

Abstract
Alteration in gene expression along the pain signaling pathway is a key mechanism contributing to the genesis of neuropathic pain. Accumulating studies have shown that epigenetic regulation plays a crucial role in nociceptive process in the spinal dorsal horn. In this present study, we investigated the role of enhancer of zeste homolog-2 (EZH2), a subunit of the polycomb repressive complex 2, in the spinal dorsal horn in the genesis of neuropathic pain in rats induced by partial sciatic nerve ligation. EZH2 is a histone methyltransferase, which catalyzes the methylation of histone H3 on K27 (H3K27), resulting in gene silencing. We found that levels of EZH2 and tri-methylated H3K27 (H3K27TM) in the spinal dorsal horn were increased in rats with neuropathic pain on day 3 and day 10 post nerve injuries. EZH2 was predominantly expressed in neurons in the spinal dorsal horn under normal conditions. The number of neurons with EZH2 expression was increased after nerve injury. More strikingly, nerve injury drastically increased the number of microglia with EZH2 expression by more than 7 fold. Intrathecal injection of the EZH2 inhibitor attenuated the development and maintenance of mechanical and thermal hyperalgesia in rats with nerve injury. Such analgesic effects were concurrently associated with the reduced levels of EZH2, H3K27TM, Iba1, GFAP, TNF-α, IL-1β, and MCP-1 in the spinal dorsal horn in rats with nerve injury. Our results highly suggest that targeting the EZH2 signaling pathway could be an effective approach for the management of neuropathic pain.
Keywords: Nociception, cytokine, glial, PRC2, DZnep, GSK126
Graphical abstract
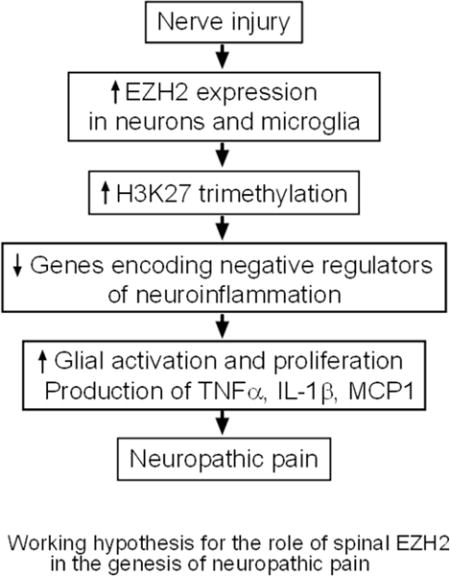
Introduction
Neuropathic pain is a debilitating condition affecting millions of people and poorly managed by the current therapeutics such as opioids or nonsteroidal anti-inflammatory drugs. Developing more potent and safe analgesics is an urgent need to combat such devastating condition. It is known that alterations of protein expression via transcriptional and post transcriptional regulation in spinal neurons and glial cells are critical mechanisms contributing to the genesis of neuropathic pain (Befort, Costigan, & Woolf, 2001; Suzuki & Dickenson, 2000; Taylor, 2001; C. J. Woolf & Salter, 2000). Identifying signaling molecules regulating gene expression would provide potential targets for the development of analgesics.
One key component leading to the development and maintenance of neuropathic pain is excessive activation of spinal dorsal horn neurons following nerve injury (Suzuki & Dickenson, 2000; Taylor, 2001). Activation and proliferation of microglia and astrocytes, and subsequent over-production of inflammatory mediators such as TNF-α, IL-1β and MCP-1 are causative factors causing excessive activation of spinal dorsal horn neurons (Grace, Hutchinson, Maier, & Watkins, 2014; Ren & Dubner, 2016). In this context, TNF-α, IL-1β (Kawasaki, Zhang, Cheng, & Ji, 2008; Yan & Weng, 2013) and MCP-1 (Gao et al., 2009) enhance excitatory glutamatergic synaptic activities in the spinal dorsal horn while inhibitory GABAergic synaptic activities in the same region are suppressed by TNF-α, IL-1β (Kawasaki et al., 2008; Yan, Jiang, & Weng, 2015). Suppression of microglial activation with minocycline (Nie, Zhang, & Weng, 2010), or astrocytic activation with propentofylline (Tawfik et al., 2008) can effectively attenuate neuropathic pain. Recent studies have demonstrated that regulation of gene expression profile via epigenetic mechanisms play an important role in the genesis of chronic pain conditions (Descalzi et al., 2015; Ligon, Moloney, & Greenwood-Van Meerveld, 2016). Currently, it remains unclear whether and how epigenetic mechanisms are engaged in the regulation of glial activation and production of TNF-α, IL-β and MCP-1 in the spinal dorsal horn in animals with neuropathic pain.
Epigenetics mechanisms including DNA methylation, histone modification, and non-coding RNAs are implicated in activation and suppression of various gene expressions in the development and maintenance of chronic pain in animals (Bai, Ren, & Dubner, 2015; Liang, Lutz, Bekker, & Tao, 2015; Ligon et al., 2016). Among the many histone modification mechanisms, N-terminal tail of histone is subjected to various post translation modification such as acetylation, methylation, phosphorylation, ubiquitination. In general, histone acetylation at the lysine residue promotes gene transcription. Whereas, histone methylation can either activate or represses gene transcription, depending upon which histone lysine residue is methylated (Margueron et al., 2009; Martinez & Simeonov, 2015). It is generally believed that methylation of histone H3 on K27 results in gene silencing (Margueron et al., 2009). Polycomb complex 2 (PRC2) is one class of gene repressive epigenetic transcriptional regulators. PRC2 induces gene silencing by transferring the mono, di or tri-methyl groups onto lysine 27 of histone3 (H3K27) via its subunit the enhancer of zeste homolog-2 (EZH2), a histone methyltransferase (Alam, Gu, & Lee, 2015; Burgold et al., 2008). Previous studies have shown that EZH2 plays an important role in controlling cell differentiation and proliferation. Increased levels of tri-methylated H3K27 (H3K27TM) and EZH2 promote the differentiation and proliferation of neurons, astrocytes and oligodendrocytes cells (Pereira et al., 2010). During neocortical development PRC2 complex checks the multipotent neuronal progenitor cells and controls their early neurogenic- and astrogenic cellular fate (Hirabayashi et al., 2009). EZH2 protein prevents the premature onset of neurogenesis in a developing brain (Sparmann et al., 2013). More recently, it was shown that EZH2 is engaged in the production of inflammatory cytokines and proliferation of cells in various diseases such as rheumatoid arthritis (Miao et al., 2013) and breast cancer (Hartman et al., 2013; Kim & Roberts, 2016). Given that glial cell proliferation and over-production of inflammatory mediators are salient mechanisms implicated in the generation of chronic pain, the role of EZH2 in the spinal pain system was investigated in this study.
We demonstrated that increased global levels of EZH2 and H3K27TM in the L4–L5 spinal dorsal horn are critical epigenetic mechanisms contributing to the regulation of activation of microglia and astrocytes, and over-production of TNF-α, IL1-β, and MCP-1 in the spinal dorsal horn, which plays a key role in the genesis of neuropathic pain induced by partial sciatic nerve ligation.
Methods and materials
Animals
Adult male Sprague-Dawley rats (body weight range: 210 to 280 g, Harlan Laboratories) were used. All experiments were approved by the Institutional Animal Care and Use Committee at the University of Georgia and were fully compliant with the National Institutes of Health Guidelines for the Use and Care of Laboratory Animals.
Partial sciatic nerve ligation (pSNL) neuropathic pain model
Partial sciatic nerve ligation was made as previously described (Seltzer, Dubner, & Shir, 1990; Yan, Yadav, Gao, & Weng, 2014). Briefly, animals were anesthetized with 2–3% isoflurane. Under sterile conditions, the left sciatic nerve was exposed at the upper thigh and ligated approximately to one-thirds to one-half of its diameter with a 5-0 silk suture. In sham-operated rats, the left sciatic nerve was exposed but not ligated. Following the surgery, the wounds were closed with skin staples.
Behavioral tests
Behavioral tests were conducted in a quiet room with the room temperature at 22°C as we previously described (Weng et al., 2006; Yan et al., 2013). To verify the changes in mechanical sensitivity before and after surgery, rats were placed on a wire mesh under a plexiglass cage (12 × 20 × 15 cm3), loosely restrained and allowed to acclimate for at least 30 min. A series of von Frey monofilaments (bending force from 0.6 to 26.00 g) were tested in ascending order to generate response-frequency function for each animal. Each von Frey filament was applied 10 times to the mid planter area of the left hind paw for about 1 s to measure hind paw withdrawal response. The response-frequency function for each von Frey filament was defined as [(number of withdrawal responses of the hind paw/10) × 100%]. Withdrawal responses mechanical threshold was determined by the lowest force filament that evoked a 50% or greater withdrawal responses frequency (Weng et al., 2006; Yan et al., 2013). This value was later averaged across all animals in each group to yield the group response threshold (g).
