

Abstract
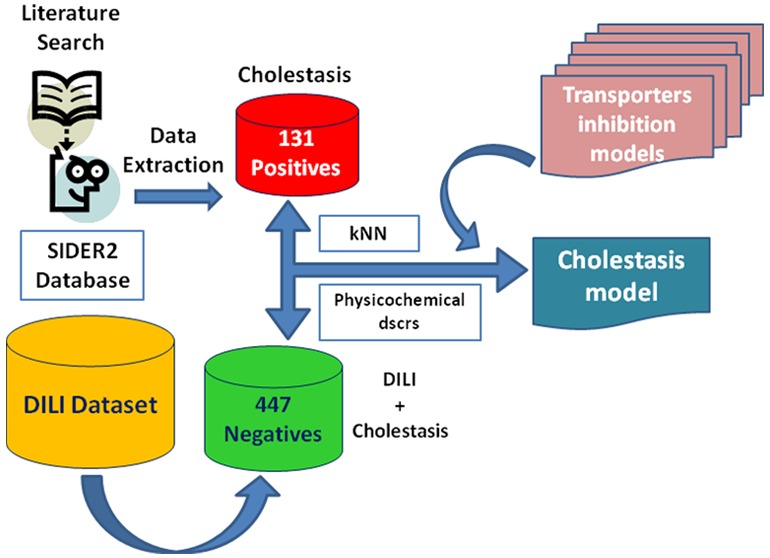
Cholestasis represents one out of three types of drug induced liver injury (DILI), which comprises a major challenge in drug development. In this study we applied a two-class classification scheme based on k-nearest neighbors in order to predict cholestasis, using a set of 93 two-dimensional (2D) physicochemical descriptors and predictions of selected hepatic transporters’ inhibition (BSEP, BCRP, P-gp, OATP1B1, and OATP1B3). In order to assess the potential contribution of transporter inhibition, we compared whether the inclusion of the transporters’ inhibition predictions contributes to a significant increase in model performance in comparison to the plain use of the 93 2D physicochemical descriptors. Our findings were in agreement with literature findings, indicating a contribution not only from BSEP inhibition but a rather synergistic effect deriving from the whole set of transporters. The final optimal model was validated via both 10-fold cross validation and external validation. It performs quite satisfactorily resulting in 0.686 ± 0.013 for accuracy and 0.722 ± 0.014 for area under the receiver operating characteristic curve (AUC) for 10-fold cross-validation (mean ± standard deviation from 50 iterations).
Introduction
Drug induced liver injury (DILI) is a major issue worldwide, both for patients and health providers.1,2 It is one of the primary causes for attrition during clinical and preclinical studies and the main reason for drug withdrawal from the market.3−6 DILI is divided into types, (i) hepatocellular, (ii) cholestatic, or (iii) mixed (hepatocellular and cholestatic), according to the type of liver damage and the clinical chemistry biomarker alterations.7 The cholestatic and mixed hepatocellular and cholestatic type are the two most severe manifestations of DILI and yield almost half of the recorded cases of DILI.8,9
Cholestatic liver injury, or more simply cholestasis, is the disruption of the bile flow, which might be either due to biliary tract obstruction or to complications in bile acid uptake. While the mechanistic basis for hepatocellular DILI is still a mystery for the majority of the cases, more knowledge exists for cholestatic DILI. There is growing evidence for a vast amount of cholestasis cases pinpointing the important role of hepatic transporters.10
Hepatic transporters are classified into basolateral and canalicular ones. Basolateral transporters are responsible for the uptake of drugs and other endobiotics and xenobiotics from the blood, influencing the exposure of the hepatocyte to potential damage. Canalicular transporters regulate the hepatic clearance, as well as the secretion of bile salts and bile conjugates into bile.10−15 Any disturbance of the transporters’ physiological function may result in the accumulation of potentially harmful bile products that can finally cause cholestasis.10Figure 1 provides an overview on the respective location of hepatocyte transporters.
Figure 1.
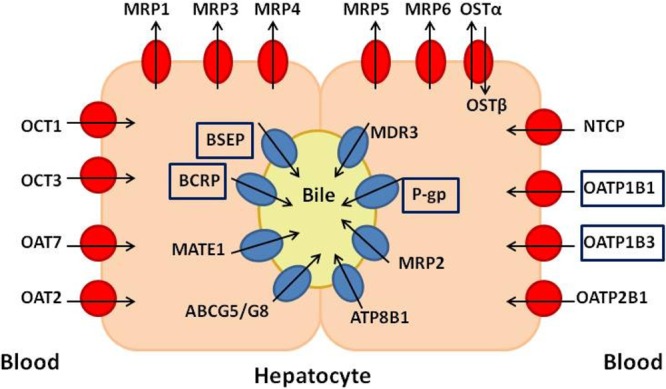
Transporters located on the hepatocyte. Blue symbols represent mainly the canalicular transporters, and red symbols, the basolateral ones. The arrows define the direction of transport. The transporters used in this study are presented within rectangular frames. MRP1–6 multidrug resistance-associated proteins 1–6, OSTα/OSTβ organic solute transporter, BSEP bile salt export pump, BCRP breast cancer resistance protein, MATE1 multidrug and toxin extrusion transporter 1, ABCG5/G8 ATP-binding cassette subfamily G member 5/8, MDR3 multidrug resistance protein 3, P-gp P-glycoprotein, ATP8B1 ATPase-aminophospholipid transporter, OATP organic anion transporting polypeptide, NTCP sodium (Na+) taurocolate cotransporting polypeptide, OCT organic cation transporter 1, OAT organic anion transporter.
Several transporters’ malfunctions have been associated with cholestasis. The most important one, due to its pivotal role in bile salts clearance, is the bile salt export pump (BSEP).8,10,16−21 Apart from BSEP, there is evidence for the implication of other canalicular efflux transporters such as the multidrug resistance-associated protein 2 (MRP2),8−10,22 breast cancer resistance protein (BCRP),8−10 multidrug resistance protein 3 (MDR3),8,10 and P-glycoprotein (P-gp).8−10 MDR3 functions as an ATP-dependent phospholipid flippase, translocating phosphatidylcholine from the inner to the outer canalicular membrane. Canalicular phospholipids are then solubilized by canalicular bile salts to form mixed micelles, protecting cholangiocytes from the detergent properties of bile salts. While P-gp is also not transporting bile salts, it is implicated in cholestasis because of its large amount of substrates and inhibitors which cause drug–drug interactions that disrupt the smooth function of the hepatocyte.10 The basolateral transporters play also an important role, both the uptake transporters, such as organic anion transporting polypeptides 1B1, 1B3, and 2B1 (OATP1B1, 1B3, and 2B1)8−10 and sodium (Na+) taurocolate cotransporter (NTCP),8−10,23,24 and the efflux transporters, like multidrug resistance-associated protein 38−10 and 48−10,20 (MRP3 and MRP4). In particular, in cases of cholestasis, the basolateral uptake transporters NTCP and OATP1B1 have been found down-regulated.10,11 However, in this case, OATP1B3 is up-regulated as a compensatory mechanism for the elimination of xenobiotics from sinusoidal blood.10,25,26 On the contrary, in cases of cholestasis, MRP3 and MRP4 are up-regulated to facilitate the efflux of the toxic bile salts out of the hepatocyte.27 Thus, simultaneous inhibition of several of these transporters could induce drug toxicity due to inadequate elimination from the blood or increase the cholestatic effect due to accumulation of bile salts in the hepatocyte.
