

Abstract
Motivation
Variant calling from next-generation sequencing (NGS) data is susceptible to false positive calls due to sequencing, mapping and other errors. To better distinguish true from false positive calls, we present a method that uses genotype array data from the sequenced samples, rather than public data such as HapMap or dbSNP, to train an accurate classifier using Random Forests. We demonstrate our method on a set of variant calls obtained from 642 African-ancestry genomes from the Consortium on Asthma among African-ancestry Populations in the Americas (CAAPA), sequenced to high depth (30X).
Results
We have applied our classifier to compare call sets generated with different calling methods, including both single-sample and multi-sample callers. At a False Positive Rate of 5%, our method determines true positive rates of 97.5%, 95% and 99% on variant calls obtained using Illuminas single-sample caller CASAVA, Real Time Genomics multisample variant caller, and the GATK UnifiedGenotyper, respectively. Since NGS sequencing data may be accompanied by genotype data for the same samples, either collected concurrent to sequencing or from a previous study, our method can be trained on each dataset to provide a more accurate computational validation of site calls compared to generic methods. Moreover, our method allows for adjustment based on allele frequency (e.g. a different set of criteria to determine quality for rare versus common variants) and thereby provides insight into sequencing characteristics that indicate call quality for variants of different frequencies.
Availability and Implementation
Code is available on Github at: https://github.com/suyashss/variant_validation
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Whole-genome sequencing has become increasingly common as a method to query genetic differences between individuals, both for population genetic studies and studies of genetic factors contributing to clinical phenotypes (Koboldt et al., 2013). Methods for translating sequenced fragments into individual genotype calls have gone through a period of active development, and many different options are available (Cleary et al., 2014; DePristo et al., 2011; Liu et al., 2013). Each of them must account for the occurrence of sequencing errors (Cheng et al., 2014) in determining whether a genetic variant is present in a particular sample, a condition that becomes especially challenging with lower sequencing depth, or in the case of a variant that has either never been seen in a given population or that is very rare (Reuter et al., 2015).
One key decision researchers make when choosing a variant caller is whether to use a single-sample or multi-sample calling algorithm. The argument in favor of multi-sample calling includes borrowing information across individuals at sites of shared genetic variation. While several pipeline comparison analyses have been conducted (Cornish and Guda, 2015; Hwang et al., 2015; Yu and Sun, 2013), limited work has been done to characterize the differences between the sets of variants generated by different calling algorithms, making it challenging for researchers to make a principled choice when designing an analysis pipeline. The increased computational burden of performing multi-sample calling across a large cohort means that benefits of such a calling method should be understood before carrying out this phase of a study. In addition, while genotype callers usually include some measure of quality or call confidence as part of their output, room for improvement remains in terms of better characterizing true variant calls from false positives.
In this manuscript, we present a method for characterizing variant call sets produced with different calling algorithms in part to illustrate that certain characteristics of individual variants make them more or less likely to be called by different algorithms. We also present a method for assessing variant call quality that incorporates external genotyping array data for each subject, in order to build and train a classifier which distinguishes true variant calls from false positives. Such external array data is sometimes or even frequently available to accompany whole-genome sequencing data, either collected concurrent to sequencing for sample quality control purposes, or existing from a previous study of the same samples. We leverage this additional resource to improve sequencing-based variant call quality.
While other work has used Random Forests to classify variants according to suites of characteristics, the focus has mainly been on de novo (Francioli et al., 2015; T. G. of the Netherlands Consortium, 2014) or somatic (Löwer et al., 2012) mutations. In contrast, we focus here on using validation data generated on a genotyping array, rather than through costly resequencing-based validation.
In this presentation, because of our use of genotyping array data as our gold standard, we limit ourselves to Single Nucleotide Variants (SNVs) and not short insertions or deletions, although those can be detected with sequencing data. We demonstrate our method on a set of variant calls obtained from 642 high-coverage African-ancestry genomes from the Consortium on Asthma among African-ancestry Populations in the Americas (CAAPA) (Mathias et al., 2016), sequenced to high depth (30X).
