

Abstract
Therapeutic antibodies or inhibitors targeting CSF-1R block colony stimulating factor-1/colony stimulating factor-1 receptor (CSF-1/CSF-R) signaling, and have shown remarkable efficacy in the treatment of cancer. However, little is known about tumor cell-intrinsic CSF-1R effects. Here, we show that human osteosarcomas contain CSF-1R-expressing cancer subpopulations, and demonstrate that osteosarcoma cell-intrinsic CSF-1R promotes growth in vitro and in vivo. CSF-1R inhibition in osteosarcoma cells by RNA interference suppresses cell proliferation and tumor growth in mice. Conversely, CSF-1R overexpression enhances cell proliferation and accelerates tumor growth. CSF-1R overexpression can significantly enhance osteosarcoma cell migration, invasion, and epithelial-mesenchymal transition (EMT), whereas silencing CSF-1R inhibits these processes. Microarray analysis suggests that jagged 1 (JAG1) can function as a downstream mediator of CSF-1R. Moreover, we report a signaling pathway involving CSF-1R and JAG1 that sustains osteosarcoma cell migration and invasion. Our results identify osteosarcoma cell intrinsic functions of the CSF-1R/JAG1 axis in dissemination of osteosarcoma cells.
Keywords: CSF-1R, osteosarcoma, EMT, metastasis, JAG1
Introduction
Established solid tumors consist of both transformed neoplastic cells and non-transformed host cells such as stromal cells, lymphocytes, dendritic cells, and macrophages [1]. Stromal cells in the surrounding tumor microenvironment sustain tumor cell growth and metastasis by producing cytokines. Colony-stimulating factor 1 (CSF-1) is a cytokine frequently produced by several stromal cells and cancers, including melanoma and lung cancer [2]. The secreted CSF-1 binds to the tyrosine kinase receptor on the tumor cells, resulting in increased proliferation and survival of cancer cells. Colony stimulating factor 1 receptor (CSF-1R) is a class III transmembrane tyrosine kinase receptor and its expression is frequently detected in a broad range of cancers. Indeed, the expression of CSF-1 is associated with reduced survival and an unfavorable prognosis in a number of cancers, including lung, renal, pancreatic, and ovarian [3]. Data reported in the literature for solid tumors indicate that the oncogenic potential of CSF-1/CSF-1R is due to the coexpression of this growth factor/receptor pair, rather than CSF-1R overexpression or mutations activating CSF-1R independently of ligand. Activation of CSF-1R by its ligand triggers a series of rapid events, including receptor dimerization and tyrosine phosphorylation of its intracellular domain. Phosphorylation at particular CSF-1R tyrosines creates binding sites for a variety of cytoplasmic proteins that activate signal transduction pathways, including that of extracellular regulated protein kinase 1/2 (ERK1/2) and phosphatidylinositol 3-kinase (PI3K) [4]. Targeting receptor tyrosine kinases with kinase inhibitors (e.g., imatinib, dasatinib or nilotinib) has recently opened a new era in the treatment of hematologic malignancies and solid tumors such as gastrointestinal stromal tumors [5]. These drugs are effective on CSF-1R, and other CSF-1R-specific inhibitors have been developed. More importantly, several drugs targeting CSF-1 and CSF-1R are currently in Phase I/II trials (www.clinicaltrials.org). PLX3397 is a potent tyrosine kinase inhibitor selected for its ability to inhibit CSF-1R, and is currently in clinical development as a single agent or in combination therapy for the treatment of patients with glioblastoma, breast cancer, and other cancers through its inhibition of the CSF-1R [6].
Osteosarcoma is the second most common primary malignant bone cancer. It is a rare disease with a poor prognosis, usually occurs in adults, and the cure rate for this disease has not improved over the last several decades [7]. For high-grade tumors the cure rate has remained at 10-25%. The treatment for osteosarcoma is surgical resection; chemotherapy and radiation therapy are not ordinarily used since osteosarcoma is resistant to these adjuvant modalities. In contrast to osteosarcoma, chondrosarcoma, which usually occurs in adolescents, is sensitive to chemotherapy and the cure rate has increased from 20% to 75% with the advent of multi-agent chemotherapy. However, in patients with either tumor, the majority of those who are not cured succumb to lung metastases. Our efforts are directed at elucidating the mechanisms of osteosarcoma invasion and metastasis. Ultimately, the cancer cells spread to distant sites including liver, bone, brain, and other organs. Unfortunately, no major clinical breakthrough that can efficiently prevent tumor-cell invasion has been made, although this is a major obstacle to the therapeutic management of lung cancer. Epithelial-mesenchymal transition (EMT) refers to specific morphological and phenotypic alterations in epithelial cells during embryonic development [8]. In the EMT process, the polarity and adhesion of cells surrounding the epithelial cells and matrix are reduced, and the cells become similar in morphology to fibroblasts; thus, this process enhances migratory ability [9]. The occurrence of EMT is accompanied by different expression-specific epithelial and mesenchymal molecular markers. In EMT, epithelial cell markers such as E-cadherin, α-catenin, β-catenin, and γ-catenin are down-regulated, whereas the expression levels of mesenchymal tissue markers such as N-cadherin, α-smooth muscle actin, vimentin, and fibronectin protein are up-regulated. EMT is closely associated with tumor progression and resistance to chemotherapy. This finding has drawn the attention of academics, clinicians, and pharmaceutical researchers to EMT [10].