Consequently, drug-induced liver injury and cholestasis are important toxicity alerts to be considered in drug development. Interestingly, there are only a few computational studies for the prediction of cholestasis reported in literature.28,29 With respect to the involvement of hepatic transporter, there are some in vitro studies correlating cholestasis with transporter inhibition, such as BSEP,17,18,30 MRP3, MRP4,20 and NTCP.31 Also several in silico studies for the identification of potentially cholestatic compounds via modeling of transporters and then associating them with the cholestatic effect of their inhibitors have been conducted. A characteristic example is the study by Greupink et al. in 2012, who developed a pharmacophore approach for NTCP24 in order to identify potentially NTCP inhibitors. Under the same principles, in 2014 Ritschel and colleagues71 performed a 3D ligand-based pharmacophore model for BSEP inhibition. However, in most of these cases the amount of validated drugs is small and what is basically described is the association between transporter inhibition and cholestasis. Thus, as the respective is associated with cholestasis, it is assumed that an inhibitor is causing cholestasis. Most recently, Muller et al.,32 in order to model DILI, also modeled some more hepatotoxicity end points, including cholestasis. Moreover, Mulliner et al.33 presented a multilevel modeling approach for DILI, where cholestasis was also included as a morphological hepatobiliary finding. However, examining the liver transporters contribution was not within the scope of their work. Finally, it is noteworthy to mention some multiscale modeling approaches for DILI. DILIsym (www.dilisym.com) is a mechanistic mathematical model of DILI, that has been used to investigate the effects of BSEP inhibition on drug-induced liver injury,34 as well as on bile acid-mediated DILI.35 Additionally, Sluka et al. developed a multiscale, liver-centric in silico modeling framework for acetaminophen pharmacology and metabolism that can be extended in predicting hepatotoxicity due to acetaminophen overdosing.36
In this study we present a classification scheme in order to predict cholestasis from a public data set, using physicochemical descriptors as well as predicted transporter inhibition profiles. For the latter we used our in house classification models for BSEP,37 BCRP,38 P-gp,38 OATP1B1, and OATP1B3.39
Methods
Data Compilation
Training Set
For compiling the DILI training data set we searched in PubMed (http://www.ncbi.nlm.nih.gov/pubmed),40,41 Google,42 Scopus (https://www.scopus.com/),43 and the SIDER database v244,45 using the search terms: “drug-induced cholestasis” or “cholestasis”. The retrieved publications8,31,46,47 were then investigated manually for human data, i.e. compounds that are positive or negative for drug-induced cholestasis in humans. Those compounds—in principle drugs—were added to those obtained from the SIDER v244,45 database. Unfortunately, cholestasis is an end point that is not widely examined in terms of experimental or in silico studies that would potentially guide us to big data sets. Thus, even though we were able to compile several drugs positive for cholestasis, there was almost no information in terms of the negatives. On the other hand, DILI in general is studied quite extensively and there are several respective data sets. Since choleastasis is a possible manifestation for DILI, we can consider safely that any compound negative for DILI will definitely be also negative for cholestasis. Thus, the negative compounds for DILI that we had compiled and curated in a previous work were also used as negatives for this study. The data set was carefully curated according to the following rules:
All inorganic compounds were removed based on their chemical formula in MOE 2014.09.48
Salt parts and compounds containing metals and/or rare or special atoms were removed and the chemical structures were standardized using the Standardiser tool created by Francis Atkinson.49
Duplicates and permanently charged compounds were removed using MOE 2014.09.48 Here we have to note that stereoisomers, even if biologically can be considered as different compounds, were considered as duplicates, since they give the exactly same vector of descriptors. If two (or more) stereoisomers are of the same class, only one was kept. If they were of different classes, they were all removed.
3D structures were generated using CORINA (version 3.4),50 and their energy was minimized with MOE 2014.09,48 using default settings, but changing the gradient to 0.05 RMS kcal/(mol A2). In addition, the existing chirality was preserved.
After these curation steps 152 compounds remained as positives for cholestasis. The negatives for DILI, and subsequently for cholestasis, were 466 compounds. However, when uniting the data, there were compounds with contradictory class assignments. These compounds were removed from the data set, yielding a data set of in total 578 compounds (131 positives and 447 negatives). The compiled data set is provided in the Supporting Information.
External Test Set
Recently—and after having already compiled our training set for cholestasis and developed the respective model—a data set covering multiple levels of hepatotoxicity was published by Mulliner and co-workers.33 The data are hierarchically clustered by the authors into three levels of hepatotoxicity: level 0 corresponds to general hepatotoxicity, level 1 corresponds to clinical chemistry findings and morphological finding as distinguished parts of general hepatotoxicity, and level 2 discriminates both clinical chemistry and morphological findings into hepatocellular and hepatobiliary injury. We use only clinical data, i.e. the human data, of morphological findings for hepatobiliary injury as an external test set for validating the developed cholestasis model. Once more, we performed chemical data curation and removed the compounds overlapping with the training set, which led to 1347 compounds (230 positives and 1117 negatives) as an external test set.
Merged Data Set
We also merged the training with the test set and tried to generate a model based on the united data. The merged data set comprises 1904 compounds: 355 positives and 1549 negatives for cholestasis. Here we must note that the total number of compounds in the merged data set is not the same as the sum of the compounds of training set and test set. The reason is that when removing the overlapping compounds between training set and test set, all of the compounds were removed from the test set, since we had already selected the final model for cholestasis. From those overlapping compounds, 21 had contradictory class labels. This did not matter so much, since they were totally removed from the test set. However, when merging the two data sets for modeling, we did not want to decide regarding the class of those compounds.