To determine thermal sensitivity, rats were placed on a smooth glass plate pre-heated at 30 °C. A radiant heat beam (diameter 5 mm) was directed onto the mid-plantar area of the left hind paw to evoke a paw withdrawal response from beneath (Hargreaves, Dubner, Brown, Flores, & Joris, 1988). The withdrawal latency (defined as the time between the onset of radiant heat and the time when the rat withdrew his paw) was recorded. Each hind paw was stimulated three times with an interval of at least 2 min. A cutoff time of 20 s was used to avoid damage to the skin. Three paw withdrawal latencies recorded from each individual rat were averaged.
Intrathecal catheter implantation and drug administration
Intrathecal (i.t.) injection of tested drugs to the spinal enlargement was made through the pre-implanted intrathecal catheter. Briefly, a polyethylene (PE-10) catheter that ended at the spinal L4 segment was intrathecally placed as previously described (Yadav, Yan, Maixner, Gao, & Weng, 2015; Yaksh & Rudy, 1976). Rats were anesthetized with 2–3% isoflurane and a PE-10 catheter was carefully inserted through the atlanto-occipital membrane opening and advanced to the lumbar enlargement (L4–L5). The muscles were then sutured in layers and the skin edges were closed with skin staples. The animals were allowed to recover for 7 days before the baseline behavior test were conducted. Animals with the i.t. catheter were then randomly divided into pSNL or sham groups to receive pSNL and sham operation respectively. Drugs or vehicle in a volume of 10 μL was injected into the spinal lumbar enlargement region through the intrathecal catheter, followed by 20 μL of saline to flush. The EZH2 inhibitors (DZNep and GSK126) were prepared in DMSO and saline. The final concentration of DMSO in the injected solution was less than 1%. Saline with the same concentration of DMSO was used as a vehicle control. When the drug administration fell on same day as the behavior analysis, the behavior tests were completed prior to the drug administration. At the end of the behavior experiments, rats were intrathecally injected with 50 μL of 2% lidocaine. If hind paw paralysis did not occur, rats were removed from the experiment. The experimenter who conducted the behavioral tests was blinded to the treatments given to the animals.
Western blotting
Animals were deeply anesthetized with urethane (1.3–1.5 g/kg, i.p.). The L4–L5 spinal segment was exposed and removed from the rats. The dorsal quadrant of the spinal segment ipsilateral to the operation side was isolated and quickly frozen in liquid nitrogen and stored at −80 °C for later use. Frozen tissues were homogenized with a hand-held pellet in lysis buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.1% SDS, 1% deoxycholic acid, 2 mM orthovanadate, 100 mM NaF, 1% Triton X-100, 0.5 mM phenylmethylsulfonyl fluoride, 20 μM leupeptin, 100 IU mL-1 aprotinin) for about 30 min on ice. The samples were then centrifuged for 20 min at 12,000 g at 4 °C and the supernatants containing proteins were collected. The quantification of protein contents was made by the BCA method. Protein samples (50 μg) were electrophoresed in 12.5 % SDS polyacrylamide gels and transferred to polyvinylidene difluoride membrane (Millipore, Bedford, MA). The membranes were blocked with 5% milk or 5% BSA in TBST, and then incubated respectively overnight at 4 °C with polyclonal rabbit anti-GFAP (1:1,000, cell signaling), polyclonal rabbit anti-Iba1 (1:1,000; Wako), rabbit anti-TNF-α (1:500; Millipore), rabbit anti-IL-1β (1:500; Millipore), rabbit anti-MCP-1 (1:500; Abcam), rabbit anti-EZH2 (1:500; Abcam), rabbit anti-H3K27TM (1:500; Epigentek), rabbit anti-total histone H3 (1;1,000; cell signaling) primary antibodies, or a monoclonal rabbit anti-β-actin (1:2,000; Millipore) primary antibody as a loading control. The blots were then incubated for 2 hr at room temperature with corresponding HRP-conjugated secondary antibodies (1:5,000; Santa Cruz Biotechnology, CA, USA), visualized in ECL solution (Super Signal West Pico Chemiluminescent Substrate, Pierce, Rockford, IL, USA) for 2.5 min, and exposed onto Odyssey imaging system-LI-COR biosciences. The bands intensity of immunoreactivity was quantified using ImageJ 1.46 software (NIH). Levels of each biomarker were expressed as the relative ratio to the loading control protein (β-actin) unless otherwise indicated.
Immunohistochemical Analysis
Animals were deeply anesthetized with urethane (1.3–1.5 g/kg, i.p.) and perfused through the ascending aorta with 200 ml heparinized phosphate-buffered solution (0.1 M PBS, pH = 7.35) followed by a solution of 4% formaldehyde in (0.1 M PBS, pH = 7.35). The L4 and L5 spinal cord was removed and fixed for next 24 hrs at 4 °C in fresh 4% formaldehyde. The L4–L5 spinal lumbar region was dehydrated with gradient ethanol, and embedded in paraffin. The transverse sections of the spinal cord were sliced at a 10 μM thickness and mounted on superfrost plus slides. After dewaxing and hydration, slices were subjected for an antigen-retrieval method by sodium citrate (pH = 6) at 90°C for 30 min followed by bench-cooling for 20 min. After washing with 0.1 M PBS, slices were then blocked in 10% normal goat serum with 0.05% BSA, 0.3% Triton X-100 in 0.1 M PBS for 3 hrs at room temperature. The sections were then incubated overnight at 4 °C with primary antibodies. Either simultaneous or sequential incubation of primary antibodies mixture in 2% normal goat serum with 0.0 5% BSA, 0.3% Triton X-100 in 0.1 M PBS against the following targets: mouse anti-NeuN (a marker for neuronal cells,1:200, Cell Signaling); mouse anti-GFAP (a marker for astrocytes, 1:500, Cell Signaling); mouse anti-Iba1 (1:200, Millipore) antibodies. After washing three times with 0.1 M PBS, the sections were incubated for 4 hrs at room temperature with the corresponding Texas Red antibody (1:500, Vector Laboratories) or Alexa Fluor 488 antibody (1:500, Life Technologies). After rinsing three times with 0.1 M PBS, the sections were air-dried and cover-slipped with Vectashield mounting medium (Vector Laboratories). Non-adjacent sections were randomly selected, and the immunostaining for each antibody were viewed under an Olympus BX43 microscope with an Olympus U-CMAD3 camera. Images were processed using Olympus-cell Sens Dimensions. Four to five sections from each rodent’s L4–L5 spinal cord segments (4–5 animals per group) were randomly selected and used for analysis. The number of GFAP+/EZH2+, Iba1+/EZH2+ microglia and NeuN+/EZH2+ neurons with clear, visible cell body and nuclei in the spinal dorsal horn were counted.
Materials
DZnep and GSK126 were purchased from Cayman chemicals (USA).
Data analysis
All data are presented as the mean ± S.E. One-way and two-way ANOVA with repeated measures were used to respectively detect difference in behavioral data over different time points in the same group, and detect difference in behavioral data between groups over different time points. Once difference was found, the Bonferroni post hoc test was used to determine sources of differences. Non-paired Student’s t-tests were used to make comparison between groups for data collected from western blots. A p value less than 0.05 was considered statistically significant. All statistical analyses were performed using GraphPad Prism (v5.03, GraphPad Software, Inc.).
Results
Nerve Injury induced allodynia and thermal hyperalgesia
In order to define the role of EZH2 in the genesis of neuropathic pain, expressions of multiple molecules in the spinal dorsal horn ipsilateral to the operation side were analyzed in rats on days 3 and 10 after pSNL or sham operation. Adult male animals were randomly assigned into 4 groups, two groups receiving pSNL, and two groups receiving sham operation. Behavioral tests were performed to confirm that animals receiving pSNL developed mechanical allodynia and thermal hyperalgesia before the animals were used for any biochemical assays. The mechanical thresholds of hind paw withdrawal responses (5.75 ± 2.7 g, n = 10) in rats with pSNL on day 3 after surgery were significantly lower than their own baseline (prior to the surgery) (14.18 ± 2.3 g, n = 10, p < 0.001) or the sham operated rats (14.96 ± 2.4 g, n = 8, p < 0.001) on the same day. At the same time, latencies of hind paw withdrawal responses to radiant heat stimuli in rats with pSNL (7.49 ± 0.71 s, n = 10) were significantly shorter than their own baseline values (14.29 ± .64 s, n = 10, p < 0.001) or the sham-operated rats (15.82 ± 1.17 s, n = 8, p < 0.001). We also found that mechanical thresholds (6.05 ± 1.18 g, n = 15) in rats with pSNL on day 10 after surgery were significantly reduced in comparison with their baseline values (15.45 ± 0.90 g, n = 15, p < 0.001) or rats with sham-operation (17.33 ± 0.64 g, n =12, p < 0.001). Meanwhile, latencies of hind paw withdrawal responses to radiant heat stimuli in rats with pSNL were significantly (p < 0.01) reduced in comparison with their baseline 15.23 ± 0.50 s to 6.91 ± 0.32 s (n = 15) and with the sham-operated rats (16.15 ± 0.77 s, n = 12). Mechanical thresholds (n = 8) or thermal thresholds (n = 12) in rats with sham-operation measured on day 3 or day 10 after surgery were similar to their own baseline prior to the surgery. These data indicate that rats receiving pSNL had clear signs of neuropathic pain (mechanical allodynia and thermal hyperalgesia) both on day 3 and day 10 after the surgery. Unless otherwise stated, all data in the rest of this paper were obtained from animals that completed the behavioral assessment stated above.