A previous study characterized CSF-1R as a promoter of proliferation, migration, and invasion of canine mammary cancer cells. In a study of noninvasive and invasive breast adenocarcinomas (grades 1-3), CSF-1 expression was evaluated and found predominantly in invasive tumor cells [11]. Given the association between CSF-1R and breast cancer metastasis, blocking of CSF-1 or CSF-1R would deplete tumor-associated macrophages (TAMs) and subsequently limit metastasis and primary tumor growth as well [12]. Targeting of TAMs through the inhibition of CSF-1R, the key macrophage signaling pathway, has been reported to reduce tumor growth and metastasis, and these treatments are now in clinical trials [13]. PLX3397, the small molecule inhibitor of CSF-1R, is in phase I trials for solid tumors and for metastatic breast cancer in combination with chemotherapy. Coexpression of CSF-1 and its receptor in metastatic ovarian cancer specimens is a predictor of poor outcome in epithelial ovarian cancer [14]. Clinical specimens from ovarian cancer metastasis display strong immunostaining for both CSF-1 and its receptor in contrast to noninvasive borderline tumors, which do not coexpress CSF-1, and to benign ovarian tissue, which expresses little to no CSF-1/CSF-1R [15]. Human lung carcinoma cell lines that express CSF-1R, but not the ligand, show increased invasion in vitro into an amniotic basement membrane upon stimulation with CSF-1. On the basis of the results from these studies, CSF-1/CSF-1R signaling is considered a potential target for cancer metastasis [16]. While previous studies indicated that CSF-1R and CSF-1 are expressed in several cancer cell lines and tumors and demonstrated the relevance of CSF-1/CSF-1R signaling in the metastasis of cancer cells, few studies have focused on the biological role of CSF-1/CSF-1R signaling in the metastasis of osteosarcoma [17].
Here, we report that established human osteosarcoma cell lines frequently contain CSF-1R-expressing cancer subpopulations. Enforced CSF-1R expression enhanced osteosarcoma growth and conversely, osteosarcoma-specific CSF-1R knockdown inhibited tumor growth. In addition, we showed that CSF-1R activity was important for the epithelial-mesenchymal transition (EMT) and osteosarcoma cells metastasis. Moreover, our data demonstrated that JAG1, the essential regulator of the process of epithelial mesenchymal transition, is the direct target gene and functional mediator of CSF-1R in osteosarcoma cells. Down-regulation of JAG1 could also inhibit migration and invasion of osteosarcoma cells. Our results expand our current understanding of CSF-1R/JAG1 pathway functions in osteosarcoma, and suggest that targeting cancer cell-intrinsic CSF-1R might significantly contribute to reducing osteosarcoma growth and metastasis.
Materials and methods
Cell culture and cell lines
The human osteosarcoma lines (SJSA-1, SW1353, U-2 OS and MG-63) were obtained from the Chinese Academy of Sciences Cell Bank of Type Culture Collection (CBTCCCAS, Shanghai, China). The cancer cell lines were cultured as monolayers in RPMI 1640 culture media supplemented with 10% fetal bovine serum (Wisent, Quebec, Canada), 100 μg/mL penicillin, 100 μg/mL streptomycin and maintained in an incubator with a humidified atmosphere of 95% air and 5% CO2 at 37°C.
Cell viability assay
Cells (1×105) suspended in RPMI 1640 were plated in 96-well cell culture plates. After 24 h, 48 h, 72 h or 96 h, cell viability was measured by CellTiter 96® AQueous One Solution cell proliferation assay (Promega, Madison, WI, USA). Absorbance measurements were recorded using a SpectraMax 340 (Molecular Devices) at 490 nm on the indicated days (Cell Proliferation Kit I; Roche).
Generation of CSF-1R knockdown or overexpressing cell line variants and RNA interference
Human shRNA constructs against CSF-1R were purchased from Santa Cruz Biotechnology. Plasmids coding for His-tagged CSF-1R proteins, His-JAG1 or control vector pReceiver-M01 were purchased from GeneCopoeia. Cells were transfected with plasmids and shRNAs by Lipofectamine 2000 reagent (Thermo Fischer Scientific). Oligonucleotides for human CSF-1R or JAG1 shRNA kit were purchased from OriGene (Rockville, MD, USA). The kit contains three predesigned duplexes targeting a specific gene of interest, and we used a pool of three target siRNAs to ensure work efficiency. Cells were transfected with specific shRNA or non-specific shRNA using the opti-MEM plus X-tremeGENE shRNA transfection reagent (Roche, Mannheim, Germany) according to the instruction manual.
Wound healing assay
Wound healing assay was performed as previously described [18]. Briefly, cells were seeded in 60 mm dishes and cultured at 37°C overnight to produce a confluent monolayer. After starvation in serum-free medium for 24 hours, a wound was created by scratching the monolayer with a 200 µl yellow sterile pipette tip. The wounded monolayer was then washed twice to remove cell debris and incubated with fresh normal medium. The area of cell-free scratch was photographed at 0 h 24 h after scratching respectively. The wound healing effect was determined by measuring the percentage of the remaining cell-free area compared with the area of the initial wound.
Invasion assay
The invasion assay was performed as previously described. Briefly, 1×105 cells suspended in serum-free medium were seeded onto the upper chamber. Medium containing 10% FBS as a chemoattractant was added to the lower chamber. After 22 h, invaded cells were counted at 200× magnification in at least 5 randomly selected fields [19].
Flow cytometry
Cells were detached by PBS1X/EDTA 2 mM and fixed with 4% PFA (10 min on ice), washed twice with PBS1X and resuspended for antibody staining at 1×106 cells/100 uL in PBS1X/BSA 1%. Live/dead cell discrimination was performed with the SYTOX-orange dye (Thermo Scientific), according to the manufacturer instructions. Gates (in the live cell population) were drawn in order to exclude > 99% of background staining (based on the isotype-stained samples). Data were acquired using a FACS CALIBUR instrument (BD Biosciences) and analysis was performed by using Summit 5.0.0 (Dako, Agilent Technologies, CA, USA). For CSF-1R staining, anti-CD115-APC conjugated and its highly adsorbed isotype matched control (Biolegend) were used.
Cytokine quantification
ELISA-based cytokine quantification kits for CSF-1 (Abnova, Taipei City, Taiwan) secreted in the conditioned media was commercially available.