Generation of Statistical Models
Molecular Descriptors
For both data sets, several types of molecular descriptors have been calculated, such as all 192 2D MOE descriptors, the 3D VolSurf series of descriptors,32 as well as ECFPs (extended connectivity fingerprints; ECFP6), using RDKit (http://www.rdkit.org/).51 The list of the final descriptors used for the proposed model is given in the Supporting Information (Table S1). In addition to this, predicted hepatic transporter inhibition profiles were also included in the list of descriptors. The transporters investigated comprise BSEP, P-gp, BCRP, OATP1B1, and OATP1B3.
In particular, for basolateral transporters we calculated the predictions for four in silico classification models built upon PaDEL descriptors52 for OATP1B1 and OATP1B339 inhibition. For obtaining the predictions we use the models’ version implemented in eTOXlab,53 an open source modeling framework for implementing predictive models. Out of each model we got a binary result: positive or negative. For each transporter we use the sum of these binary scores, denoted “Sum binary score”. The sum binary score can take values between 0 (if all models predict the compound as negative) and 4 (if all models predict the compound as positive). For basolateral transporters, we used the continuous score obtained by the BSEP37 inhibition prediction model. Float prediction-scores were also retrieved for P-glycoprotein38 and BCRPinhibition.38 A more thorough description of the transporters inhibition models is provided in the Supporting Information (Table S2), where the size of the training set, the inhibition threshold of the training set, and the algorithm and performance of each individual model based on AUC values are provided.
Algorithms Used
The two-class classification models were built using the software package WEKA (version 3.7.12).54 We investigated the performance of several base classifiers, such as logistic regression, tree methods (random forest and J48 tree), support vector machines (SMO in WEKA with polynomial, RBF, and Puk kernels), naïve Bayes, and k-nearest neighbors. Moreover, because the data set is slightly imbalanced, in order to equilibrate the effect of the majority class on model performance, we also applied the cost-sensitive meta-classifier MetaCost.55 The cost matrix applied corresponds to the imbalance ratio of the data (3:1). Additionally, several meta-classifiers were explored for attribute selection (AttributeSelectedClassifier), as well as for improving the statistical performance, such as Bagging56 and Boosting.57,58
Model Validation
The models were originally validated via 10-fold cross validation, which is considered a quite trustworthy method of validation.59 The best models—according to 10-fold cross-validation evaluation—were further validated via using the data set by Mulliner.33 Subsequently, for the best obtained models, 50 iterations were performed by changing the cross-validation seed (for splitting the data within cross validation) and the respective performance parameters were calculated. In order to compare whether the inclusion of the transporters predictions in the descriptors set improves significantly the model’s performance, a two-sample t test was performed in R.60
The statistics metrics taken into consideration were accuracy, sensitivity, specificity, Matthews correlation coefficient (MCC), area under the curve (AUC), precision and weighted average precision. Apart from the conventional metrics of accuracy, sensitivity, and specificity, we also use AUC, since it is a measure of the models capability to rank the predictions, while it decouples classifiers from class imbalance and error costs. Moreover receiver operating characteristic (ROC) graphs are very useful for visualization of the models results.61 The MCC value, considering its formula, takes into account all values of the confusion matrix:
Where, TP is true positives, FP is false positives, TN is true negatives, and FN is false negatives. Thus, it is considered more balanced and informative than the column- or linewise metrics.62 Weighted average precision is the average precision obtained for the two classes but weighted from the total number of instances of the classes.54 It is a quite helpful parameter in multiclass classification problems, as well as for imbalanced data sets where the number of negatives is greater than the number of positives. Especially for the latter case, due to the definition of precision [PPV = TP/(TP + FP)], its value for the positive class would be low, which not necessarily means that the total performance of the model is bad. Of course, since we are dealing with a toxicity classification problem, like cholestasis, the metrics that is of particular interest and that should by no means drop below 0.5 is sensitivity or true positive rate.
Defining Applicability Domain of the Models
In order to be confident regarding the validity of the models we used, we investigated the coverage of the transporters models for the cholestasis data. Additionally, we checked how reliable the predictions of the cholestasis model for the cholestasis test set are. The applicability domain was checked on KNIME with the Enalos nodes63,64 that compute the applicability domain on the basis of the Euclidean distances.65 The number of compounds within the model’s applicability domain for each model and for each cholestasis data set is provided in the Supporting Information (Table S3).
Results and Discussion
Generation of a Cholestasis Classification Model
Several combinations of descriptors and classifiers were investigated and the optimal classification model was selected on the basis of the results of 10-fold cross validation. With respect to the classifier, the best results were obtained using as base classifier IBk—the k-nearest neighbors implementation in WEKA—with k = 5. The meta-classifier MetaCost was also applied, with the application of the cost matrix [0.0, 1.0; 3.0, 0.0], i.e. weighting the minority class 3 times more than the majority class, in order to cope with the slightly imbalanced training set. 2D MOE descriptors were performing better than fingerprints and/or VolSurf descriptors, especially for sensitivity, MCC and AUC. Combining the VolSurf descriptors with 2D MOE descriptors also did not provide any significant improvement of the results. From the whole set of 2D MOE descriptors we decided to use a subset of 93 interpretable descriptors that give almost the same performance compared to using all 2D MOE descriptors. Apart from the 93 2D descriptors, we also included the predicted transporter inhibition profiles. In order to assess the importance and significance of this additional information individually, we used them in different combinations: all transporters, only BSEP, all transporters excluding either BSEP, or P-gp, or BCRP, or the OATPs. This led to in total seven models (Table 1).
Table 1. Performance of the Model for MetaCost [0.0, 1.0; 3.0, 0.0] + IBk (k = 5), Changing the Descriptor Settings via Including or Excluding Particular Transporters.