Global levels of EZH2 and H3K27TM were increased in the spinal dorsal horn in rats with neuropathic pain
To determine the role of EZH2 in the genesis of neuropathic pain, we examined global EZH2 protein expression in the spinal dorsal horn ipsilateral to the operation side in rats receiving pSNL and rats receiving sham operation on days 3 and 10 after the surgery. Using western blot techniques, we found that in comparison with sham-operated rats (n = 4) EZH2 protein expression in the spinal dorsal horn was significantly (p < 0.05) increased in animals (n = 5) with pSNL on days 3 (Fig. 1A) and 10 (Fig. 1B) after surgery. EZH2 is a methyltransferase that causes methylation of H3K27 (Kuzmichev, Nishioka, Erdjument-Bromage, Tempst, & Reinberg, 2002). To determine whether the increased protein expression of EZH2 is accompanied with an increase of its functional activity, we measured levels of H3K27TM using western blots. We found that in comparison with the sham-operated groups, animals in the pSNL groups had a higher level of global H3K27TM protein in the spinal dorsal horn on days 3 (Fig. 1A) and 10 (Fig. 1B) after surgery. Together these data indicate that the EZH2 activity is increased in the spinal dorsal horn of rats with neuropathic pain.
Figure 1. Nerve injury increases EZH2 protein expression and H3K27TM levels in spinal dorsal horn.

Bar graphs show the mean (+S.E.) of relative density ratio of EZH2 over β-actin and H3K27TM over total-histone H3 in the spinal dorsal horn of rats receiving either sham operation or pSNL on day 3 (A) and day 10 (B) after surgery. Western blot samples of each molecular protein in each group are displayed. *p < 0.05; **p < 0.01.
EZH2 was predominantly expressed in neurons in the spinal dorsal horn under normal conditions, and nerve injury increased the number of neurons and microglia with EZH2 expression
To identify the cellular types responsible for the globally increased expression of EZH2, we investigated the cellular location of EZH2 since the cellular location of EZH2 in the spinal cord has not been reported. To define the cellular location of EZH2 protein, the L4–L5 spinal segment obtained from rats receiving sham operation and pSNL on day 3 and day 10 (four to five rats per group) after surgery were used. We observed a stronger EZH2 staining in the dorsal horn of rats with pSNL on days 3 and 10 than that in rats with sham-operation (Fig. 2A). Furthermore, microglia, astrocytes, and neurons in spinal slices were respectively labeled with Iba1, GFAP, and NeuN antibodies. The slices were doubled-labeled with the EZH2 antibody. We found that EZH2 was predominantly co-localized with NeuN in sham operated rats (Fig. 2B and C). The number of neurons with EZH2 expression was significantly (p < 0.001) increased after nerve injury (Fig. 2B and C). More strikingly, nerve injury drastically increased (p < 0.001) the number of microglia with EZH2 expression by more than 7 fold on day 3 and day 10 after nerve injury in comparison with the sham operated rats on the same days (Fig. 2B and C). Consequently, the ratios between cells with NeuN+ and EZH2+ and cells with Iba1+ and EZH2+ were significantly reduced from 8.77 ± 2.04 (n = 5) in the sham operated rats to 1.23 ± 0.06 (n = 5, p < 0.01) in rats on day 3 after nerve injury, and from 7.86 ± 1.23 (n = 5) in the sham operated rats to 2.21 ± 0.18 (n = 5, p < 0.01) in rats on day 10 after nerve injury.
Figure 2. EZH2 is predominantly expressed in neurons in the spinal dorsal horn under normal conditions, and nerve injury increases the number of neurons with EZH2 expression and microglia with EZH2 expression.
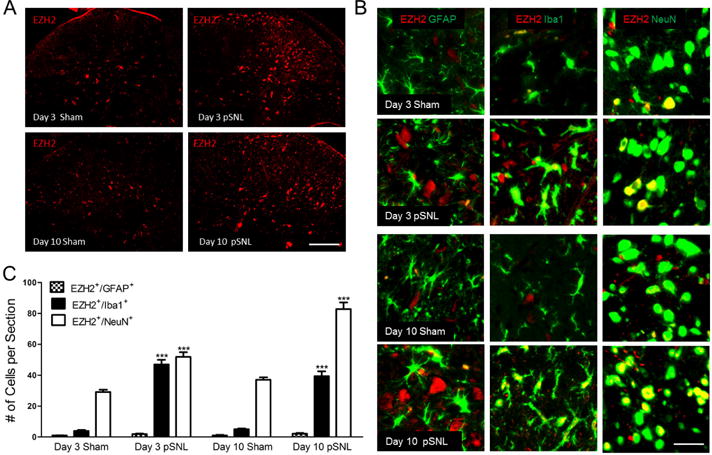
(A) shows samples of EZH2 staining in L4–L5 spinal dorsal horn ipsilateral to the operation site in sham and pSNL rats on days 3 and 10 after surgery. Scale bar: 200 μm. (B) shows co-localization of EZH2 staining (red) with different cellular markers (in green): Iba1 for microglia, GFAP for astrocytes, and NeuN for neurons. Scale bar: 50 μm. (C) Bar graphs show the mean (+ S.E.) numbers of EZH2 +/Iba1+, EZH2+/GFAP+, and EZH2+/NeuN+ cells in the spinal dorsal horn per section in sham operated and pSNL rats on days 3 and 10 after surgery. Four to five rats per group were used for analysis. Comparisons of the same cellular types on the same days after surgery between sham-operated and pSNL rats are shown. *** p < 0.001.
No significant change in the number of GFAP+/EZH2+ cells was observed in rats with pSNL on days 3 and 10 after surgery compared to rats with sham operation (Fig. 2B and C). These data indicate that: a. EZH2 is mainly expressed in neurons under normal conditions; b. nerve injury increases the number of both microglia and neurons with EZH2 expression, with a much more drastic increase in microglia with EZH2 expression. Furthermore, the expression of Iba1 on day 3 and day 10 after nerve injury were about 2 fold of those in sham operated animals on the same day, suggesting that the number of microglia in animals with nerve injury was about 2 fold of that in sham animals. The more than 7 fold increase in number of microglia with EZH2 expression in animals on day 3 and day 10 after nerve injury suggests that the increase in number of microglia with EZH2 expression is not purely due to the increase in the number of microglia, rather, it suggests that at least some of the original microglia prior to nerve injury have EZH2 expression increased.
The increased levels of EZH2 and H3K27TM was temporally associated with neuroinflammation in the spinal cord in neuropathic rats
The drastic increased number of microglia with EZH2 expression prompted us to explore the potential link between EZH2 and neuroinflammation in the spinal dorsal horn. We examined whether the increased levels of EZH2 and H3K27TM in the spinal cord are temporally associated with activation of microglia and astrocytes, and over-production of TNF-α, IL-1β and MCP-1. Protein expressions of the microglial marker (Iba1), astrocytic marker (GFAP), TNF-α, IL-1β and MCP-1 in the spinal dorsal horn of rats with pSNL on day 3 (Fig. 3A; n = 5) and day 10 (Fig. 3B; n = 5) were significantly higher than the sham-operated animals on day 3 (Fig. 3A; n = 4) and day 10 (Fig. 3B; n = 4). The temporal association between the increased levels of EZH2 and H3K27TM and spinal neuroinflammation highly suggest that EZH2 and H3K27TM may be implicated in the spinal neuroinflammation process under neuropathic conditions.
Figure 3. Nerve injury increases protein expressions of Iba1, GFAP, TNF-α, IL-1β, and MCP-1 in the spinal dorsal horn.