Immunofluorescence staining
Cells were fixed in 4% paraformaldehyde for 20 min and permeabilized in 0.1% Triton X-100 for 5 min. Rhodamine-conjugated phalloidin (Molecular Probes: Eugene, OR) was employed to stain actin stress fiber (30 min at room temperature). JAG1 was detected by staining with anti-JAG1 antibody (1 μg/ml) overnight at 4°C followed by incubating with Alexa 488-conjugated anti-mouse antibody (Molecular Probes) for 1 hr at room temperature. Cell nuclei were stained with Hoechst for 3 min.
Colony formation assay
Growth medium (0.4 ml) containing 1% agarose gel was added to a 25 cm2 cell culture plate in triplicates and allowed to harden. 2000 breast cancer cells were suspended in 0.2 ml medium containing 0.1% agarose gel and overlaid on the hardened bottom layer. Fresh medium (0.2 ml) was added to each well once a week for 4 weeks. The colonies were visualized by staining with 0.1% crystal violet for 4 hours at 37°C [20].
Microarray analysis
Total RNA was extracted from parental SW1353 and SW1353 CSF-1R knock-down cells, and microarray analysis was performed using SurePrint G3 Human Gene Expression 8×60K v2 (Agilent Array®). Gene expression was analyzed in SW1353 versus SW1353 CSF-1R knock-down cells.
RNA extraction and quantitative real-time RT-PCR
Total RNA isolation and first-strand cDNA synthesis were performed as previously described. The primer pairs used for PCR were as follows (sense and antisense, respectively): β-tubulin (forward, 5’-AACGAGCTGTGCTACAAGGTC-3’; reverse, 5’-GCGTGGTCGATGAGGAAGA-3’) was used as an internal control; CSF-1R (forward, 5’-GGGAATCCCAGTGATAGAGCC-3’; reverse, 5’-TTGGAAGGTAGCGTTGTTGGT-3’). CSF-1 (forward, 5’-TGGCGAGCAGGAGTATCAC-3’; reverse, 5’-AGGTCTCCATCTGACTGTCAAT-3’). cDNAs amplification and relative expression values were obtained from three independent experiments. Quantitative real-time polymerase chain reaction (qRT-PCR) was performed using the StepOne real-time PCR system (Applied Biosystems, Foster City, CA). Data were normalized to the β-tubulin expression level and are expressed as the fold change relative to control.
Western blot analysis
Protein concentrations were determined using the BCA assay kit (Pierce). Equivalent amounts of each protein sample were mixed with loading buffer, heated at 100°C for 10 min, and subjected to SDS-PAGE using 10% separation gel and 5% spacer gel. Then protein was transferred to a PVDF membrane (Solarbio) by electro blotting (Bio-RAD). The membrane was blocked for 3 hours at room temperature in TBST (25 mM Tris/HCl, pH 7.5, 150 mM NaCl, 0.1% Tween 20) containing 5% nonfat dry milk. The membranes were incubated overnight at 4°C in 1:200 dilution of anti-CSF-1R monoclonal antibody, 1:1000 dilution of E-cadherin monoclonal antibody, 1:2000 dilution of N-cadherin monoclonal antibody, 1:1000 dilution of Vimentin monoclonal antibody, 1:1000 dilution of JAG1 monoclonal antibody, 1:2000 dilution of Twist monoclonal antibody and 1:2500 dilution of β-tubulin polyclonal antibody with WB Antibody Diluent (P0023A, Beyotime), washed six times in TBST, and incubated for 35 min in a 1:5000 dilution of goat anti-rabbit Ig-HRP (EK020, Zhuangzhibio) with WB Secondary Antibody Diluent (P0023D, Beyotime). Immunoreactive bands were revealed by the enhanced chemiluminescence system (Santa Cruz Biotechnology, Santa Cruz, CA). Pictures were photographed and analyzed by Gel Dox XR system (Bio-Rad, Philadelphia, PA) [21].
Subcutaneous xenograft models
All animal experiments were approved and conducted by the Institutional Animal Care and Treatment Committee of College of Medicine, Zhejiang University. SW1353 tumors were established by injecting 1×107 cells (100 μL) into the dorsal area of 7-8 week old female athymic nude mice (balb/c, nu/nu). Tumor growth and body weight was measured every three days during the treatment. Tumor volumes were calculated using the formula as follow: volume (mm3) = 0.5× length (mm) × width (mm)2 [22].
Statistical analysis
The data were presented as mean ± SD. Differences in the results of two groups were evaluated using either two-tailed Student’s t test or one-way ANOVA followed by post hoc Dunnett’s test. The differences with P < 0.05 were considered statistically significant.
Results
Osteosarcomas frequently contain CSF-1R expressing cancer subpopulations
Previous studies have reported that the expression of CSF-1R is associated with tumor progression in multiple types of cancer. To investigate whether CSF-1R and its associated factors are involved in human osteosarcoma progression, real-time quantitative RT-PCR (qRT-PCR) analysis of the human CSF-1R gene revealed CSF-1R mRNA expression (Figure 1A), and immunoblot analysis demonstrated CSF-1R protein expression by human SW1353, SJSA-1, U-2 OS, and MG-63 osteosarcoma cells (Figure 1B), which is consistent with the previous demonstration of CSF-1R expression by multiple types of cancer. Flow cytometric analyses showed CSF-1R surface protein expression by 4/4 osteosarcoma cell lines tested, with CSF-1R+ tumor cell frequencies ranging from 23.6% ± 5.2% to 42.5% ± 4.7% (Figure 1C), and revealed preferential CSF-1R expression by osteosarcoma cell subsets. SW1353 osteosarcoma grafts grown in mice lacking adaptive immunity also demonstrated CSF-1R expression by osteosarcoma cells (Figure 1D). We also assessed the expression of the CSF-1R ligand (CSF-1) by qRT-PCR and enzyme-linked immunosorbent assay (ELISA), and these revealed that most of the cell lines expressed CSF-1 mRNA (Figure 1E). ELISA assay of conditioned medium from the analyzed cells revealed detectable levels of secreted CSF-1 (Figure 1F). These data confirm that the level of CSF-1R protein increases during osteosarcoma progression.