| model settings | validation | accuracy | sensitivity | specificity | MCC | AUC | precision | weighted average precision |
|---|---|---|---|---|---|---|---|---|
| 93 2D MOE dscrs | 10 CV | 0.656 | 0.496 | 0.702 | 0.175 | 0.657 | 0.328 | 0.713 |
| test set | 0.611 | 0.609 | 0.611 | 0.168 | 0.617 | 0.244 | 0.774 | |
| 93 2D MOE dscrs + all transporters pred. | 10 CV | 0.704 | 0.611 | 0.732 | 0.301 | 0.726 | 0.400 | 0.760 |
| test set | 0.564 | 0.591 | 0.559 | 0.113 | 0.585 | 0.216 | 0.758 | |
| 93 2D MOE dscrs + BSEP pred | 10 CV | 0.682 | 0.481 | 0.740 | 0.200 | 0.665 | 0.352 | 0.721 |
| test set | 0.629 | 0.517 | 0.652 | 0.131 | 0.604 | 0.234 | 0.760 | |
| 93 2D MOE dscrs + all transporters pred without BSEP | 10 CV | 0.683 | 0.603 | 0.707 | 0.270 | 0.702 | 0.376 | 0.749 |
| test set | 0.558 | 0.587 | 0.551 | 0.104 | 0.595 | 0.212 | 0.755 | |
| 93 2D MOE dscrs + all transporters pred without P-gp | 10 CV | 0.725 | 0.522 | 0.766 | 0.241 | 0.706 | 0.315 | 0.789 |
| test set | 0.572 | 0.574 | 0.571 | 0.110 | 0.589 | 0.216 | 0.756 | |
| 93 2D MOE dscrs + all transporters pred without BCRP | 10 CV | 0.696 | 0.618 | 0.718 | 0.294 | 0.719 | 0.391 | 0.758 |
| test set | 0.569 | 0.604 | 0.561 | 0.125 | 0.599 | 0.221 | 0.762 | |
| 93 2D MOE dscrs + all transporters pred. without OATPs | 10 CV | 0.654 | 0.496 | 0.700 | 0.173 | 0.660 | 0.327 | 0.713 |
| test set | 0.612 | 0.522 | 0.63 | 0.117 | 0.606 | 0.225 | 0.756 |
It is interesting to mention that we also exchanged the training-test set roles between the two data sets and tried to generate a model for cholestasis based on the bigger data set from the work of Mulliner et al.33 However, even though the results for 10-fold cross validation were equivalent to the model we propose (generated on the compiled training set of 578 compounds), the results for the external validation of the 578 compounds were rather poor (results not shown). Thus, we decided to stay with our original model.
Furthermore, we tried to generate a cholestasis model on the merged data for the subset of 93 2D MOE descriptors, with or without the transporters interaction profiles. Interestingly, the k-nearest neighbor settings (k = 5), which gave quite satisfactory results for 10-fold cross validation while modeling either the training or the test set standalone, did not have the same effect for the united data. For the merged data set SVM (SMO implementation in WEKA) using a polynomial kernel, with exponent equal to 2, performs better. The use of MetaCost with a cost matrix of [0.0, 1.0; 5.0, 0.0], due to the new imbalance ratio of the data, is also necessary. Additionally, under these settings, the performance of the model is significantly better when using the transporters predictions as additional descriptors. The obtained performance of this model, as well as the respective p-values of the performed two-sample paired t test out of 50 iterations, is presented in the Supporting Information (Table S4).
Inspecting the obtained results in Table 1, it becomes obvious that the best settings for the model for 10-fold cross validation are achieved with the inclusion of all transporter inhibition predictions in the list of descriptors. Nevertheless, this is not the case for the external validation, where including predicted inhibitor profiles for all transporters yields lower accuracy and specificity values, while sensitivity remains almost the same. Interestingly, the use of BSEP inhibition prediction stand-alone does not seem to be sufficient. There is a drop in the statistics—especially for sensitivity—in comparison to the use of the whole set of transporter predictions, both for 10-fold cross-validation and for the external test set.
Statistical Analysis of Transporter Predictions on the Model’s Performance
In order to assess if the predicted transporter inhibition profiles indeed statistically significantly improve the models, we performed 50 iterations of 10-fold cross validation followed by a two sample t test on the performance parameters. For this we used the models with 2D MOE descriptors, 2D MOE plus all transporters, 2D MOE plus BSEP, and 2D MOE plus all transporters without BSEP (Table 2).
Table 2. Mean Standard Deviation Values Obtained from 50 Iterations of 10-fold Cross-Validation for the Statistics Metrics of Accuracy, Sensitivity, Specificity, MCC, AUC, Precision, and Weighted Average Precision.
| model settings | accuracy | sensitivity | specificity | MCC | AUC | precision | weighted average precision | |
|---|---|---|---|---|---|---|---|---|
| 93 2D MOE dscrs | mean | 0.657 | 0.523 | 0.697 | 0.192 | 0.661 | 0.336 | 0.720 |
| sd | ±0.011 | ±0.030 | ±0.012 | ±0.027 | ±0.015 | ±0.015 | ±0.010 | |
| 93 2D MOE dscrs + all transporters pred | mean | 0.686 | 0.621 | 0.706 | 0.284 | 0.722 | 0.382 | 0.755 |
| sd | ±0.013 | ±0.030 | ±0.015 | ±0.027 | ±0.014 | ±0.016 | ±0.011 | |
| 93 2D MOE dscrs + BSEP pred | mean | 0.669 | 0.508 | 0.716 | 0.199 | 0.660 | 0.345 | 0.722 |
| sd | ±0.012 | ±0.029 | ±0.015 | ±0.025 | ±0.014 | ±0.014 | ±0.009 | |
| 93 2D MOE dscrs + all transporters pred without BSEP | mean | 0.677 | 0.632 | 0.690 | 0.278 | 0.707 | 0.374 | 0.754 |
| sd | ±0.013 | ±0.031 | ±0.015 | ±0.028 | ±0.015 | ±0.015 | ±0.011 |
Analyzing the p-values for the pairwise comparisons (Supporting Information Table S5) the main conclusion is that indeed the use of liver transporter inhibition predictions contributes significantly to the models performance when compared to the use of only 2D physicochemical descriptors. Interestingly, it is not only the BSEP inhibition contribution, which matters. On the contrary, when only BSEP predictions are used, there is only a slight increase in sensitivity and specificity of the model, while sensitivity decreases. The other way round, if all transporters predictions are used, sensitivity increases, but accuracy and specificity reach their peak only after the inclusion of BSEP predictions. This suggests that BSEP apparently contains important information. Nevertheless, it does not contain all the important information, despite the fact that it is the most widely discussed transporter in literature with respect to cholestasis.8,10,16−21 A possible explanation for this interesting result could be the different thresholds required for BSEP inhibition to cause cholestasis versus the threshold of the BSEP inhibition model. For the BSEP model, every compounds with an IC50 > 50 μM was classified as noninhibitors, while IC50 < 10 μM was the threshold for characterizing a compound as inhibitor. However, for the development of cholestasis, an IC50 < 300 μM has been reported as sufficient.17 Thus, potentially predicted noninhibitors could actually be cholestatic compounds. The other transporters included in our study are in general not well described in literature via experimental procedures, but they are rather pinpointed due to the fact that they are transporting bile salts or bile conjugates (with the exception of P-gp, whose role is attributed mainly to drug–drug interactions). Thus, our study gives extra weight to literature indications concerning BCRP, P-gp, OATP1B1, and 1B3.