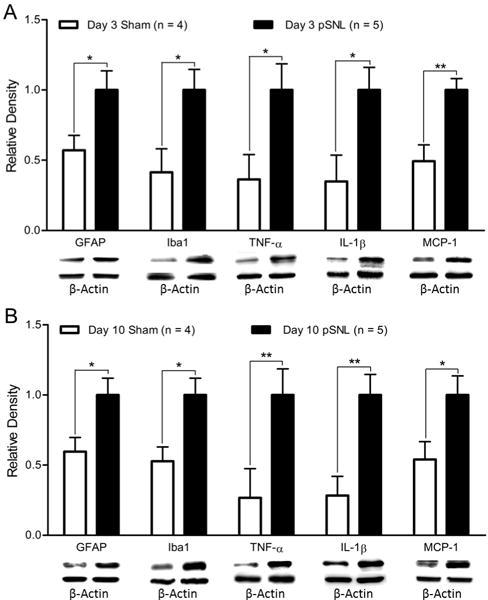
Bar graphs show the mean (+ S.E.) density of Iba1, GFAP, TNF-α, IL-1β, and MCP-1 relative to β-actin in the spinal dorsal horn in rats receiving either sham operation or pSNL on day 3 (A) and day 10 (B) after surgery. Samples of each molecular protein expression in each group are displayed. *p < 0.05; **p < 0.01.
Pharmacological inhibition of EZH2 prevented the development of neuropathic pain
To determine whether the increased EZH2 activity contributes to the development of chronic pain induced by pSNL, we examined whether pharmacological inhibition of EZH2 can prevent the development of neuropathic pain. In the first set of experiments, a widely used EZH2 inhibitor, 3-Deazaneplanocin (DZNep) (Girard et al., 2014; Glazer, Knode, Tseng, Haines, & Marquez, 1986; Miranda et al., 2009; Tan et al., 2007) was used. DZNep is an S-adenosylhomocysteine hydrolase inhibitor, known to suppress the protein expression of EZH2 and levels of H3K27TM without effects on DNA methyltransferases (Fiskus et al., 2009; Fujiwara et al., 2014; Tan et al., 2007). Rats were randomly assigned into four groups: Sham + Vehicle, Sham + DZNep, pSNL + Vehicle, and pSNL + DZNep groups. Behavior analysis was performed on day 1 before the surgery and then on days 2, 4, 6, 8, and 10 after the surgery. Rats in the pSNL + DZNep group and Sham + DZNep group received DZNep (20 nM in 10 μL) through the pre-implanted intrathecal catheter on day 1 immediately prior to the surgery and then daily till day 9 after the surgery. Vehicles (10 μL) were administered to rats in the pSNL + Vehicle and Sham + Vehicle groups in the same fashion. As shown in Figure 4A, mechanical thresholds of withdrawal responses were significantly (p < 0.001) reduced in the pSNL + vehicle group (n = 12) starting from day 2 through day 10 following the nerve injury compared to their baseline prior to the surgery and those from the sham + vehicle group (n = 9). Mechanical thresholds of withdrawal responses in the pSNL + DZNep group (n = 11) at baseline, and on days 2 and 4 were similar to those in the pSNL + Vehicle group (n = 12), but became significantly higher than those in the pSNL + vehicle group from day 6 through day 10 (p < 0.001). Meanwhile, mechanical thresholds in the Sham + Vehicle group and Sham + DZNep group remained stable during the 10 day observation period. Similarly, we also observed that latencies of withdrawal responses to radiant heat stimuli were significantly prolonged at the same time point in the pSNL + DZNep group than those in the pSNL + Vehicle group (Fig. 4B).
Figure 4. EZH2 inhibitors prevent the pain hypersensitivity induced by nerve injury.
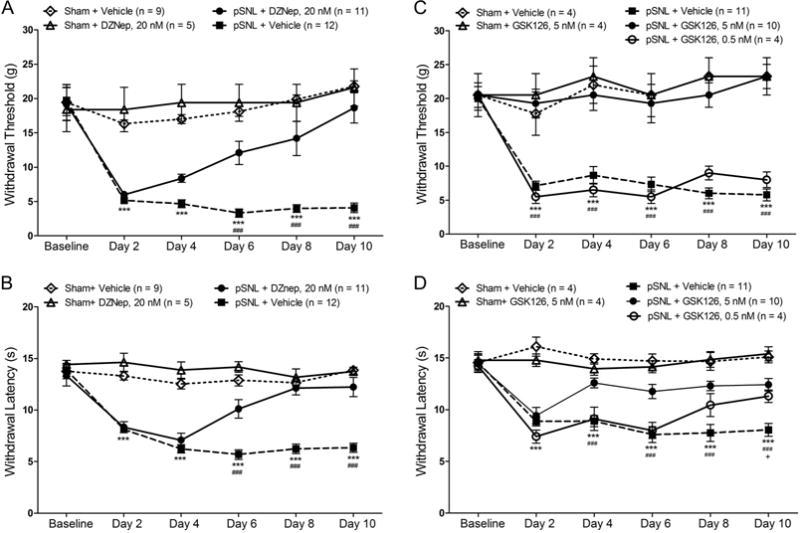
(A) and (B) show the mean (± S.E.) mechanical withdrawal threshold and thermal withdrawal latency during the 10-day observation period in four groups of rats treated with daily intrathecal injection of either 20 nM DZNep (in 10 μL) or vehicle (10 μL) for 9 days. Comparisons between baseline and each time point in the pSNL+ vehicle group are indicated with *; comparisons between the pSNL + 20 nM DEZNep and pSNL + Vehicle groups are indicated with #. (C) and (D) show the mean (± S.E.) mechanical withdrawal threshold and thermal withdrawal latency during the 10-day observation period in five groups of rats treated with daily intrathecal injection of either 5 nM GSK-126 (in 10 μL), 0.5 nM GSK-126 (in 10 μL), or vehicle (10 μL) for 9 days. Comparisons between baseline and each time point in the pSNL+ vehicle group are indicated with *; comparisons between the pSNL + 5 nM GSK-126 and pSNL + Vehicle group are indicated with #; comparisons between the pSNL + 0.5 nM GSK-126 and pSNL + vehicle group are indicated with +. One symbol: p < 0.05; three symbols: p < 0.001.
In the second set of experiments, we used another agent GSK126, which is a selective, S-adenosyl-methionine-competitive small molecule inhibitor of EZH2 methyltransferase activity (McCabe et al., 2012). Rats were randomly assigned into five groups: Sham + Vehicle, Sham + GSK-126 (5 nM), pSNL + Vehicle, pSNL + GSK-126 (5 nM), and pSNL + GSK-126 (0.5 nM). Drugs and vehicle were administered in the same fashion and time schedule as those described for the DZNep treatment. We found that mechanical thresholds of withdrawal responses in the pSNL + GSK-126 (5 nM) group (n = 10) but not in the GSK-126 (0.5 nM) group (n = 4) were significantly elevated in comparison with those in the pSNL + vehicle group (n = 11) from day 2 through day 10 (p < 0.001) (Fig. 4C). Mechanical thresholds in the Sham + vehicle group and sham + GSK-126 (5 nM) group were not significantly altered during the same period. We also found that GSK-126 prolonged latencies of withdrawal responses to radiant heat stimuli in rats with pSNL in a dose-dependent manner (Fig. 4D) from day 4 to day 10 after the surgery. These data demonstrate that inhibition of EZH2 can prevent the development of neuropathic pain.
Pharmacological inhibition of EZH2 attenuated the pre-existing neuropathic pain
Next, we determined whether inhibition of EZH2 can attenuate the pre-existing neuropathic pain. Four groups of rats were used: Sham + Vehicle, Sham + DZNep, pSNL + Vehicle, and pSNL + DZNep groups. DZNep (20 nM in 10 μL) were administered intrathecally daily from day 3 to day 9 after the nerve injury. Vehicles (10 μL) were administered to rats in the pSNL + Vehicle and Sham + Vehicle in the same fashion. We found that mechanical thresholds and thermal latencies of withdrawal responses in the pSNL + DZNep (n = 6) group were significantly increased (p < 0.05 to p < 0.01) from day 7 to day 10 after the surgery in comparison with their own values on day 3 and those in the pSNL + Vehicle group (n = 6) (Fig. 5). These data indicate that blocking spinal EZH2 activity can attenuate the pre-existing neuropathic pain.
Figure 5. EZH2 inhibitors attenuate the pre-existing pain hypersensitivity induced by nerve injury.