2 Results
To illustrate our method, we first identified SNVs on chromosome 22 from 642 high-coverage samples from CAAPA using three variant calling algorithms:
CASAVA from Illumina
Population caller from Real Time Genomics (RTG)
UnifiedGenotyper from the Genome Analysis Toolkit (GATK)
For Illumina, variants were called on each sample individually and then merged into a combined-sample call set whereas for RTG and GATK, variants were identified jointly across all 642 samples. In all cases, the same set of 642 aligned read files (BAM files) was used. The filters used to obtain the final call sets from the raw calls are described in more detail in the Methods section.
Experimental validation of putative variants is expensive and difficult to perform for thousands of variants. Therefore, we focused on variants identified in a single sample for which we have a technical replicate, referred to as Sample 1 and Sample 1R, allowing variants to be partially validated. We characterize our results based on whether a variant call appears in both replicates, with calls appearing in both assumed to be correct and those appearing in only one potentially false positives. While such technical replicates are not available for all our samples, we performed similar analysis on an additional nine subjects, referred to as Samples 2–10, selected to have characteristics spanning a range of values for sequencing quality (i.e. average depth and fraction of missing calls) and to have a variety of ethnic backgrounds (see Supplementary Table S1 for a summary of these characteristics across all 10 samples relative to the full dataset).
2.1 Differentiating between call sets from different algorithms
Figure 1 shows a Venn diagram of the overlap between the three call sets for a single individual. From the figure, we can see that about 65.3% of all variants (43 291 out of 66 335) are found by all three methods. Nearly 19.1% of all variants (10 385 out of 66 335) are found by only two out of three methods while 15.6% (12 658 out of 66 335) are found by only one method. To determine if any variant features affected the ability of methods to detect a given variant, we trained a Random Forest classifier to identify which subset of methods would identify a variant with given variant features. One purpose of this exercise is to characterize the way different calling algorithms incorporate information from different sources, so we developed classifiers using both a limited set of features which rely only on individual-level data, and a set of features that incorporates information across samples, including allele frequencies and quality metrics. A detailed description of the variant features used for the limited and the full classifier, which each include both features specific to the individual dataset and those related to the location of the variant in the genome, can be found in the Methods section and in the Supplementary Table S2, including sources of each measurement used.
Fig. 1.
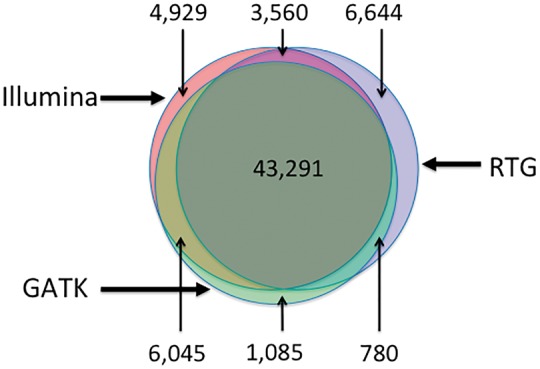
Overlap between the three call sets for variants on chromosome 22 for our individual of interest
Using strictly individual-level features without frequency information produces an error rate of 25.69%, which is lower than the error rate of a classifier that always predicts the majority class (error rate 34.7%). Figure 2 shows which variant features are important for classifying a given variant, including mapping quality, indel proximity and allele balance. Interestingly, not only the coverage in the chosen individual but also the total coverage at the site across all individuals is important. Expanding to a classifier that allows a full set of cross-sample summary information, including allele frequency, overall variant quality and quality by depth, our classifier achieves a 1.2% error rate, suggesting that the features examined do in fact allow for distinctions between the call sets. Supplementary Table S3 shows the result of this full classifier on the union of variants from the three calling methods for our single individual. While using the frequency of the alternate allele in the full set of individuals to determine whether a variant is present in one specific call set is circular, this frequency is important for the classifier, especially for the GATK and RTG callers, which combine information across multiple samples when making a call (Supplementary Fig. S1). Overall, the fact that variant features can be used to determine which caller will call a particular variant motivates the development of our call set-specific call quality assessment models presented below.