It was quite curious that even though the transporters predictions significantly increase the performance both for the model built on the training set of 578 compounds and for the model trained with the merged data set of 1904 compounds (but with a different base classifier); this was not the case for the prediction on the test set. The performance of all statistics metrics decrease, when transporters predictions are used as descriptors. We are unable to provide a solid explanation for this phenomenon. We can only speculate a different way of class assignment between the two data sets, since they are coming from different sources. Another plausible explanation could be the fact that the external validation set had a contradiction rate of almost 20% regarding the class labels of those compounds shared with the training set (71 out of 419 shared compounds had contradictory class labels). We assume that this is due to the drawbacks of the toxicity reporting system: under-reporting,66−68 voluntarily basis,68−70 difficulties to obtain the data, which are often proprietary,66 as well as the lack of the prerequisite of a causal relationship between drug and adverse event.68 In any case, despite these contradictions between the training and the test set, the model retained its satisfactory performance.
Furthermore, we would like to mention that there is also experimental evidence for the implication of the basolateral efflux transporters MRP3 and MRP4,8−10,20 as well as for the canalicular efflux transporter MRP2.8−10,22 We are aware of this fact, but for these transporters there are currently not sufficient data available to develop high quality models that can be further used for contributing to our cholestasis model. Additionally, we should pinpoint the fact that we are using predictions for the inhibition of transporters rather than real in vitro data. This could potentially accumulate some additional error in the final predictions of the cholestasis model. In any case, despite any deteriorating factors, our final in silico models for cholestasis were extensively validated with 10-fold cross validation and statistical tests. Furthermore, the external validation set was of a significant size being even bigger than the training set. For both cases the results were quite satisfactory. Moreover, for both training sets (regular and merged one) there is a trend indicating the importance of transporter predictions in the development of cholestasis. The performance of the classification models from which we obtained the predictions provides us with enough confidence to use them in our input matrix. Moreover, the percentage of the cholestasis data that are within the applicability domain of the transporters models ranges between 93.1% and 99.5% (Table S3, Supporting Information), which is quite satisfactory and further enhances our confidence in using predicted transporter interaction profiles as descriptors.
Conclusions
In this study we present a two-class classification model for the prediction of cholestasis (or cholestatic DILI) based on a public data set of 578 compounds. The model is incorporating information both from 2D physicochemical descriptors, as well as predictions of inhibition of the hepatic transporters BSEP, BCRP, P-gp, OATP1B1, and OATP1B3. The performance of the resulting model is rather satisfactory and is validated both via 50 iterations of different 10-fold cross validations, as well as an external test set of over 1300 compounds.33 Our results demonstrate that adding transporter predictions as additional descriptors to the list of 2D physicochemical descriptors is improving model performance. This is in alignment with evidence from literature which shows that inhibition of selected hepatic transporters contributes to cholestasis.
Interestingly, the increase in model performance cannot be attributed solely to BSEP inhibition, which is the transporter that is most correlated to cholestasis in literature. The performance increases only when the whole list of transporter inhibition predictions is included. This result points toward a rather synergistic effect of several transporters, including the less elucidated role of OATPs, BCRP, and P-gp in cholestasis.
Our study is the first of its kind regarding combining physicochemical descriptors and predicted transporter information in order to predict cholestasis. This provides a useful extension to previous approaches for the prediction of cholestasis.
Acknowledgments
The research leading to these results has received support from the Innovative Medicines Initiative Joint Undertaking under grant agreements No. 115002 (eTOX) resources of which are composed of financial contribution from the European Union’s Seventh Framework Programme (FP7/2007-2013) and EFPIA companies’ in kind contribution. We also acknowledge financial support provided by the Austrian Science Fund, Grant F3502. We are thankful to ChemAxon (https://www.chemaxon.com/) for providing us with an Academic License of Marvin Suite. Marvin was used for drawing, displaying, and characterizing chemical structures, substructures, and reactions, Marvin 6.1.3., 2013, ChemAxon (http://www.chemaxon.com). We are thankful to Dr. Alexander Amberg from Sanofi-Aventis Deutschland GmbH, coauthor of the Mulliner et al. publication, for providing us with the Supporting Information before it was available online from the journal. Finally, E.K. is cordially thankful to Dr. Floriane Montanari for the fruitful discussions throughout the project and her useful feedback from revising the manuscript.
Glossary
ABBREVIATIONS
- AUC
area under the ROC curve
- BCRP
breast cancer resistance protein
- cpd(s)
compound(s)
- DILI
drug-induced liver injury
- dscr(s)
descriptors
- MCC
Matthews correlation coefficient
- MDR3
multidrug resistance protein 3
- MRP2
multidrug resistance-associated protein 2
- MRP3
multidrug resistance-associated protein 3
- OATP1B1
organic anion transporting polypeptide 1B1
- OATP1B3
organic anion transporting polypeptide 1B3
- P-gp
P-glycoprotein
- transp
transporters
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jcim.6b00518.