Figure 1.
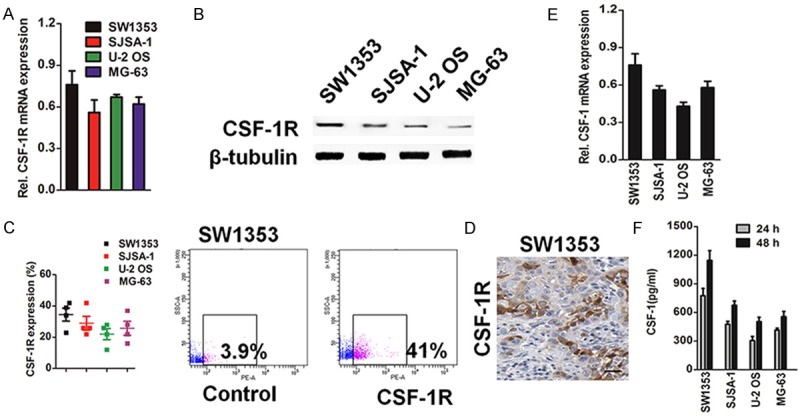
CSF-1R expression by osteosarcoma cells. A: qRT-PCR expression analysis of CSF-1R mRNA expression by human osteosarcoma cell lines. PCR values were normalized to the levels of β-tubulin. Data were presented as the mean ± SD from three independent measurements. B: Western blotting analysis of the levels of CSF-1R in various osteosarcoma cell lines. β-tubulin was used as loading controls. Band intensities were measured by densitometry and normalised to β-tubulin expression. C: Percentages and representative flow cytometry plots of CSF-1R surface protein expression by osteosarcoma cells (n = 3 independent experiments, respectively). D: Representative CSF-1R immunohistochemistry staining for expression of CSF-1R from SW1353 osteosarcoma graft grown in nude mice (size bar, 100 μm). E: qRT-PCR analysis of CSF-1 mRNA level in various osteosarcoma cell lines. PCR values were normalized to the levels of β-tubulin. Data were presented as the mean ± SD from three independent measurements. F: The various osteosarcoma cell lines (SW1353, SJSA-1, U-2 OS and MG-63) secret CSF-1. Level of in the medium conditioned from the indicated cells and harvested at 24 h, and 48 h, respectively. The levels of CSF-1 in the cell free medium were medium were subtracted as a back-group control. Values shown are the mean ± SD from three wells from a representative of three independent experiments.
Osteosarcoma-expressed CSF-1R promotes tumor growth
To functionally dissect the precise role of osteosarcoma-expressed CSF-1R in tumor growth, we generated CSF-1R knockdown and CSF-1R-overexpressing SW1353 osteosarcoma cells. Transduction of SW1353 cells with two distinct short hairpin (sh) RNAs targeting CSF-1R inhibited CSF-1R mRNA expression and significantly blocked CSF-1R protein expression compared to controls (Figure 2A). Conversely, transduction of SW1353 cells with CSF-1R-encoding constructs resulted in up-regulation of CSF-1R, both at the mRNA and protein level (Figure 2D). Osteosarcoma-specific CSF-1R knockdown resulted in decreased SW1353 osteosarcoma growth in nude mice compared to that of vector controls (Figure 2B), and CSF-1R overexpression resulted in increased growth (Figure 2E). CSF-1R-shRNA osteosarcoma grafts demonstrated diminished CSF-1R mRNA and CSF-1R protein expression compared to control tumors at the experimental endpoint (Figure 2C), and CSF-1R over-expression osteosarcomas significantly enhanced expression of CSF-1R mRNA and protein (Figure 2F). Together, these findings identify osteosarcoma-expressed CSF-1R as protumorigenic. We next examined whether osteosarcoma-specific CSF-1R silencing or overexpression affects SW1353 cell growth in vitro. Consistent with our in vivo findings, CSF-1R knockdown impaired proliferation of SW1353 cells (Figure 3A) and colony forming ability in soft agar (Figure 3B), whereas CSF-1R overexpression promoted in vitro growth (Figure 3C) and colony formation (Figure 3D) compared to the respective controls. Together, these findings suggest that the cancer cell-intrinsic functions of osteosarcoma-expressed CSF-1R promote tumor growth.
Figure 2.