Fig. 2.

Feature importance for the Random Forest classifier distinguishing calls made by different calling algorithms, limited to individual-level features. Scale on the x-axis is unitless but indicates relative importance of the different features
2.2 Identifying true variant sites within a single call set
One feature of our dataset is that in addition to whole-genome sequencing data, we obtained genotype microarray data from the Illumina HumanOmni2.5 BeadChip (Omni). For each of the three call sets, we used the Omni genotype data to learn a Random Forest classifier that could predict whether or not a variant site detected in a call set was truly variable or not. Using the Omni data, we declare a site to be truly variable if it is variable in both the sequencing call set and based on the Illumina array data; if the site is only variable in the sequencing dataset but not based on the array data, we declare it to be a false positive. For this task, we used a slightly different set of features to that used earlier (see Methods and Supplementary Table S2). Figure 3 shows the importance of various features for the three Random Forest classifiers trained. As before, allele frequency was the most important determinant for the Illumina call set and the second most important for the GATK call set, while haplotype score, a measure of evidence for more than two segregating haplotypes in the sample, was the most important feature for the RTG call set. Features of just slightly less importance were allele balance (AB, highest relative importance for GATK), total coverage, haplotype score, GC content and coverage. Figure 4 shows the receiver-operating characteristic (ROC) curves for the three classifiers. We see that all three classifiers have high sensitivity and specificity (true positive rate > 0.95 with false positive rate =0.05), with area under the curve (AUC) greater than 0.99. This suggests that truly variant sites for each call set can be determined statistically using a machine-learning algorithm (similar to the Gaussian mixture model used by GATKs variant quality score recalibration (VQSR) scoring scheme, but on an individual basis).
Fig. 3.
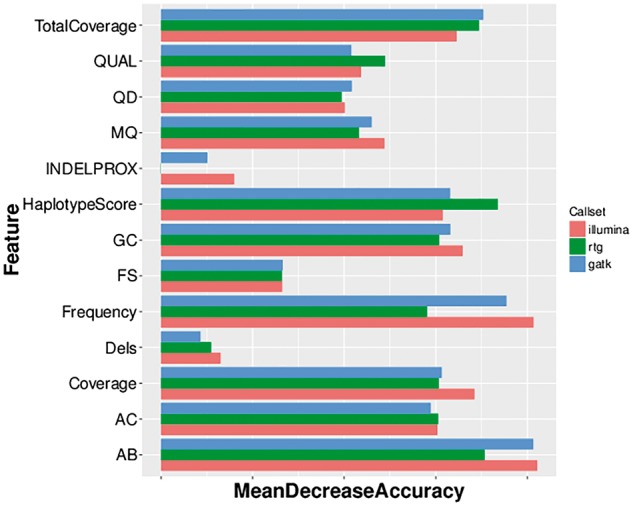
Feature importance for call set-specific classifiers based on Omni genotype data. Note that the frequency features refer to the estimates of the allele frequency from the call set being studied. Also note that the INDELPROX variable has a value of 0 for RTG
Fig. 4.
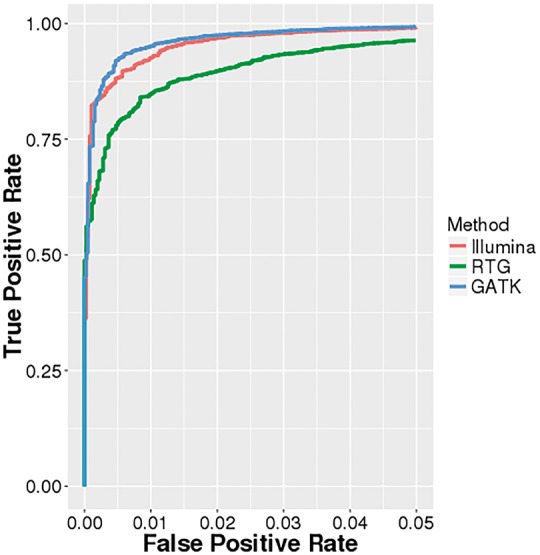
Zoomed-in detail of ROC curves for call set-specific classifiers based on Omni genotype data
Results for the complete set of 10 subjects considered are included in Supplementary Figures S2 (feature importance values), S3 and S4 (ROC plots). From these figures, we can see that relative importance of features in the classifier relative remain consistent across individuals. The ROC plots look very similar across subjects, and in all cases the performance is strong with a True Positive Rate of over 0.9 at a False Positive Rate of 0.05. Moreover, the relative ordering of the results for the different callers is consistent. Taken together, these results confirm that conclusions drawn from the analysis of Sample 1 hold across samples with a variety of sequencing characteristics.