List with the subset of 93 2D MOE descriptors (Table S1), a brief description of the models used for the transporters predictions (Table S2), the level of applicability domain coverage of the transporters models on cholestasis data and the cholestasis models for the cholestasis test set (Table S3), the performance of the model trained on the merged data set of 1904 compounds (Table S4), and the p-values of the pairwise statistical comparison of the models (Table S5) for 10-fold cross validation (PDF)
training set, external test set, and merged data set for cholestasis (chemical structures, compound names, and descriptors) provided in .csv format (ZIP)
Author Contributions
E.K. performed all statistical analyses and contributed to writing. G.F.E. designed the study and contributed to writing. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Holt M. P.; Ju C. Mechanisms of Drug-Induced Liver Injury. AAPS J. 2006, 8 (1), E48–54. 10.1208/aapsj080106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins P. B.; Seeff L. B. Drug-Induced Liver Injury: Summary of a Single Topic Clinical Research Conference. Hepatology 2006, 43 (3), 618–31. 10.1002/hep.21095. [DOI] [PubMed] [Google Scholar]
- O’Brien P. J.; Irwin W.; Diaz D.; Howard-Cofield E.; Krejsa C. M.; Slaughter M. R.; Gao B.; Kaludercic N.; Angeline A.; Bernardi P.; et al. High Concordance of Drug-Induced human Hepatotoxicity with in Vitro Cytotoxicity Measured in a Novel Cell-Based Model Using High Content Screening. Arch. Toxicol. 2006, 80 (9), 580–604. 10.1007/s00204-006-0091-3. [DOI] [PubMed] [Google Scholar]
- Ballet F. Hepatotoxicity in Drug Development: Detection, Significance and Solutions. J. Hepatol. 1997, 26, 26–36. 10.1016/S0168-8278(97)80494-1. [DOI] [PubMed] [Google Scholar]
- Chen M.; Vijay V.; Shi Q.; Liu Z.; Fang H.; Tong W. FDA-Approved Drug Labeling for the Study of Drug-Induced Liver Injury. Drug Discovery Today 2011, 16 (15–16), 697–703. 10.1016/j.drudis.2011.05.007. [DOI] [PubMed] [Google Scholar]
- Regev A. Drug-Induced Liver Injury and Drug Development: Industry Perspective. Semin. Liver Dis. 2014, 34 (2), 227–39. 10.1055/s-0034-1375962. [DOI] [PubMed] [Google Scholar]
- Benichou C. Criteria of Drug-Induced Liver Disorders. Report of an International Consensus Meeting. J. Hepatol. 1990, 11 (2), 272–6. 10.1016/0168-8278(90)90124-A. [DOI] [PubMed] [Google Scholar]
- Padda M. S.; Sanchez M.; Akhtar A. J.; Boyer J. L. Drug-Induced Cholestasis. Hepatology 2011, 53 (4), 1377–87. 10.1002/hep.24229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K.; Kock K.; Sedykh A.; Tropsha A.; Brouwer K. L. An Updated Review on Drug-Induced Cholestasis: Mechanisms and Investigation of Physicochemical Properties and Pharmacokinetic Parameters. J. Pharm. Sci. 2013, 102 (9), 3037–57. 10.1002/jps.23584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauli-Magnus C.; Meier P. J. Hepatobiliary Transporters and Drug-Induced Cholestasis. Hepatology 2006, 44 (4), 778–87. 10.1002/hep.21359. [DOI] [PubMed] [Google Scholar]
- Faber K. N.; Müller M.; Jansen P. L. M. Drug Transport Proteins in the Liver. Adv. Drug Delivery Rev. 2003, 55 (1), 107–124. 10.1016/S0169-409X(02)00173-4. [DOI] [PubMed] [Google Scholar]
- Roth M.; Obaidat A.; Hagenbuch B. OATPs, OATs and OCTs: The Organic Anion and Cation Transporters of the SLCO and SLC22A Gene Superfamilies. Br. J. Pharmacol. 2012, 165 (5), 1260–87. 10.1111/j.1476-5381.2011.01724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roma M. G.; Crocenzi F. A.; Mottino A. D. Dynamic Localization of Hepatocellular Transporters in Health and Disease. World J. Gastroenterol 2008, 14 (44), 6786–801. 10.3748/wjg.14.6786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacomini K. M.; Huang S. M.; Tweedie D. J.; Benet L. Z.; Brouwer K. L.; Chu X.; Dahlin A.; Evers R.; Fischer V.; Hillgren K. M.; et al. Membrane Transporters in Drug Development. Nat. Rev. Drug Discovery 2010, 9 (3), 215–36. 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halilbasic E.; Claudel T.; Trauner M. Bile Acid Transporters and Regulatory Nuclear Receptors in the Liver and Beyond. J. Hepatol. 2013, 58 (1), 155–68. 10.1016/j.jhep.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J.; Vandeberg J. L. Hepatobiliary Transport in Health and Disease. Clin Lipidol 2012, 7 (2), 189–202. 10.2217/clp.12.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson S.; Stahl S.; Paul N.; Barber J.; Kenna J. G. In Vitro Inhibition of the Bile Salt Export Pump Correlates with Risk of Cholestatic Liver Injury in Humans. Drug Metab. Dispos. 2012, 40 (1), 130–8. 10.1124/dmd.111.040758. [DOI] [PubMed] [Google Scholar]
- Kis E.; Ioja E.; Rajnai Z.; Jani M.; Mehn D.; Heredi-Szabo K.; Krajcsi P. BSEP Inhibition: In Vitro Screens to Assess Cholestatic Potential of Drugs. Toxicol. In Vitro 2012, 26 (8), 1294–9. 10.1016/j.tiv.2011.11.002. [DOI] [PubMed] [Google Scholar]
- Ogimura E.; Sekine S.; Horie T. Bile Salt Export Pump Inhibitors are Associated with Bile Acid-Dependent Drug-Induced Toxicity in Sandwich-Cultured Hepatocytes. Biochem. Biophys. Res. Commun. 2011, 416 (3–4), 313–7. 10.1016/j.bbrc.2011.11.032. [DOI] [PubMed] [Google Scholar]
- Köck K.; Ferslew B. C.; Netterberg I.; Yang K.; Urban T. J.; Swaan P. W.; Stewart P. W.; Brouwer K. L. Risk Factors for Development of Cholestatic Drug-Induced Liver Injury: Inhibition of Hepatic Basolateral Bile Acid Transporters Multidrug Resistance-Associated Proteins 3 and 4. Drug Metab. Dispos. 2014, 42 (4), 665–74. 10.1124/dmd.113.054304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner D. J.; Chen H.; Cantin L. D.; Kenna J. G.; Stahl S.; Walker C. L.; Noeske T. Mitigating the Inhibition of Human Bile Salt Export Pump by Drugs: Opportunities Provided by Physicochemical Property Modulation, in Silico Modeling, and Structural Modification. Drug Metab. Dispos. 2012, 40 (12), 2332–41. 10.1124/dmd.112.047068. [DOI] [PubMed] [Google Scholar]
- Payen L.; Sparfel L.; Courtois A.; Vernhet L.; Guillouzo A.; Fardel O. The Drug Efflux Pump MRP2: Regulation of Expression in Physiopathological Situations and by Endogenous and Exogenous Compounds. Cell Biol. Toxicol. 2002, 18 (4), 221–33. 10.1023/A:1016020626941. [DOI] [PubMed] [Google Scholar]
- Erlinger S. NTCP Deficiency: a New Inherited Disease of Bile Acid Transport. Clin. Res. Hepatol. Gastroenterol. 2015, 39 (1), 7–8. 10.1016/j.clinre.2014.07.011. [DOI] [PubMed] [Google Scholar]