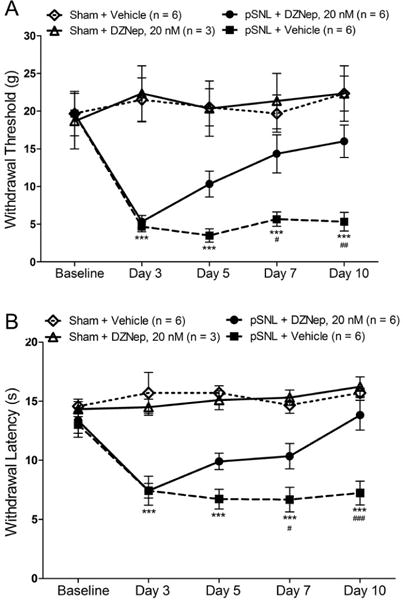
Line plots show the mean (± S.E.) mechanical withdrawal threshold (A) and thermal withdrawal latency (B) during the 10-day observation period in four groups of rats. Daily intrathecal injection of either 20 nM DZNep (in 10 μL) or vehicle (10 μL) was made from days 3 to 9. Comparisons between baseline and each time point in the pSNL+ Vehicle group are indicated with *; comparisons between the pSNL + 20 nM DEZNep and pSNL + vehicle group are indicated with #. One symbol: p < 0.05; two symbols: p < 0.01; three symbols: p < 0.001.
Global levels of EZH2 and H3K27TM in the spinal dorsal horn of neuropathic rats were restored by inhibition of EZH2
DZNep treatment is known to suppress protein expression of EZH2 and reduce levels of H3K27TM (Fiskus et al., 2009; Fujiwara et al., 2014; Tan et al., 2007). To determine levels of EZH2 and H3K27TM in the spinal dorsal horn, the spinal dorsal horn of the same animals that had finished the 9 day DZNep and vehicle treatments as well as behavioral tests on day 10 as described in Fig. 4A and B were used. Using western blot techniques, we found that 9 day treatment of DZNep restored basal protein expressions of EZH2 in rats with pSNL (n = 7) (Fig. 6A). At the same time, the H3K27TM level in the neuropathic rats was also restored as demonstrated by western blots (n = 7) (Fig. 6A). Similarly, we determined spinal levels of EZH2 and H3K27TM in the rats that had completed the 9 day GSK126 and vehicle treatments as well as behavioral tests on day 10 as described in Fig. 4C and D. We found that GKS126 treatment normalized the levels of EZH2 and H3K27TM in rats with pSNL (Fig. 6B).
Figure 6. Pre-emptive DZNep or GSK126 treatment significantly attenuates the increased levels of EZH2 and H3K27TM in the spinal dorsal horn induced by nerve injury.
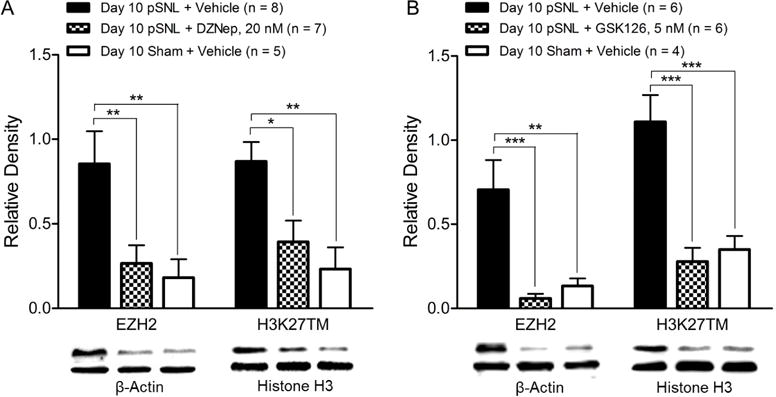
(A): Data were obtained from the spinal dorsal horn of animals treated with daily intrathecal injection of either 20 nM DZNep (in 10 μL) or vehicle (10 μL) for 9 days. (B): Data were obtained from the spinal dorsal horn of animals treated with daily intrathecal injection of either 5 nM GSK126 (in 10 μL) or vehicle (10 μL) for 9 days. Bar graphs show the mean (+ S.E.) relative density ratio of EZH2 over β-actin and H3K27TM over total-histone H3. Western blot samples of each molecular protein in each group are displayed. *p < 0.05; **p < 0.01; ***p < 0.001.
DZNep treatment suppressed neuroinflammation in the spinal dorsal horn of rats with neuropathic pain
Finally, we examined the molecular mechanisms by which EZH2 inhibitors exerts its analgesic effects. The drastic increased number of microglia with expression of EZH2 highly suggests that DZNep treatment may suppress neuroinflammation in the spinal dorsal horn in rats with pSNL. Thus, we determined protein expressions of Iba1, GFAP, TNF-α, IL-1β, and MCP-1 in the same animals that had completed with the behavioral tests as described in Figure 4A and B. We found that the increased expressions of Iba1, GFPA, and TNF-α, IL-1β, and MCP-1 in the spinal dorsal horn of rats with pSNL (n = 8) were significantly attenuated when 20 nM DEZNep was intrathecally injected into the animals immediately prior to the pSNL surgery and then daily for 9 days (n = 7) (Fig. 7). These results demonstrate that suppression of glial activation and production of TNF-α, IL-1β, and MCP-1 are important mechanisms by which DZNep produces the analgesic effects in the animals with neuropathic pain.
Figure 7. Pre-emptive treatment of the EZH2 inhibitor attenuates the increased expressions of Iba1, GFAP, TNF-α, IL-1β and MCP-1 in the spinal dorsal horn of rats with nerve injury.
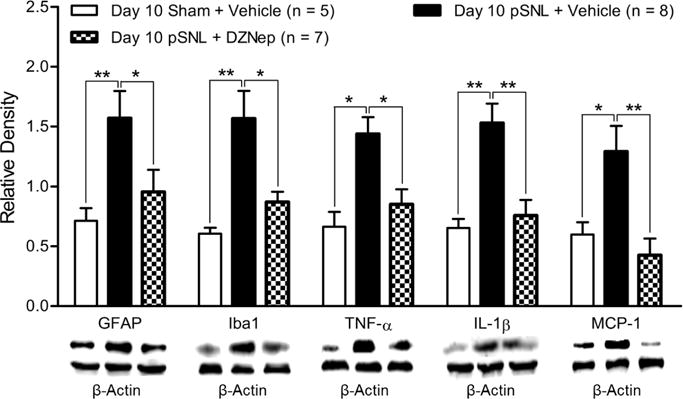
Data were obtained from the spinal dorsal horn of animals treated with daily intrathecal injection of either 20 nM DZNep (in 10 μL) or vehicle (10 μL) for 9 days. Bar graphs show the mean (+ S.E.) density of GFAP, Iba1, TNF-α, IL-1β and MCP-1 relative to β-actin in the spinal dorsal horn of sham operated rats treated vehicle, pSNL rats treated with DEZNep, and pSNL rats treated vehicle. Samples of each molecular protein in each group are displayed. *p < 0.05; **p < 0.01.
Discussion
In this study, we for the first time demonstrated that EZH2 is a key regulator in the development of neuropathic pain. We found that nerve injury caused increases in levels of EZH2 and H3K27TM, and the number of neurons and microglia with EZH2 expression in the spinal dorsal horn. Importantly, spinal pharmacological inhibition of EZH2 attenuated the development and maintenance of mechanical and thermal hyperalgesia in rats with nerve injury. Such analgesic effects are concurrently accompanied with the reduced protein expressions of Iba1, GFPA, TNF-α, IL-1β, and MCP-1 in the spinal dorsal horn induced by nerve injury. Our findings highly indicate that EZH2 is implicated in the genesis of neuropathic pain through regulating neuroinflammation in the spinal dorsal horn.
Gene activation and suppression play a critical role in the development of plasticity along the peripheral and central pain signaling pathways in chronic pain conditions (Kuner, 2010; Lacroix-Fralish, Tawfik, Tanga, Spratt, & DeLeo, 2006; Clifford J. Woolf, 2011). Emerging studies have implicated epigenetic mechanisms such as DNA methylation, histone modification, and non-coding micro RNAs in aberrant gene expression in animals with neuropathic pain (Buchheit, Van de Ven, & Shaw, 2012; Doehring, Geisslinger, & Lotsch, 2011; Gräff, Kim, Dobbin, & Tsai, 2011; Tajerian et al., 2011). For example, rats with neuropathic pain have increases in global DNA methylation and methyl-CpG-binding protein 2 (MeCP2), and suppression of global DNA methylation and MeCP2 expression with 5-azacytidine attenuate mechanical allodynia and thermal hyperalgesia induced by nerve injury (Wang et al., 2011). Suppression of miR-7a expression in the dorsal root ganglion induced by nerve injury causes an increased protein expression of β2 subunit of the voltage-gated sodium channel, which contributes importantly to the late phase of neuropathic pain (Sakai et al., 2013). Inhibition of histone acetylation with HDAC inhibitors attenuates the development of mechanical and thermal hypersensitivity induced by nerve trauma or antiretroviral drug—induced neuropathy in rats, but do not have effects on the pre-existing neuropathic pain (Denk et al., 2013)) Increased histone H3 acetylation in the promoter of MIP-2 and CXCR2 leads to the up-regulation of MIP-2 and CXCR2 following nerve injury, and neuropathic pain (Kiguchi et al., 2012). In addition to histone acetylation, histone methylation is another important mechanism regulating gene expression. Currently, our understanding about the role of histone methylation in the nervous system is limited.