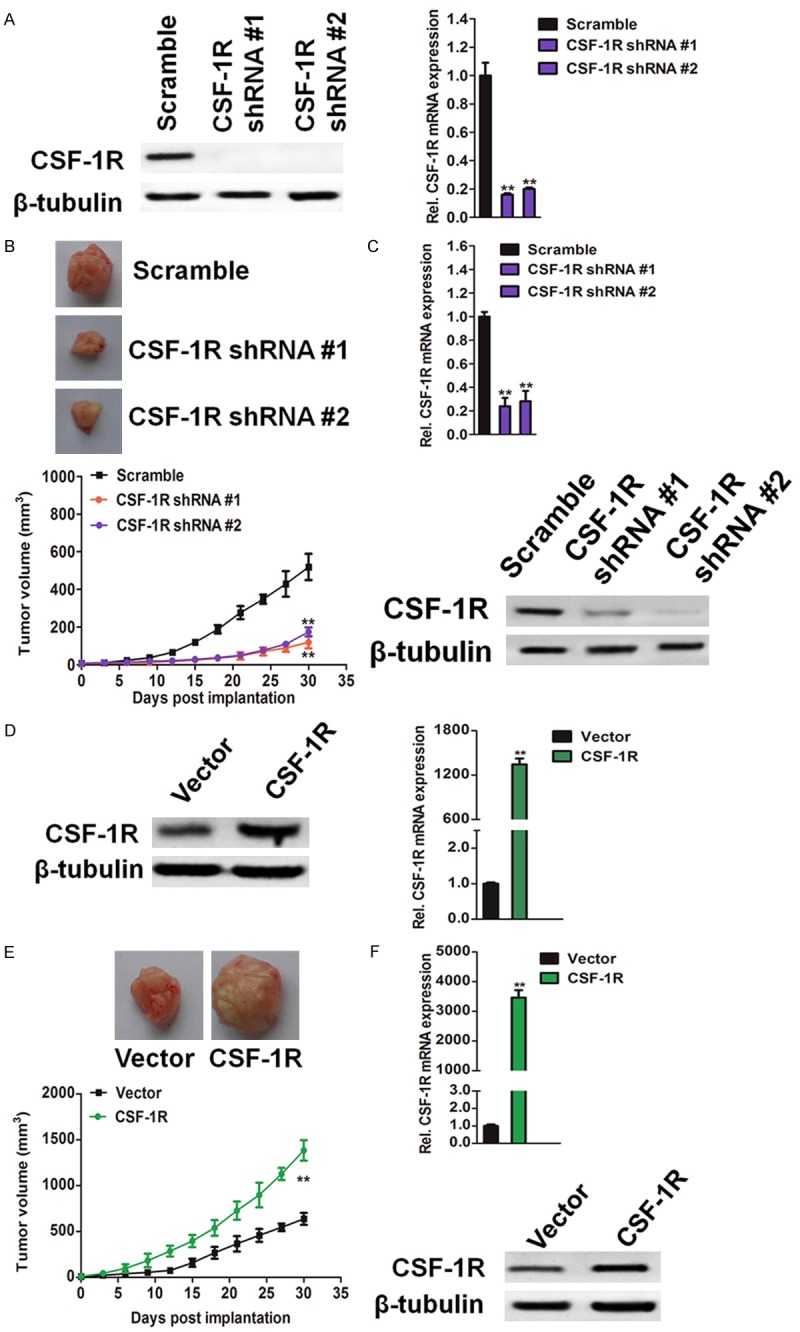
Osteosarcoma cells expressed CSF-1R promotes tumorigenicity in xenotransplanted tumor model. A: CSF-1R mRNA (left) and protein expression (right) by CSF-1R-shRNA #1 and CSF-1R-shRNA #2 versus vector control. PCR values were normalized to the levels of β-tubulin. Data were presented as the mean ± SD from three independent measurements. **P < 0.01, compared with control SW1353 cells. B: Tumor growth kinetics (mean ± SD) of SW1353-shRNA versus vector control SW1353 osteosarcoma cells in BALB/c nu/nu mice (n = 6 each). **P < 0.01, compared with mice inoculated with control cells. C: CSF-1R mRNA expression (mean ± SD) determined by qRT-PCR (upper) and western blotting analysis of CSF-1R protein expression (lower) of osteosarcoma tissues harvested post inoculation of SW1353-shRNA versus control SW1353 cells to BALB/c nu/nu mice, respectively. PCR values were normalized to the levels of β-tubulin. Data were presented as the mean ± SD from three independent measurements. **P < 0.01, compared with control SW1353 cells. D: CSF-1R mRNA (left) and protein expression (right) by CSF-1R over-expressing versus vector control. PCR values were normalized to the levels of β-tubulin. Data were presented as the mean ± SD from three independent measurements. E: Tumor growth kinetics (mean ± SD) of SW1353 over-expression versus vector control SW1353 osteosarcoma cells in BALB/c nu/nu mice (n = 6 each). **P < 0.01, compared with mice inoculated with control cells. F: CSF-1R mRNA expression (mean ± SD) determined by qRT-PCR (upper) and western blotting analysis of CSF-1R protein expression SW1353 (lower) of osteosarcoma harvested post inoculation of SW1353 over-expression versus vector control-transduced SW1353 cells to BALB/c nu/nu mice, respectively. PCR values were normalized to the levels of β-tubulin. Data were presented as the mean ± SD from three independent measurements. **P < 0.01, compared with vector control.
Figure 3.
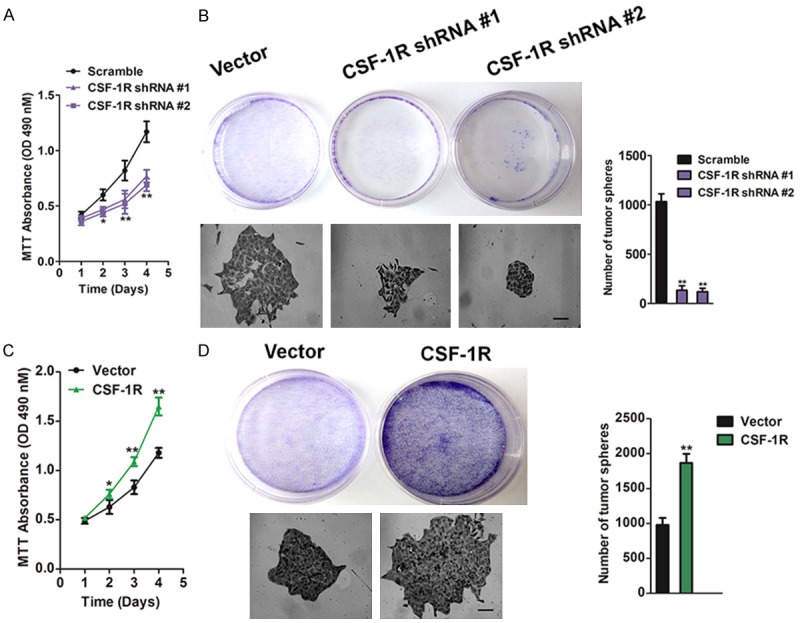
SW1353 cells expressed CSF-1R promotes cells growth in vitro. A: Scramble or shRNA against CSF-1R were transfected into SW1353 cells. After 24 h post transfections, cells were cultured for further 24 h, 48 h, 72 h, 96 h, respectively, and subjected to cell proliferation assay. MTT assays showed that knock-down CSF-1R inhibit cell viability in SW1353 cells when compared with control cells. The data are presented as mean ± SD. For indicated comparisons, *P < 0.05, **P < 0.01. B: Colony-formation assays showed that the number of clones was significantly decreased in CSF-1R knock-down SW1353. The data were presented as mean ± SD. For indicated comparisons, **P < 0.01. Size bar, 50 μm. C: CSF-1R over-expression in SW1353 cells accelerated cell proliferation in vitro. After 24 h post transfected with vector or CSF-1R, cells were cultured for further 24 h, 48 h, 72 h, 96 h, respectively, and subjected to cell proliferation assay. MTT assays showed that over-expression CSF-1R increase cell proliferation in SW1353 cells when compared with control cells. The data are presented as mean ± SD. For indicated comparisons, *P < 0.05, **P < 0.01. D: CSF-1R over-expression in SW1353 cells accelerated colony formation. The data were presented as mean ± SD. For indicated comparisons, **P < 0.01. Size bar, 50 μm.