- Greupink R.; Nabuurs S. B.; Zarzycka B.; Verweij V.; Monshouwer M.; Huisman M. T.; Russel F. G. In Silico Identification of Potential Cholestasis-Inducing Agents via Modeling of Na(+)-Dependent Taurocholate Cotransporting Polypeptide Substrate Specificity. Toxicol. Sci. 2012, 129 (1), 35–48. 10.1093/toxsci/kfs188. [DOI] [PubMed] [Google Scholar]
- Alrefai W. A.; Gill R. K. Bile Acid Transporters: Structure, Function, Regulation and Pathophysiological Implications. Pharm. Res. 2007, 24 (10), 1803–23. 10.1007/s11095-007-9289-1. [DOI] [PubMed] [Google Scholar]
- Hagenbuch B.; Meier P. Organic Anion Transporting Polypeptides of the OATP/SLC21 Family: Phylogenetic Classification as OATP/SLCO Superfamily, New Nomenclature and Molecular/Functional Properties. Pfluegers Arch. 2004, 447 (5), 653–665. 10.1007/s00424-003-1168-y. [DOI] [PubMed] [Google Scholar]
- Roma M. G.; Crocenzi F. A.; Sanchez Pozzi E. A. Hepatocellular Transport in Acquired Cholestasis: New Insights into Functional, Regulatory and Therapeutic Aspects. Clin. Sci. 2008, 114 (9), 567–88. 10.1042/CS20070227. [DOI] [PubMed] [Google Scholar]
- Chen M.; Bisgin H.; Tong L.; Hong H.; Fang H.; Borlak J.; Tong W. Toward Predictive Models for Drug-Induced Liver Injury in Humans: Are We There Yet?. Biomarkers Med. 2014, 8 (2), 201–13. 10.2217/bmm.13.146. [DOI] [PubMed] [Google Scholar]
- Ekins S. Progress in Computational Toxicology. J. Pharmacol. Toxicol. Methods 2014, 69 (2), 115–40. 10.1016/j.vascn.2013.12.003. [DOI] [PubMed] [Google Scholar]
- Byrne J. A.; Strautnieks S. S.; Mieli-Vergani G.; Higgins C. F.; Linton K. J.; Thompson R. J. The Human Bile Salt Export Pump: Characterization of Substrate Specificity and Identification of Inhibitors. Gastroenterology 2002, 123 (5), 1649–58. 10.1053/gast.2002.36591. [DOI] [PubMed] [Google Scholar]
- Mita S.; Suzuki H.; Akita H.; Hayashi H.; Onuki R.; Hofmann A. F.; Sugiyama Y. Inhibition of Bile Acid Transport across Na+/Taurocholate Cotransporting Polypeptide (SLC10A1) and Bile Salt Export Pump (ABCB 11)-Coexpressing LLC-PK1 Cells by Cholestasis-Inducing Drugs. Drug Metab. Dispos. 2006, 34 (9), 1575–81. 10.1124/dmd.105.008748. [DOI] [PubMed] [Google Scholar]
- Muller C.; Pekthong D.; Alexandre E.; Marcou G.; Horvath D.; Richert L.; Varnek A. Prediction of Drug Induced Liver Injury Using Molecular and Biological Descriptors. Comb. Chem. High Throughput Screening 2015, 18 (3), 315–22. 10.2174/1386207318666150305144650. [DOI] [PubMed] [Google Scholar]
- Mulliner D.; Schmidt F.; Stolte M.; Spirkl H. P.; Czich A.; Amberg A. Computational Models for Human and Animal Hepatotoxicity with a Global Application Scope. Chem. Res. Toxicol. 2016, 29 (5), 757–67. 10.1021/acs.chemrestox.5b00465. [DOI] [PubMed] [Google Scholar]
- Woodhead J. L.; Yang K.; Siler S. Q.; Watkins P. B.; Brouwer K. L.; Barton H. A.; Howell B. A. Exploring BSEP Inhibition-Mediated Toxicity with a Mechanistic Model of Drug-Induced Liver Injury. Front. Pharmacol. 2014, 5, 240. 10.3389/fphar.2014.00240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhead J. L.; Yang K.; Brouwer K. L.; Siler S. Q.; Stahl S. H.; Ambroso J. L.; Baker D.; Watkins P. B.; Howell B. A. Mechanistic Modeling Reveals the Critical Knowledge Gaps in Bile Acid-Mediated DILI. CPT: Pharmacometrics Syst. Pharmacol. 2014, 3, e123. 10.1038/psp.2014.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluka J. P.; Fu X.; Swat M.; Belmonte J. M.; Cosmanescu A.; Clendenon S. G.; Wambaugh J. F.; Glazier J. A. A Liver-Centric Multiscale Modeling Framework for Xenobiotics. PLoS One 2016, 11 (9), e0162428. 10.1371/journal.pone.0162428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montanari F.; Pinto M.; Khunweeraphong N.; Wlcek K.; Sohail M. I.; Noeske T.; Boyer S.; Chiba P.; Stieger B.; Kuchler K.; et al. Flagging Drugs That Inhibit the Bile Salt Export Pump. Mol. Pharmaceutics 2016, 13 (1), 163–71. 10.1021/acs.molpharmaceut.5b00594. [DOI] [PubMed] [Google Scholar]
- Schwarz T.; Montanari F.; Cseke A.; Wlcek K.; Visvader L.; Palme S.; Chiba P.; Kuchler K.; Urban E.; Ecker G. F. Subtle Structural Differences Trigger Inhibitory Activity of Propafenone Analogues at the Two Polyspecific ABC Transporters: P-Glycoprotein (P-gp) and Breast Cancer Resistance Protein (BCRP). ChemMedChem 2016, 11 (12), 1380–94. 10.1002/cmdc.201500592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotsampasakou E.; Brenner S.; Jäger W.; Ecker G. F. Identification of Novel Inhibitors of Organic Anion Transporting Polypeptides 1B1 and 1B3 (OATP1B1 and OATP1B3) Using a Consensus Vote of Six Classification Models. Mol. Pharmaceutics 2015, 12 (12), 4395–404. 10.1021/acs.molpharmaceut.5b00583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Home-PubMed-NCBI. http://www.ncbi.nlm.nih.gov/pubmed (accessed December 20, 2015).
- Wang Y.; Suzek T.; Zhang J.; Wang J.; He S.; Cheng T.; Shoemaker B. A.; Gindulyte A.; Bryant S. H. PubChem. BioAssay: 2014 update. Nucleic Acids Res. 2014, 42, D1075–82. 10.1093/nar/gkt978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Google. https://www.google.at (accessed December 20, 2015).
- Scopus. Elsevier. https://www.scopus.com/ (accessed December 20, 2015).
- Kuhn M.; Campillos M.; Letunic I.; Jensen L. J.; Bork P. A Side Effect Resource to Capture Phenotypic Effects of Drugs. Mol. Syst. Biol. 2010, 6, 343. 10.1038/msb.2009.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn M.; Letunic I.; Jensen L. J.; Bork P. The SIDER Database of Drugs and Side Effects. Nucleic Acids Res. 2016, 44 (D1), D1075–9. 10.1093/nar/gkv1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullak-Ublick G. A.Drug-Induced Cholestatic Liver Disease. In Molecular Pathogenesis of Cholestasis, Trauner M., Jansen P., Eds.; Springer, 2003; pp 271–280. [Google Scholar]
- Van den Hof W. F.; Coonen M. L.; van Herwijnen M.; Brauers K.; Wodzig W. K.; van Delft J. H.; Kleinjans J. C. Classification of Hepatotoxicants Using HepG2 Cells: A Proof of Principle Study. Chem. Res. Toxicol. 2014, 27 (3), 433–42. 10.1021/tx4004165. [DOI] [PubMed] [Google Scholar]
- Molecular Operating Environment (MOE), 2013.08.01; Chemical Computing Group Inc.: Montreal, QC, Canada, 2015. [Google Scholar]
- Atkinson F. L.Standardiser; 2014; https://github.com/flatkinson/standardiser/tree/1.0.1.