The PRC2 is an essential epigenetic machinery that suppresses the gene expression by transferring the mono, di or tri-methyl groups onto lysine 27 of histone3 (H3K27) via its catalytic subunit EZH2, a methyltransferase (Bannister & Kouzarides, 2011; Ferrari et al., 2014; Kirmizis et al., 2004). It was recently reported that a global increase of H3K27TM and EZH2 protein expression in the dorsal root ganglion of rats with neuropathic pain results in suppression of potassium channel activities in the sensory neurons (Laumet et al., 2015). Little is known about the global levels of H3K27TM and its methyltransferase (EZH2) in the spinal cord in pathological conditions. Similarly, the effects induced by pharmacological intervention targeting this signaling pathway at the spinal cord have not been explored. In this study, we found that global protein expression of EZH2 and levels of H3K27TM in the spinal dorsal horn were increased on day 3 and day 10 after never injury and spinal pharmacological EZH2 inhibitors attenuate the development and maintenance of neuropathic pain. Importantly, we validated the targets of the EZH2 inhibitors by demonstrating that the levels of EZH2 and H3K27TM were normalized in the spinal cord of neuropathic rats treated with the EZH2 inhibitors. These findings suggest that normalization of the EZH2 activity in the spinal dorsal horn is effective to attenuate neuropathic pain.
Given that glial activation and over-production of inflammatory mediators are critically implicated in the genesis of neuropathic pain, molecular mechanisms regulating spinal glial activation and neuroinflammation have been highly sought-after as the basis of a new generation of therapeutics for neuropathic pain. Recent studies suggest that histone modification is engaged in epigenetic regulation of genes related to inflammation along the pain signaling pathway under pathological pain conditions. For example, systemic administration of a HDAC inhibitor, sodium butyrate, significantly attenuated the increased TNF-α level in the injured sciatic nerve (Kukkar, Singh, & Jaggi, 2014). Nerve injury promotes histone H3 and H4 acetylation and increases expression of BDNF via activating the BDNF promoter (Uchida, Matsushita, & Ueda, 2013). Pharmacological inhibition or knockdown of the histone acetyltransferase E1A binding protein p300 in the spinal cord attenuates the COX-2 expression in the spinal cord induced by nerve injury (Zhu et al., 2012). While it was reported that an upregulation of MCP-3 gene expression in the spinal cord of neuropathic rats was ascribed to the decreased H3K27TM at the MCP-3 promoter (Imai et al., 2013), it remains unknown whether global spinal neuroinflammation under pathological pain conditions can be modulated by altering the global levels of EZH2 and H3K27TM in the spinal dorsal horn. Our results show that nerve injury significantly increases the global levels of EZH2 and H3K27TM in the spinal dorsal horn. Neuropathic rats treated with the EZH2 inhibitors had significant less mechanical allodynia and thermal hyperalgesia and less protein expressions of Iba1, GFAP, and TNF-α, IL-1β, and MCP-1 in the spinal dorsal horn in comparison with those in neuropathic rats receiving vehicle treatment. These data strongly indicate that pharmacological suppression of the increased activities of EZH2 attenuate the development of neuropathic pain via suppressing activation of microglia and astrocytes, and production of TNF-α, IL-1β, and MCP-1. Our findings also reiterate the importance to investigate the global levels and function of a given molecule when considering the therapeutic use of the target molecule.
Mechanisms by which EZH2 regulates neuroinflammation remain not well understood. Neuroinflammation is characterized by glial activation and proliferation and their subsequent over-production of proinflammatory mediators (including TNF-α, IL-1β, and MCP-1). Given that trimethylation of H3K27 by EZH2 results in gene repression (Cao and Zhang, 2004; Hansen et al., 2008; Marqueron et al., 2009), the types of genes repressed by H3K27TM/EZH2 must be negative regulators of neuroinflammation. One gene known to be suppressed by EZH2/H3K27TM is the cyclin-dependent kinase inhibitor p16 (Maertens et al., 2009). P16 is a well-known cell-cycle-regulatory protein, which inhibits cyclin dependent kinase (like CDK4) activities and suppresses cell proliferation (Traves, Luque, & Hortelano, 2012). Previous studies have shown that cell proliferation and inflammation are regulated positively by EZH2/H3K27TM but negatively by P16 gene expression. For example, embryos with EZH2 deletion display severe defects during gastrulation (O’Carroll et al., 2001; Zhang et al., 2015) Over expression of EZH2 in differentiating neural stem cells promotes neural stem cells to be differentiated into oligodendrocytes (Sher et al., 2008). EZH2 suppression in dorsal root ganglion cocultures interferes with in vitro myelination by Schwann cells (Heinen et al., 2012; Zhang et al., 2015). Down-regulation of P16 leads to cell proliferation (Traves et al., 2012). More recent studies show that EZH2 and P16 are involved in the inflammation processes. For example, EZH2 is overexpressed in fibroblast-like synoviocytes in the rheumatoid joint and EZH2 causes changes in fibroblast-like synoviocytes through the Wnt signaling pathway, which plays an important role in regulating rheumatoid arthritis (Miao et al., 2013; Trenkmann et al., 2011). Activation of NF-kB by lysophosphatidic acid and production of IL-6, IL-8 in breast cancer cells is dependent on high EZH2 expression (Hartman et al., 2013). Enhancement of gene expression of P16 results in reduction of rheumatoid arthritis in animal models, and attenuation in production of proinflammatory mediators from synovial fibroblasts of rheumatoid arthritis, and macrophages in response to lipopolysaccharide (LPS) treatment (Murakami, Mizoguchi, Saito, Miyasaka, & Kohsaka, 2012). Currently, it is unknown about the role of EZH2 in the regulation of inflammation in the CNS. Our present study fills this gap by investigating the role of EZH2 in the spinal cord. The cellular location of EZH2 in the spinal cord was defined for the first time. We revealed that EZH2 is predominantly expressed in neurons under normal conditions. This is in line with a previous report that EZH2 is only expressed in neurons in the spinal dorsal root ganglion (Laumet et al., 2015). We found that nerve injury causes an increase in the global EZH2 expression and the number of neurons with EZH2 expression in the spinal dorsal horn. Intriguingly, nerve injury results in a tremendous increase in the number of microglia with EZH2 expression on 3 day and 10 day after nerve injury. Given that microglial activation, as evident by an increased expression of Iba1 protein, was found at the same time, it is conceivable that the increased expression of EZH2 in microglia contributes importantly to the microglial activation and its production of inflammatory mediators, which ultimately brings about neuroinflammation in the spinal dorsal horn. This notion is indirectly supported by several previous findings. First, cell proliferation is associated with production of inflammatory mediators (Gong et al., 2011; Turgeon, Blais, Delabre, & Asselin, 2013). Second, microglial proliferation is a form of microglial activation, which has been recently demonstrated to be a critical mechanism contributing to the genesis of neuroinflammation and neuropathic pain in rodents (Guan et al., 2016; Okubo et al., 2016).
In conclusion, our current study reveals that EZH2 plays a critical role in the development and maintenance of neuropathic pain through regulating the neuroinflammation process in the spinal dorsal horn. Hence, suppressing the spinal EZH2 and H3K27TM levels may be a novel strategy for the development of new analgesics for the treatment of neuropathic pain.
Highlights.
Nerve injury increases protein expressions of EZH2 and H3K27TM in the dorsal horn.
Number of neurons and microglia with EZH2 expression is increased after nerve injury.
Inhibition of EZH2 attenuates the development and maintenance of neuropathic pain.
Inhibition of EZH2 suppresses spinal neuroinflammation induced by nerve injury.
Acknowledgments
This project was supported by the NIH RO1 grant (RO1NS064289) to H.R.W.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no conflict of interest.