CSF-1R knockdown in osteosarcoma cells inhibits EMT
To determine the potential role of CSF-1R in osteosarcoma cell metastasis, we noticed a change in cell morphology from the cobble stone-like shape typical of epithelial cells to a more spindle-like and scattered appearance (Figure 4A), indicating that these cells may be undergoing the epithelial-mesenchymal transition (EMT). EMT is characterized by a number of functional and molecular changes, including marked increases in cell migration and invasion, actin stress fiber formation, up-regulation of mesenchymal markers, and down-regulation of epithelial markers. We therefore measured these characteristics in the SW1353 CSF-1R knockdown cells in order to determine whether the knockdown inhibits EMT. In the wound healing assay, the control cells showed markedly faster wound closure than the knockdown cells (Figure 4B). Consistently, CSF-1R depletion significantly reduced invasion in the Transwell system (Figure 4C). In addition, the CSF-1R knockdown cells displayed a marked increase in epithelial markers and a loss of mesenchymal markers (Figure 4D). Re-expression of CSF-1R in the shCSF-1R cells effectively restored the loss of CSF-1R (Figure 4E), restored the epithelial morphology (Figure 4F), and rescued the other EMT phenotypes (Figure 4G), confirming that the inhibited EMT of shCSF-1R cells is due to the loss of CSF-1R expression. Together, these data indicate that reducing CSF-1R suppressed EMT in SW1353 cells. Taken together, our results suggest that knockdown of CSF-1R inhibits osteosarcoma cancer EMT in vitro and metastasis in vivo.
Figure 4.

CSF-1R KD suppresses EMT in SW1353 cells. A: Phase-contrast images of control and two CSF-1R knock-down pools. Scale bars represent 50 μm. B: Wound healing assay. Confluent cell monolayers were wounded, and wound closure was monitored at 0 hour and 24 hour. Scale bars represent 100 μm. C: Invasion assay. SW1353 control or CSF-1R knock-down cells were subjected to a Transwell invasion assay. The invasived cells were stained with 1% crystal violet and counted. Data were collected from five fields in three independent experiments. Scale bars represent 100 μm. For indicated comparisons, **P < 0.01. D: Western blot analysis showed that silencing CSF-1R causes up-regulation of the epithelial cell markers and down-regulation of mesenchymal cell markers. E: The levels of CSF-1R protein in control, CSF-1R knock-down cells and rescue cells (CSF-1R-2 knock-down cells transfected with CSF-1R cDNA) were examined by western blotting (left panel) and qRT-PCR (right panel), respectively. The mRNA level was normalized to the level of β-tubulin mRNA. **P < 0.01, compared with control cells, ##P < 0.01, compared with CSF-1R shRNA cells. F: Phase contrast pictures of control, CSF-1R-2 knock-down, and rescue cells. G: The wound healing assay. Data were presented as mean ± SD. **P < 0.01, compared with control cells, ##P < 0.01, compared with CSF-1R shRNA cells. Scale bars represent 50 μm. H: Transwell invasion assay. SW1353 control or CSF-1R overexpression cells were subjected to a Transwell invasion assay. The invasived cells were stained with 1% crystal violet and counted. Data were collected from five fields in three independent experiments. Scale bars represent 100 μm. **P < 0.01, compared with control cells, ##P < 0.01, compared with CSF-1R shRNA cells. I: Western blot analysis showed that over-expression CSF-1R causes up-regulation of the mesenchymal cell markers and down-regulation of the epithelial cell markers.
Identification of JAG1 as a CSF-1R target gene
In order to unravel the cellular pathways involved in CSF-1R-mediated migration and invasion, we performed gene expression analysis in control and CSF-1R-depleted SW1353 cells (Figure 5A). We selected a panel of important genes involved in regulation of migration and invasion (Z score ≥ 2 or ≤ -2, p value < 0.05). The most down-regulated gene was JAG1, which activates the notch receptor signaling pathway (Figure 5B). JAG1 regulates multiple cancer-associated processes including proliferation, survival, EMT, metastasis, and angiogenesis. Interestingly, lung cancers generally have higher levels of JAG1 expression, which is associated with reduced disease-free survival. We confirmed that CSF-1R knockdown down-regulated both JAG1 mRNA and protein (Figure 5C, 5D). Similar results were obtained by the immunofluorescence assay. Notably, shRNA-based CSF-1R depletion remarkably diminished the expression of JAG1 in SW1353 cells (Figure 5E). We therefore evaluated migration and invasion of SW1353 cells upon JAG1 depletion (Figure 5F). JAG1 knockdown in SW1353 cells reduced migration by 45% (Figure 5G) and invasion potential was significantly suppressed (Figure 5H). Furthermore, SW1353 JAG1 shRNA cells had a spindle shape, suggesting mesenchymal features, whereas the control cells had a flat, cobblestone-like morphology (Figure 5I). These results identify JAG1 as a CSF-1R target gene that regulates SW1353 cancer cell migration and invasion.
