinated proteins), in addition to neurodegeneration and synaptic loss. These neuropathological defects resulted in a number of neurotoxic phenotypes, including reduced lifespan, locomotor abnormalities and decreased resistance to oxidative stress. Likely as a compensatory response to the autophagy block, mammalian target of rapamycin (mTOR)-complex 1 (mTORC1) activity was decreased and Mitf/TFEB gene expression was up-regulated in the brains of GCase-deficient flies. The mTOR inhibitor rapamycin was able to functionally rescue the lifespan, locomotor and oxidative stress phenotypes of the GCase-deficient flies, highlighting the potential therapeutic benefits of rapamycin and other inhibitors of mTORC1 in Gaucher disease (GD) and Parkinson's disease (PD) (Kinghorn et al., 2016). LAMP-2A: Lysosomal-associated membrane protein 2.