
Figure 4. Mechansisms of action of the fast acting antidepressant ketamine in the medial prefrontal cortex.
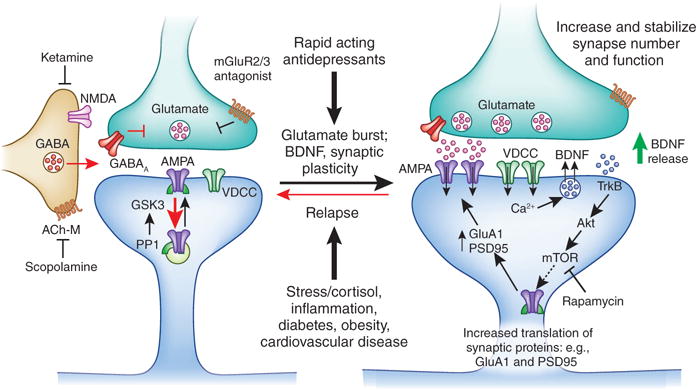
Ketamine causes a burst of glutamate that is thought to occur via disinhibition of GABA interneurons; the tonic firing of these GABA interneurons is driven by NMDA receptors, and the active, open channel state allows ketamine to enter and block channel activity. The resulting glutamate burst stimulates AMPA receptors causing depolarization and activation of voltage dependent Ca2+ channels, leading to release of BDNF and stimulation of TrkB-Akt that activates mTORC1 signaling leading to increased synthesis of proteins required for synapse maturation and formation (i.e., GluA1 and PSD95). Under conditions where BDNF release is blocked (BDNF Met knockin mice) or neutralized (BDNF neutralizing antibody) or when mTORC1 signaling is blocked (rapamycin infusion into the mPFC) the synaptic and behavioral actions of ketamine are blocked. Scopolamine also causes a glutamate burst via blockade of acetylcholine muscarinic M1 (ACh-M1) receptors on GABA interneurons. Antagonists of glutamate metabotropic 2/3 receptors (mGluR2/3) also produce rapid antidepressant actions via blockade of presynaptic autoreceptors that inhibit the release of glutamate. Relapse to a depressive state is associated with reduction of synapses on mPFC neurons, which could occur via stress and imbalance of endocrine (cortisol), estrogen, inflammatory cytokines, metabolic, and cardiovascular illnesses.