Figure 5.
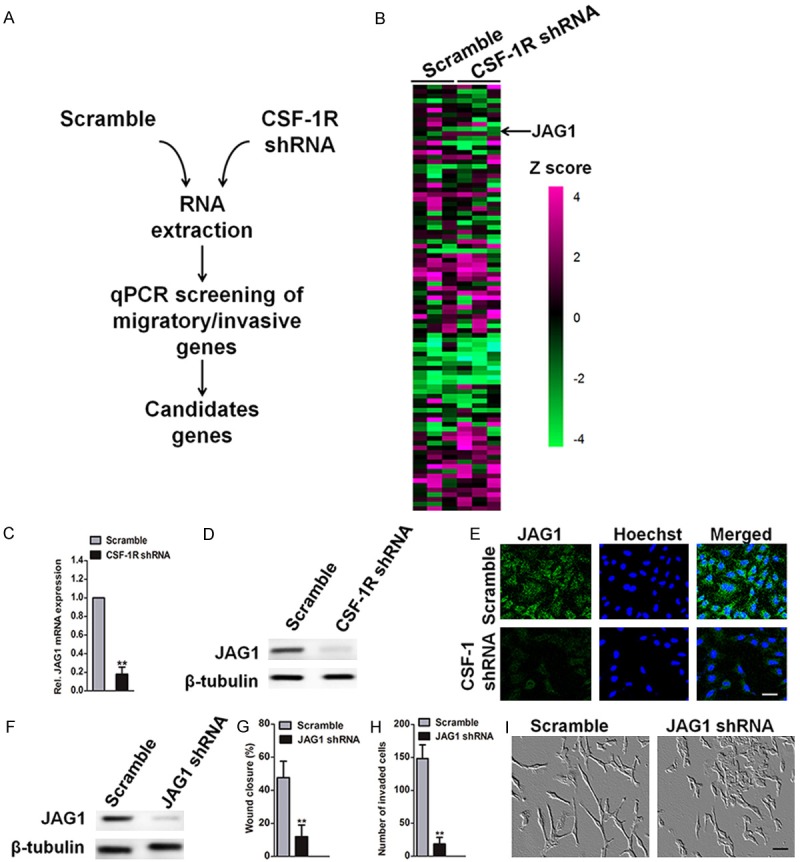
Identification of JAG1 as a CSF-1R-dependent gene. A: Screening strategy. B: Heatmap representing relative expression of genes involved in regulation of migration and invasion in response to CSF-1R depletion in SW1353 cells. Z scores are represented using a color code. Results for separate knockdown experiments were shown (n = 3). C: JAG1 mRNA level was measured in SW1353 cells 72 h after transfection with the indicated shRNAs and normalized to the level of β-tubulin mRNA. D: JAG1 protein expression was measured by western blotting analysis after SW1353 cells transfected with the indicated shRNAs. E: Immunofluorescence of JAG1 was performed in SW1353-control and CSF-1R knock-down cells. Scale bar: 50 um. F: Immunoblot validates JAG1 depletion in SW1353 24 h after transfection with the indicated shRNAs. G: Migration assay was conducted with scramble or JAG1 shRNA SW1353 cells. Bars show means ± SD of three independent experiments. Statistical analyses were performed by using one-way ANOVA. H: SW1353 cells were treated with control scramble or shRNA targeting JAG1, which was followed by the Transwell invasion assay. The following symbols were used to indicate significant differences: **P < 0.01 vs. control. I: Photomicrographs of scrambled or JAG1 shRNA SW1353 cells. Scale bars represent 50 μm.
CSF-1R regulates migration and invasion through JAG1
CSF-1R regulation of JAG1 expression led us to hypothesize that CSF-1R regulates osteosarcoma cell migration and invasion through JAG1 signaling. CSF-1R and JAG1 may act in a single signaling pathway. If CSF-1R acts upstream of JAG1, we would expect that ablation of CSF-1R or JAG1 individually impairs migration and invasion to a similar degree, compared to co-depletion. We conducted migration and invasion assays under these conditions (Figure 6A, 6B). As shown above, individual ablation of CSF-1R or JAG1 significantly reduced migration and invasion. In support of the hypothesis, co-depletion of CSF-1R and JAG1 did not cause additive inhibition. Furthermore, if CSF-1R acts upstream of JAG1, JAG1 over-expression in CSF-1R-depleted cells should restore SW1353 cell migration and invasion. However, CSF-1R over-expression in JAG1-depleted SW1353 cells should fail to rescue. We found that over-expression of either CSF-1R or JAG1 increased migration and invasion by about 49-53% in control cells (Figure 6C, 6D). Critically, JAG1 overexpression rescued migration (Figure 6C) and invasion (Figure 6D) in CSF-1R-depleted cells. CSF-1R overexpression in JAG1-depleted cells failed to restore migration and invasion, confirming that JAG1 acts downstream from CSF-1R. Taken together, our results indicate that CSF-1R regulates migration and invasion through JAG1 signaling.
Figure 6.