We evaluated the validity of our classifier scores by stratifying sites by the call sets they were found in to compare score distributions for sites with varying degrees of concordance across call sets. Figure 5 shows the results of this analysis. Considering the results for the Illumina classifier (left panel in Fig. 5), we can see that sites found in all three call sets have higher classifier scores (n = 43 291, median score = 0.96) than sites found in two of three call sets (n = 9605, median score = 0.92), with for a one-sided t-test comparing these groups. Sites found only by the Illumina call set have the lowest classifier scores (n = 4929, median score = 0.78), with for both one-sided t-tests comparing them to the other two categories. Assuming discovery by many variant callers to be a signal that a site is truly variable, this suggests that our learned classifiers can predict truly variable sites accurately.
Fig. 5.

Call set-specific classifier scores for all sites, stratified by the call sets in which the site was found. Shown are calls from Illumina (left), RTG (center) and GATK (right). Colors represent the number of call sets a particular variant was observed in, with pink for one call set, green for two call sets and blue for all three call sets
For orthogonal validation, we used a technical replicate and generated variants using the three variant callers. For each site in the original call set, we were therefore able to ascertain whether it was found in both the original and replicate call sets or just one of the two. Figure 6 shows the results of this analysis. We can see that sites appearing in both replicates have a higher classifier score than those appearing only in one replicate (for the one-sided t-test, for illumina, for RTG and GATK).
Fig. 6.

Call set-specific classifier scores for all sites stratified by whether the site was found in both Sample 1 and Sample 1R or only one sample. Shown are calls from Illumina (left), RTG (center) and GATK (right). Colors represent the number of replicates a particular variant was observed in, with pink for one replicate only and green for both replicates
For each call set, we used the fitted call set-specific classifer to predict whether all discovered sites within the call set were variable. Table 1 shows the results of this prediction. We see that of the three methods, the Illumina call set has the largest number of sites predicted to be variable. The GATK call set has the highest proportion of sites predicted to be variable, which is expected since the call set was filtered using VQSR before our analysis. Overall, all three call sets have a high proportion of variants predicted to be true calls by our classifier.
Table 1.
Predicted number of true variant sites from three different call sets using fitted call set-specific classifiers
| Variant caller | Total sites | Predicted variant sites | Rate |
|---|---|---|---|
| Illumina | 57 825 | 54 023 | 93% |
| RTG | 54 275 | 45 297 | 83% |
| GATK | 51 201 | 49 485 | 97% |
2.3 Adaptation of classifier to rare variants
Since rare variants are considered to be particularly difficult to call accurately, we examined the performance of our classfiers on rare variants, focusing only on the Illumina call set. First, we separated our validation dataset into two groups according to allele frequency. For Sample 1, there were 50 547 variable sites on the genotyping array with minor allele frequency (MAF) of at least 5% in the Illumina call set and 7278 sites with MAF < 5%.
We then compared how well we detected these variants in three settings: (i) using the classifier built on all data, and the cutoff corresponding to a false positive rate of 5% on all variants (no stratification); (ii) using the classifier built on all data, but allowing for separate cutoffs for common and rare variants, to ensure a 5% false positive rate separately for each group (variable thresholds); and (iii) refitting the Random Forests classifier separately for the two MAF groups, and then picking the cutoff that corresponds to a 5% false positive rate separately for each group (separate training).