Reference List
- Alam H, Gu B, Lee MG. Histone methylation modifiers in cellular signaling pathways. Cell Mol Life Sci. 2015;72(23):4577–4592. doi: 10.1007/s00018-015-2023-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai G, Ren K, Dubner R. Epigenetic regulation of persistent pain. Transl Res. 2015;165(1):177–199. doi: 10.1016/j.trsl.2014.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21(3):381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Befort K, Costigan M, Woolf CJ. Differential gene expression–how to find new analgesic targets. Curr Opin Investig Drugs. 2001;2(3):396–398. [PubMed] [Google Scholar]
- Buchheit T, Van de Ven T, Shaw A. Epigenetics and the transition from acute to chronic pain. Pain Med. 2012;13(11):1474–1490. doi: 10.1111/j.1526-4637.2012.01488.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgold T, Spreafico F, De Santa F, Totaro MG, Prosperini E, Natoli G, Testa G. The histone H3 lysine 27-specific demethylase Jmjd3 is required for neural commitment. PLoS One. 2008;3(8):e3034. doi: 10.1371/journal.pone.0003034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denk F, Huang W, Sidders B, Bithell A, Crow M, Grist J, McMahon SB. HDAC inhibitors attenuate the development of hypersensitivity in models of neuropathic pain. Pain. 2013;154(9):1668–1679. doi: 10.1016/j.pain.2013.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Descalzi G, Ikegami D, Ushijima T, Nestler EJ, Zachariou V, Narita M. Epigenetic mechanisms of chronic pain. Trends in neurosciences. 2015;38(4):237–246. doi: 10.1016/j.tins.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doehring A, Geisslinger G, Lotsch J. Epigenetics in pain and analgesia: an imminent research field. Eur J Pain. 2011;15(1):11–16. doi: 10.1016/j.ejpain.2010.06.004. [DOI] [PubMed] [Google Scholar]
- Ferrari KJ, Scelfo A, Jammula S, Cuomo A, Barozzi I, Stutzer A, Pasini D. Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol Cell. 2014;53(1):49–62. doi: 10.1016/j.molcel.2013.10.030. [DOI] [PubMed] [Google Scholar]
- Fiskus W, Wang Y, Sreekumar A, Buckley KM, Shi H, Jillella A, Bhalla KN. Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor panobinostat against human AML cells. Blood. 2009;114(13):2733–2743. doi: 10.1182/blood-2009-03-213496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara T, Saitoh H, Inoue A, Kobayashi M, Okitsu Y, Katsuoka Y, Harigae H. 3-Deazaneplanocin A (DZNep), an inhibitor of S-adenosylmethionine-dependent methyltransferase, promotes erythroid differentiation. The Journal of biological chemistry. 2014;289(12):8121–8134. doi: 10.1074/jbc.M114.548651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Zhang L, Samad OA, Suter MR, Yasuhiko K, Xu ZZ, Ji RR. JNK-induced MCP-1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29(13):4096–4108. doi: 10.1523/JNEUROSCI.3623-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard N, Bazille C, Lhuissier E, Benateau H, Llombart-Bosch A, Boumediene K, Bauge C. 3-Deazaneplanocin A (DZNep), an inhibitor of the histone methyltransferase EZH2, induces apoptosis and reduces cell migration in chondrosarcoma cells. PloS one. 2014;9(5):e98176. doi: 10.1371/journal.pone.0098176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glazer RI, Knode MC, Tseng CK, Haines DR, Marquez VE. 3-Deazaneplanocin A: a new inhibitor of S-adenosylhomocysteine synthesis and its effects in human colon carcinoma cells. Biochemical pharmacology. 1986;35(24):4523–4527. doi: 10.1016/0006-2952(86)90774-4. [DOI] [PubMed] [Google Scholar]
- Gong Y, Huo L, Liu P, Sneige N, Sun X, Ueno NT, Cristofanilli M. Polycomb group protein EZH2 is frequently expressed in inflammatory breast cancer and is predictive of worse clinical outcome. Cancer. 2011;117(24):5476–5484. doi: 10.1002/cncr.26179. [DOI] [PubMed] [Google Scholar]
- Grace PM, Hutchinson MR, Maier SF, Watkins LR. Pathological pain and the neuroimmune interface. Nature reviews Immunology. 2014 doi: 10.1038/nri3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff J, Kim D, Dobbin MM, Tsai LH. Epigenetic Regulation of Gene Expression in Physiological and Pathological Brain Processes. Physiological Reviews. 2011;91(2):603–649. doi: 10.1152/physrev.00012.2010. [DOI] [PubMed] [Google Scholar]
- Guan Z, Kuhn JA, Wang X, Colquitt B, Solorzano C, Vaman S, Basbaum AI. Injured sensory neuron-derived CSF1 induces microglial proliferation and DAP12-dependent pain. Nature neuroscience. 2016;19(1):94–101. doi: 10.1038/nn.4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32(1):77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Hartman ZC, Poage GM, den Hollander P, Tsimelzon A, Hill J, Panupinthu N, Brown PH. Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8. Cancer research. 2013;73(11):3470–3480. doi: 10.1158/0008-5472.CAN-12-4524-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinen A, Tzekova N, Graffmann N, Torres KJ, Uhrberg M, Hartung HP, Kury P. Histone methyltransferase enhancer of zeste homolog 2 regulates Schwann cell differentiation. Glia. 2012;60(11):1696–1708. doi: 10.1002/glia.22388. [DOI] [PubMed] [Google Scholar]
- Hirabayashi Y, Suzki N, Tsuboi M, Endo TA, Toyoda T, Shinga J, Gotoh Y. Polycomb limits the neurogenic competence of neural precursor cells to promote astrogenic fate transition. Neuron. 2009;63(5):600–613. doi: 10.1016/j.neuron.2009.08.021. [DOI] [PubMed] [Google Scholar]
- Imai S, Ikegami D, Yamashita A, Shimizu T, Narita M, Niikura K, Narita M. Epigenetic transcriptional activation of monocyte chemotactic protein 3 contributes to long-lasting neuropathic pain. Brain. 2013;136(Pt 3):828–843. doi: 10.1093/brain/aws330. [DOI] [PubMed] [Google Scholar]
- Kawasaki Y, Zhang L, Cheng JK, Ji RR. Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008;28(20):5189–5194. doi: 10.1523/JNEUROSCI.3338-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiguchi N, Kobayashi Y, Maeda T, Fukazawa Y, Tohya K, Kimura M, Kishioka S. Epigenetic augmentation of the macrophage inflammatory protein 2/C-X-C chemokine receptor type 2 axis through histone H3 acetylation in injured peripheral nerves elicits neuropathic pain. The Journal of pharmacology and experimental therapeutics. 2012;340(3):577–587. doi: 10.1124/jpet.111.187724. [DOI] [PubMed] [Google Scholar]
- Kim KH, Roberts CW. Targeting EZH2 in cancer. Nature medicine. 2016;22(2):128–134. doi: 10.1038/nm.4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirmizis A, Bartley SM, Kuzmichev A, Margueron R, Reinberg D, Green R, Farnham PJ. Silencing of human polycomb target genes is associated with methylation of histone H3 Lys 27. Genes & development. 2004;18(13):1592–1605. doi: 10.1101/gad.1200204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukkar A, Singh N, Jaggi AS. Attenuation of neuropathic pain by sodium butyrate in an experimental model of chronic constriction injury in rats. J Formos Med Assoc. 2014;113(12):921–928. doi: 10.1016/j.jfma.2013.05.013. [DOI] [PubMed] [Google Scholar]
- Kuner R. Central mechanisms of pathological pain. Nature medicine. 2010;16(11):1258–1266. doi: 10.1038/nm.2231. [DOI] [PubMed] [Google Scholar]
- Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes & development. 2002;16(22):2893–2905. doi: 10.1101/gad.1035902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroix-Fralish ML, Tawfik VL, Tanga FY, Spratt KF, DeLeo JA. Differential spinal cord gene expression in rodent models of radicular and neuropathic pain. Anesthesiology. 2006;104(6):1283–1292. doi: 10.1097/00000542-200606000-00025. [DOI] [PubMed] [Google Scholar]
- Laumet G, Garriga J, Chen SR, Zhang Y, Li DP, Smith TM, Pan HL. G9a is essential for epigenetic silencing of K(+) channel genes in acute-to-chronic pain transition. Nat Neurosci. 2015;18(12):1746–1755. doi: 10.1038/nn.4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang L, Lutz BM, Bekker A, Tao YX. Epigenetic regulation of chronic pain. Epigenomics. 2015;7(2):235–245. doi: 10.2217/epi.14.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligon CO, Moloney RD, Greenwood-Van Meerveld B. Targeting Epigenetic Mechanisms for Chronic Pain: A Valid Approach for the Development of Novel Therapeutics. The Journal of pharmacology and experimental therapeutics. 2016;357(1):84–93. doi: 10.1124/jpet.115.231670. [DOI] [PubMed] [Google Scholar]