- Sadowski J.; Gasteiger J.; Klebe G. Comparison of Automatic Three-Dimensional Model Builders Using 639 X-ray Structures. J. Chem. Inf. Model. 1994, 34 (4), 1000–1008. 10.1021/ci00020a039. [DOI] [Google Scholar]
- Landrum G.RDKit: Open-Source Cheminformatics Software; 2015.
- Yap C. W. PaDEL-Descriptor: An Open Source Software to Calculate Molecular Descriptors and Fingerprints. J. Comput. Chem. 2011, 32 (7), 1466–1474. 10.1002/jcc.21707. [DOI] [PubMed] [Google Scholar]
- Carrio P.; Lopez O.; Sanz F.; Pastor M. eTOXlab, an Open Source Modeling Framework for Implementing Predictive Models in Production Environments. J. Cheminf. 2015, 7, 8. 10.1186/s13321-015-0058-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall M.; Frank E.; Holmes G.; Pfahringer B.; Reutemann P.; Witten I. H. The WEKA Data Mining Software: An Update. SIGKDD Explor. Newsl. 2009, 11 (1), 10–18. 10.1145/1656274.1656278. [DOI] [Google Scholar]
- Pedro D.MetaCost: A General Method for Making Classifiers Cost-Sensitive. In Proceedings of the fifth ACM SIGKDD international conference on Knowledge discovery and data mining, ACM: San Diego, California, USA, August 15−18, 1999. [Google Scholar]
- Breiman L. Bagging predictors. Machine Learning 1996, 24 (2), 123–140. 10.1007/BF00058655. [DOI] [Google Scholar]
- Freund Y.; Schaphire R. E.. Experiments with a new boosting algorithm. In 13th International Conference on Machine Learning, San Francisco, July 2−6, 1996, 1996; pp 148–156.
- Friedman J.; Tibshirani H.; Hastie T. Additive Logistic Regression: A Statistical View of Boosting. Ann. Stat 2000, 28 (2), 337–407. 10.1214/aos/1016120463. [DOI] [Google Scholar]
- Gutlein M.; Helma C.; Karwath A.; Kramer S. A Large-Scale Empirical Evaluation of Cross-Validation and External Test Set Validation in (Q) SAR (vol 32, 2013). Mol. Inf. 2013, 32 (9–10), 866–866. 10.1002/minf.201380841. [DOI] [PubMed] [Google Scholar]
- R Core Team. R: A language and Environment for Statistical Computing; R Foundation for Statistical Computing, Vienna, Austria, 2013; http://www.R-project.org/. [Google Scholar]
- Fawcett T. An Introduction to ROC Analysis. Pattern Recogn Lett. 2006, 27 (8), 861–874. 10.1016/j.patrec.2005.10.010. [DOI] [Google Scholar]
- Vihinen M. How to Evaluate Performance of Prediction methods? Measures and Their Interpretation in Variation Effect Analysis. BMC Genomics 2012, 13 (Suppl 4), S2. 10.1186/1471-2164-13-S4-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melagraki G.; Afantitis A.; Sarimveis H.; Igglessi-Markopoulou O.; Koutentis P. A.; Kollias G. In silico Exploration for Identifying Structure-Activity Relationship of MEK Inhibition and Oral Bioavailability for Isothiazole Derivatives. Chem. Biol. Drug Des. 2010, 76 (5), 397–406. 10.1111/j.1747-0285.2010.01029.x. [DOI] [PubMed] [Google Scholar]
- Afantitis A.; Melagraki G.; Koutentis P. A.; Sarimveis H.; Kollias G. Ligand-Based Virtual Screening Procedure for the Prediction and the Identification of Novel Beta-Amyloid Aggregation Inhibitors Using Kohonen Maps and Counterpropagation Artificial Neural Networks. Eur. J. Med. Chem. 2011, 46 (2), 497–508. 10.1016/j.ejmech.2010.11.029. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Golbraikh A.; Oloff S.; Kohn H.; Tropsha A. A Novel Automated Lazy Learning QSAR (ALL-QSAR) Approach: Method Development, Applications, and Virtual Screening of Chemical Databases Using Validated ALL-QSAR Models. J. Chem. Inf. Model. 2006, 46 (5), 1984–95. 10.1021/ci060132x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers A. D.; Zhu H.; Fourches D.; Rusyn I.; Tropsha A. Modeling Liver-Related Adverse Effects of Drugs Using K-Nearest Neighbor Quantitative Structure-Activity Relationship Method. Chem. Res. Toxicol. 2010, 23 (4), 724–32. 10.1021/tx900451r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo E.; Leporini C.; Chimirri S.; Marrazzo G.; Sacchetta S.; Bruno L.; Lista R. M.; Staltari O.; Scuteri A.; Scicchitano F.; et al. Limitations and Obstacles of the Spontaneous Adverse Drugs Reactions Reporting: Two ″Challenging″ Case Reports. J. Pharmacol. Pharmacother. 2013, 4, S66–72. 10.4103/0976-500X.120955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X.; Kruhlak N. L. Construction and Analysis of a Human Hepatotoxicity Database Suitable for QSAR Modeling Using Post-Market Safety Data. Toxicology 2014, 321, 62–72. 10.1016/j.tox.2014.03.009. [DOI] [PubMed] [Google Scholar]
- Hauben M. Early Postmarketing Drug Safety Surveillance: Data Mining Points to Consider. Ann. Pharmacother. 2004, 38 (10), 1625–1630. 10.1345/aph.1E023. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Guo J. J.; Healy D. P.; Lin X.; Patel N. C. Risk of Hepatotoxicity Associated with the Use of Telithromycin: A Signal Detection Using Data Mining Algorithms. Ann. Pharmacother. 2008, 42 (12), 1791–1796. 10.1345/aph.1L315. [DOI] [PubMed] [Google Scholar]
- Ritschel T.; Hermans S.; Schreurs M.; van den Heuvel J.; Koenderink J.; Greupink R.; Russel F. In silico identification and in vitro validation of potential cholestatic compounds through 3D ligand-based pharmacophore modeling of BSEP inhibitors. Chem. Res. Toxicol. 2014, 27 (5), 873–81. 10.1021/tx5000393. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.