Finally, we examined how many of the 50 547 common and 7278 rare sites are declared to be variable in each setting (Table 2). Interestingly, the detection of rare variants is highest when no special treatment is involved. This may in part be due to the fact that there are relatively few rare sites available for training, so that model accuracy suffers more from the lack of training data than from the inhomogeneity between the training data and the data to which the classifier is applied.
Table 2.
Number of predicted variant sites for the Illumina classifier, with percentages indicating fraction of total common SNPs (out of 50 547) or rare SNPs (out of 7278). The results are stratified by allele frequency, with common SNPs having frequency 5% or larger
| Method | Common SNPs | Rare SNPs |
|---|---|---|
| (n = 50 547) | (n = 7278) | |
| No stratification | 47 828 (95%) | 6354 (87%) |
| Variable thresholds | 48 870 (97%) | 6043 (83%) |
| Separate training | 47 547 (94%) | 5280 (73%) |
3 Methods
For our analyses, we used alignment files (BAMs) that we received from Illumina after sequencing for the 642 samples. Due to the number of samples included in the dataset and limits of computational resources, all analyses were restricted to variants on chromosome 22. Alignment was performed as part of Illumina’s CASAVA pipeline using their in-house ELANDv2e software, which performs multiseed and gapped alignments, as well as orphan alignments and repeat resolution. Variant calls were obtained from the alignment files using three different pipelines as follows:
Illumina: Raw calls were filtered first by removing any variants near centromeres or other high copy regions by filtering based on depth of coverage, and second by removing variants falling into regions of segmental duplication. In addition, individual genotype calls were set to missing which had call quality scores below 20 or depth of coverage below 7. The merged multi-sample VCF was then filtered for SNPs that failed Hardy–Weinberg equilibrium (HWE, ). Calling was performed at Illumina, where the sequencing was performed.
RTG: We used the population caller from RTG (version 3.2) to jointly obtain variant calls for the 642 samples. The multisample VCF was filtered using the Adapative Variant Rescoring scheme (minAVR = 0.025). The resulting VCF was then filtered for SNPs that failed HWE with .
GATK: We used the GATK UnifiedGenotyper (version 3.5) to jointly obtain variant calls for the 642 samples. The multisample VCF was filtered using VQSR with annotations MD, MQ, MQRankSum, ReadPosRankSum, FS, DP and InbreedingCoeff (sensitivity = 94%, to obtain a Ti/Tv ratio close to 2 for passing variants). The resulting VCF was then filtered for SNPs that failed HWE with .
Illumina also provided us with genotype microarray data from the Illumina HumanOmni2.5 BeadChip (Omni). There are 33 234 SNPs on the Omni array for chromosome 22. Of those, 4123 SNPs (12%) have , 10 221 SNPs (31%) have and 23 012 (69%) have .
For each of our questions of interest, we constructed a random forest classifier using the ‘randomForest’ package (Liaw and Wiener, 2002) written in R (R Core Team, 2014). Random forest classifiers are collections of decision trees that allow non-linear interactions between features and are robust to over-fitting (Breiman, 2001). We used 1000 trees for our analysis due to the large number of variants.
3.1 Distinguishing between call sets
In this analysis the objective is to predict what call sets a variant site will appear in. We used a number of variant features for the call set-specific classifiers. Many of them were chosen to be among the features used in GATK’s VQSR scoring scheme, but our approach to classification differs from that used in the GATK through our use of Random Forests. Supplementary Table S2 describes the features used and Supplementary Figure S5 illustrates how these features are correlated with one another.
3.2 Call set specific classifiers
3.2.1 Features selected
We used a number of variant features for the call set-specific classifiers. Supplementary Table S2 describes the features used. Based on results from the previous analysis, read allele imbalance measured using a RankSum test (such as BaseQualityRankSum) was not very informative for classification and were replaced by a Fisher test score of allele balance for this analysis.
3.2.2 Ground truth data
For our classification task, we used the Omni genotype calls as ground truth. Therefore, we were able to use the sites overlapping between the Omni SNPs and the variant calling output from the sequencing data as our labeled set. Sites that were heterozygous or homozygous alternate in the Omni genotype calls for the chosen individual were lableled ‘1’ while sites which were homozygous reference were lableled ‘0’.