- Maertens GN, El Messaoudi-Aubert S, Racek T, Stock JK, Nicholls J, Rodriguez-Niedenfuhr M, Peters G. Several distinct polycomb complexes regulate and co-localize on the INK4a tumor suppressor locus. PloS one. 2009;4(7):e6380. doi: 10.1371/journal.pone.0006380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ, 3rd, Gamblin SJ. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature. 2009;461(7265):762–767. doi: 10.1038/nature08398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez NJ, Simeonov A. Cell-based assays to support the profiling of small molecules with histone methyltransferase and demethylase modulatory activity. Drug Discov Today Technol. 2015;18:9–17. doi: 10.1016/j.ddtec.2015.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Creasy CL. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492(7427):108–112. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- Miao CG, Yang YY, He X, Li XF, Huang C, Huang Y, Li J. Wnt signaling pathway in rheumatoid arthritis, with special emphasis on the different roles in synovial inflammation and bone remodeling. Cellular signalling. 2013;25(10):2069–2078. doi: 10.1016/j.cellsig.2013.04.002. [DOI] [PubMed] [Google Scholar]
- Miranda TB, Cortez CC, Yoo CB, Liang G, Abe M, Kelly TK, Jones PA. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Molecular cancer therapeutics. 2009;8(6):1579–1588. doi: 10.1158/1535-7163.MCT-09-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami Y, Mizoguchi F, Saito T, Miyasaka N, Kohsaka H. p16(INK4a) exerts an anti-inflammatory effect through accelerated IRAK1 degradation in macrophages. Journal of immunology. 2012;189(10):5066–5072. doi: 10.4049/jimmunol.1103156. [DOI] [PubMed] [Google Scholar]
- Nie H, Zhang H, Weng HR. Minocycline prevents impaired glial glutamate uptake in the spinal sensory synapses of neuropathic rats. Neuroscience. 2010;170(3):901–912. doi: 10.1016/j.neuroscience.2010.07.049. doi:S0306-4522(10)01052-3 [pii] 10.1016/j.neuroscience.2010.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Carroll D, Erhardt S, Pagani M, Barton SC, Surani MA, Jenuwein T. The polycomb-group gene Ezh2 is required for early mouse development. Mol Cell Biol. 2001;21(13):4330–4336. doi: 10.1128/mcb.21.13.4330-4336.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okubo M, Yamanaka H, Kobayashi K, Dai Y, Kanda H, Yagi H, Noguchi K. Macrophage-Colony Stimulating Factor Derived from Injured Primary Afferent Induces Proliferation of Spinal Microglia and Neuropathic Pain in Rats. PLoS One. 2016;11(4):e0153375. doi: 10.1371/journal.pone.0153375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira JD, Sansom SN, Smith J, Dobenecker MW, Tarakhovsky A, Livesey FJ. Ezh2, the histone methyltransferase of PRC2, regulates the balance between self-renewal and differentiation in the cerebral cortex. Proc Natl Acad Sci U S A. 2010;107(36):15957–15962. doi: 10.1073/pnas.1002530107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren K, Dubner R. Activity-triggered tetrapartite neuron-glial interactions following peripheral injury. Current opinion in pharmacology. 2016;26:16–25. doi: 10.1016/j.coph.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai A, Saitow F, Miyake N, Miyake K, Shimada T, Suzuki H. miR-7a alleviates the maintenance of neuropathic pain through regulation of neuronal excitability. Brain. 2013;136(Pt 9):2738–2750. doi: 10.1093/brain/awt191. [DOI] [PubMed] [Google Scholar]
- Seltzer Z, Dubner R, Shir Y. A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain. 1990;43(2):205–218. doi: 10.1016/0304-3959(90)91074-S. [DOI] [PubMed] [Google Scholar]
- Sher F, Rossler R, Brouwer N, Balasubramaniyan V, Boddeke E, Copray S. Differentiation of neural stem cells into oligodendrocytes: involvement of the polycomb group protein Ezh2. Stem Cells. 2008;26(11):2875–2883. doi: 10.1634/stemcells.2008-0121. [DOI] [PubMed] [Google Scholar]
- Sparmann A, Xie Y, Verhoeven E, Vermeulen M, Lancini C, Gargiulo G, van Lohuizen M. The chromodomain helicase Chd4 is required for Polycomb-mediated inhibition of astroglial differentiation. Embo j. 2013;32(11):1598–1612. doi: 10.1038/emboj.2013.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki R, Dickenson AH. Neuropathic pain: nerves bursting with excitement. Neuroreport. 2000;11(12):R17–21. doi: 10.1097/00001756-200008210-00001. [DOI] [PubMed] [Google Scholar]
- Tajerian M, Alvarado S, Millecamps M, Dashwood T, Anderson KM, Haglund L, Stone LS. DNA methylation of SPARC and chronic low back pain. Mol Pain. 2011;7:65. doi: 10.1186/1744-8069-7-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Yang X, Zhuang L, Jiang X, Chen W, Lee PL, Yu Q. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes & development. 2007;21(9):1050–1063. doi: 10.1101/gad.1524107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawfik VL, Regan MR, Haenggeli C, Lacroix-Fralish ML, Nutile-McMenemy N, Perez N, DeLeo JA. Propentofylline-induced astrocyte modulation leads to alterations in glial glutamate promoter activation following spinal nerve transection. Neuroscience. 2008;152(4):1086–1092. doi: 10.1016/j.neuroscience.2008.01.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BK. Pathophysiologic mechanisms of neuropathic pain. Curr Pain Headache Rep. 2001;5(2):151–161. doi: 10.1007/s11916-001-0083-1. [DOI] [PubMed] [Google Scholar]
- Traves PG, Luque A, Hortelano S. Macrophages, inflammation, and tumor suppressors: ARF, a new player in the game. Mediators of inflammation. 2012;2012:568783. doi: 10.1155/2012/568783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trenkmann M, Brock M, Gay RE, Kolling C, Speich R, Michel BA, Huber LC. Expression and function of EZH2 in synovial fibroblasts: epigenetic repression of the Wnt inhibitor SFRP1 in rheumatoid arthritis. Annals of the rheumatic diseases. 2011;70(8):1482–1488. doi: 10.1136/ard.2010.143040. [DOI] [PubMed] [Google Scholar]
- Turgeon N, Blais M, Delabre JF, Asselin C. The histone H3K27 methylation mark regulates intestinal epithelial cell density-dependent proliferation and the inflammatory response. J Cell Biochem. 2013;114(5):1203–1215. doi: 10.1002/jcb.24463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida H, Matsushita Y, Ueda H. Epigenetic regulation of BDNF expression in the primary sensory neurons after peripheral nerve injury: implications in the development of neuropathic pain. Neuroscience. 2013;240:147–154. doi: 10.1016/j.neuroscience.2013.02.053. [DOI] [PubMed] [Google Scholar]
- Wang Y, Liu C, Guo QL, Yan JQ, Zhu XY, Huang CS, Zou WY. Intrathecal 5-azacytidine inhibits global DNA methylation and methyl- CpG-binding protein 2 expression and alleviates neuropathic pain in rats following chronic constriction injury. Brain Res. 2011;1418:64–69. doi: 10.1016/j.brainres.2011.08.040. [DOI] [PubMed] [Google Scholar]
- Woolf CJ. Central sensitization: Implications for the diagnosis and treatment of pain. Pain. 2011;152(3, Supplement):S2–S15. doi: 10.1016/j.pain.2010.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288(5472):1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- Yadav R, Yan X, Maixner DW, Gao M, Weng HR. Blocking the GABA transporter GAT-1 ameliorates spinal GABAergic disinhibition and neuropathic pain induced by paclitaxel. J Neurochem. 2015;133(6):857–869. doi: 10.1111/jnc.13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaksh TL, Rudy TA. Analgesia mediated by a direct spinal action of narcotics. Science. 1976;192(4246):1357–1358. doi: 10.1126/science.1273597. [DOI] [PubMed] [Google Scholar]
- Yan X, Jiang E, Weng HR. Activation of toll like receptor 4 attenuates GABA synthesis and postsynaptic GABA receptor activities in the spinal dorsal horn via releasing interleukin-1 beta. Journal of neuroinflammation. 2015;12(1):4. doi: 10.1186/s12974-014-0222-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X, Weng HR. Endogenous interleukin-1β in neuropathic rats enhances glutamate release from the primary afferents in the spinal dorsal horn through coupling with presynaptic NMDA receptors. The Journal of biological chemistry. 2013;288(42):30544–30557. doi: 10.1074/jbc.M113.495465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X, Yadav R, Gao M, Weng HR. Interleukin-1 beta enhances endocytosis of glial glutamate transporters in the spinal dorsal horn through activating protein kinase C. Glia. 2014;62(7):1093–1109. doi: 10.1002/glia.22665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Taylor RJ, La Torre A, Wilken MS, Cox KE, Reh TA, Vetter ML. Ezh2 maintains retinal progenitor proliferation, transcriptional integrity, and the timing of late differentiation. Dev Biol. 2015;403(2):128–138. doi: 10.1016/j.ydbio.2015.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu XY, Huang CS, Li Q, Chang RM, Song ZB, Zou WY, Guo QL. p300 exerts an epigenetic role in chronic neuropathic pain through its acetyltransferase activity in rats following chronic constriction injury (CCI) Mol Pain. 2012;8:84. doi: 10.1186/1744-8069-8-84. [DOI] [PMC free article] [PubMed] [Google Scholar]