3.2.3 Prevention of overfitting
Note that Random Forests incorporates protection against overfitting by only using trees not containing the site to be predicted when performing prediction after model building has been performed. In this way, it is similar to cross-validation.
3.2.4 Unbalanced class problem
On intersecting the sequencing variant calls with Omni genotype data to obtain labeled sites, we observed that the ratio of the number of sites lableled ‘1’ to those labeled ‘0’ was nearly 100:1 (11 506:184 for illumina, 10 376:170 for RTG and 11 465:184 for GATK). In classification tasks, this can be problematic since it can bias the classifier towards increasing accuracy by always predicting the majority class.
To avoid this problem we used the SMOTE method (synthetic minority over-sampling technique) by Chawla et al. (2002) implemented in the R software (Torgo, 2010). This method oversamples the minority class by creating synthetic examples of the minority class from existing examples. It also undersamples the majority class for improved performance.
4 Discussion
Here, we present a method of using random forests to both characterize different variant call sets and to assess call quality taking into account a wide variety of data features in a flexible way. We show that for sequencing data like ours, e.g. with relatively deep (30x) coverage, both single-sample and multi-sample calling methods provide calls with very good accuracy. We illustrate our method on a single sample for which we had a technical replicate, as well as 9 additional samples selected to be representative of our dataset as a whole, to provide further insight into the behavior of our classifier.
To further explore differences between the call sets, we also looked at results stratified by allele frequency bin, with three different proposed modeling choices. Here there were some differences in performance depending on which thresholds or models were applied, with different thresholds chosen for classifying rare and common sites resulting in nearly 800 additional sites being predicted as variant (Table 2). In light of these results, we conclude that the optimal calling method to apply may depend on what the intended use of the variant calls is, with different applications (e.g. population level vs variant specific analyses) best served by different calling methods. For example, recent work (Han et al., 2014) discusses potential biases in site-frequency spectrum estimation that can result from low-coverage sequencing data where rare variants are more likely to be missed. For an application like this, multi-sample calling would be most appropriate to leverage information from many samples. However, in general applications or when sequencing coverage is high as it is in our dataset, we have not observed a large impact on downstream results comparing the different call sets. As part of the analysis in the CAAPA flagship publication, we compared some downstream results generated with both the Illumina single-sample call set and the RTG multi-sample call set and found no difference in the overall patterns seen in the count of deleterious alleles by individual or by group (see Supplementary Information of Mathias et al., 2016). We do note that our false positive rate of 5% may be considered high for applications to disease genetics, and suggest that the standard practice of validating any interesting findings either through replication or through further genotyping should still be used. Finally, modifications to the method presented here to target a particular allele frequency class, such as modifying the quality threshold for different frequency bins, or potentially retraining the classifier on different subsets of the data split by frequency bin, are also possible if variants in a particular frequency range are of special interest in a particular application.
Collecting genotype array data to accompany whole-genome sequencing data, or performing sequencing on samples that have already been used in a GWAS and therefore have genotype array data available, is currently common practice for the purpose of sample verification. Our work indicates that it is also valuable to have array data as an orthogonal validation dataset for QC purposes and to assess overall call quality of the variant calls from the sequencing data. If genome-wide array data is not available for all sequenced samples, investing in such data for at least a subset of samples would allow construction of a classifier to apply across all samples to improve variant call quality.
In contrast to the frequently used VQSLOD values provided by the GATK’s VQSR algorithm, which does not require external sample-specific validation data, but may rely on publicly available data, our method provides a call-specific, rather than site-specific quality measure. This makes a direct comparison between our method and using the VQSLOD values as a filter difficult.
As an alternative to the SMOTE method, weighting the observations in the different classes to give a more balanced training set is a possibility. We also implemented this and found that performance was not as good as what we present here (Supplementary Fig. S6).
An application of our classifier to low-coverage data is a direction for possible future work. While our dataset has relatively deep average coverage at 30x, we believe our method would be potentially even more valuable on lower-coverage data. With less information available from the sequencing reads themselves, the context that the variants fall into may provide important additional information that our classification method can discover from the gold-standard array data, allowing greater discrimination of borderline variant calls.
Another area for future work is to extend our method to short insertions and deletions (indels), although this would not be straightforward due to the lack of a training dataset like the genotype array data that is available for SNVs in our study. If such a training dataset could be obtained, a similar method to ours could be used to better assess indel call quality.
A potential extension to the work presented here would be to develop a method of producing a consensus call set based on the results of the classifier output for each set of variant calls. We could produce a combined result from the different calling methods using a weighted combination (e.g. weighted by the quality score assigned by our classifier) or a latent variable model. This would be similar to work presented in DePristo et al. (2011) but with a more flexible approach to assigning weights to the various possible calls.
5 Conclusion
We demonstrate that differences between call sets generated by different variant callers can be explored and interpreted using machine learning methods like Random Forests. In addition, we show that Random Forests can be used to identify which variant calls from a specific call set are likely to be true.
Supplementary Material
Funding
Funding for this study was provided by National Institutes of Health (NIH) R01HL104608/HL/NHLBI.
Conflict of Interest: C.D.B is on the scientific advisory boards (SABs) of Ancestry.com, Personalis, Liberty Biosecurity and Etalon DX. He is also a founder and chair of the SAB of IdentifyGenomics. FMDLV was an employee of Annai Systems Inc., and later of TOMA Biosciences, Inc. during the performance of this research. None of these entities played a role in the design, interpretation, or presentation of these results. No other authors have any conflicts of interest to declare.
References
- Breiman L. (2001) Random forests. Mach. Learn., 45, 5–32. [Google Scholar]
- Chawla N.V. et al. (2002) Smote: synthetic minority over-sampling technique. J. Artif. Intell. Res., 16, 321–357. [Google Scholar]
- Cheng A.Y. et al. (2014) Assessing single nucleotide variant detection and genotype calling on whole-genome sequenced individuals. Bioinformatics, 30, 1707–1713. [DOI] [PubMed] [Google Scholar]
- Cleary J.G. et al. (2014) Joint variant and de novo mutation identification on pedigrees from high-throughput sequencing data. J. Comput. Biol., 21, 405–419. [DOI] [PubMed] [Google Scholar]
- Cornish A., Guda C. (2015) A comparison of variant calling pipelines using genome in a bottle as a reference. Biomed. Res. Int., 2015, 11 pages. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo M.A. et al. (2011) A framework for variation discovery and genotyping using next-generation dna sequencing data. Nat. Genet., 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francioli L.C. et al. (2015) Genome-wide patterns and properties of de novo mutations in humans. Nat. Genet., 47, 822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han E. et al. (2014) Characterizing bias in population genetic inferences from low-coverage sequencing data. Mol. Biol. Evol., 31, 723–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang S. et al. (2015) Systematic comparison of variant calling pipelines using gold standard personal exome variants. Sci. Rep., 5, 17875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt D.C. et al. (2013) The next-generation sequencing revolution and its impact on genomics. Cell, 155, 27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liaw A., Wiener M. (2002) Classification and regression by randomforest. R. News, 2, 18–22. [Google Scholar]
- Liu X. et al. (2013) Variant callers for next-generation sequencing data: a comparison study. PLoS One, 8, e75619.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löwer M. et al. (2012) Confidence-based somatic mutation evaluation and prioritization. PLoS Comput. Biol., 8, e1002714.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathias R.A. et al. (2016) A continuum of admixture in the western hemisphere revealed by the african diaspora genome. Nat. Commun., 7, 12522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. (2014) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- Reuter J.A. et al. (2015) High-throughput sequencing technologies. Mol. Cell, 58, 586–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- T. G. of the Netherlands Consortium. (2014) Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat. Genet., 46, 818–825. [DOI] [PubMed] [Google Scholar]
- Torgo L. (2010) Data Mining with R, Learning with Case Studies. Chapman and Hall/CRC., Boca Raton, Florida, USA. [Google Scholar]
- Yu X., Sun S. (2013) Comparing a few snp calling algorithms using low-coverage sequencing data. BMC Bioinf., 14, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.