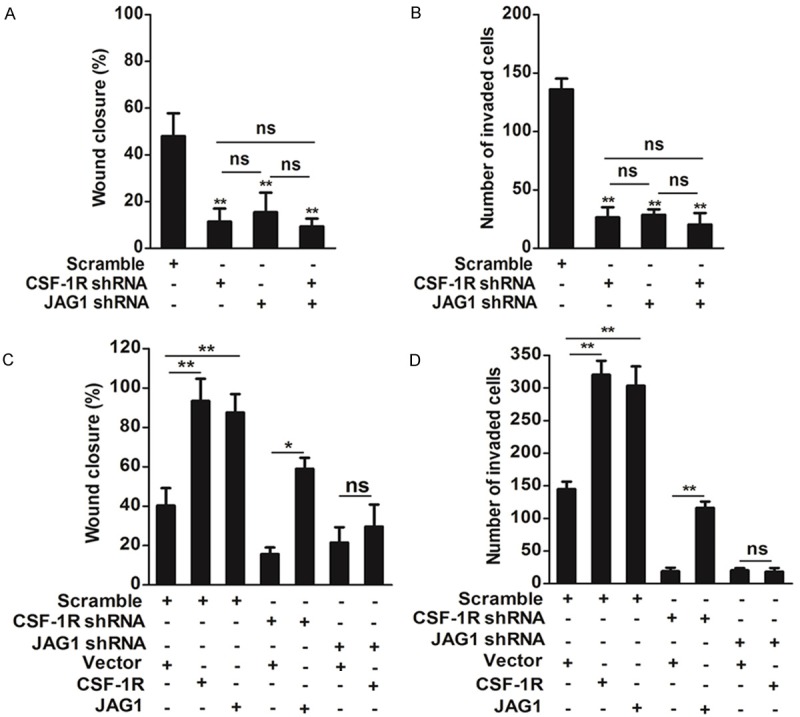
JAG1 acts downstream of CSF-1R to regulate SW1353 cells migration and invasion. A: SW1353 was transfected with the indicated siRNAs (scramble, CSF-1R shRNA or JAG1 shRNA) for 24 h prior to conduct migration assays. Bars show means ± SD of three independent experiments. Statistical analyses were performed by using one-way ANOVA. B: SW1353 was transfected with the indicated shRNAs (scramble, CSF-1R shRNA or JAG1 shRNA) for 24 h prior to conduct Transwell invasion assays. Bars show means ± SD of three independent experiments. Statistical analyses were performed by using one-way ANOVA. C: SW1353 cells were transfected with the indicated shRNAs (scramble, CSF-1R shRNA or JAG1 shRNA) for 24 h and indicated plasmids for 24 h prior to conduct migration assays. Bars show means ± SD of three independent experiments. Statistical analyses were performed by using one-way ANOVA. D: SW1353 cells were transfected with the indicated shRNAs (scramble, CSF-1R shRNA or JAG1 shRNA) for 24 h and indicated plasmids for 24 h prior to conduct invasion assays. Bars show means ± SD of three independent experiments. Statistical analyses were performed by using one-way ANOVA. The following symbols were used to indicate significant differences: ns, P > 0.05; *, P < 0.05; **, P < 0.01.
Discussion
Our study provides several insights into the functions of the colony-stimulating-factor receptor-1 (CSF-1R) pathway in osteosarcoma cell growth and metastasis. First, we have conducted a comprehensive characterization of CSF-1R transcript and protein expression by cancer cells in clinical tumor biopsies and established osteosarcoma cells [18]. CSF-1R is a type III receptor tyrosine kinase (RTK) primarily known as responsible for promoting the differentiation, survival, and homing of the monocyte-macrophage cell lineages. Data reported in the literature for solid tumors indicate that the elevated expression of CSF-1R and/or CSF-1 has been documented in several human cancers, and has been shown to correlate in some cases with adverse prognosis, including carcinomas of breast, female reproductive tract, prostate, and kidney [19]. Activation of CSF-1R by its ligand CSF-1 is likely to occur in an autocrine manner in tumor cells in which CSF-1R and CSF-1 are coexpressed, or in a paracrine manner, when CSF-1R is stimulated by CSF-1 released by fibroblasts. Taking these data together, several drugs specifically targeting CSF-1R (e.g., imatinib) have been developed and are currently in Phase I/II trials. Although the expression of CSF-1R had been previously documented in solid tumors, few studies have been performed to understand the role of CSF-1R in osteosarcoma. In the current study, qRT-PCR, immunoblot, and flow cytometric analysis revealed the presence of CSF-1R in osteosarcoma cells in all cell lines and clinical tumor samples examined. Furthermore, immunofluorescence labeling similarly showed CSF-1R expression by osteosarcoma subpopulations in clinical and biopsy specimens. Thus, using various independent methods, our work clearly establishes that osteosarcomas frequently contain CSF-1R+ tumor cell fractions.
Preclinical data indicate that TAMs represent attractive therapeutic targets as they represent key orchestrators of various tumor-promoting processes, such as escape of immune surveillance and secretion of pro-angiogenic factors [20]. To date, CSF-1R has been mainly studied in TAMs. Several approaches have been used to ablate TAMs or inhibit their tumor-promoting functions in mouse tumor models. One strategy is CSF-1R inhibition, which depleted macrophages and reduced the tumor volume in several xenografts [21]. Our study demonstrates CSF-1R signaling in a non-TAM cell type, i.e., osteosarcoma cells. Similar to the pro-tumorigenic effects of CSF-1R expressed by TAMs, our findings establish that CSF-1R expression by osteosarcoma cells functions as a mechanism promoting tumor growth in multiple independent experimental in vitro and in vivo systems. Phosphorylation at particular CSF-1R tyrosines creates binding sites for a variety of cytoplasmic proteins that activate signal transduction pathways, including those of PI3K and ERK [22]. Consistent with the interrelationship of CSF-1R and ERK1/2 signaling in breast cancer cells, the important roles of these pathways in osteosarcoma proliferation, and the herein described pro-tumorigenic effects of osteosarcoma-CSF-1R, future studies will try to determine whether osteosarcoma CSF-1R dependent tumor cell growth operates via ERK1/2 signaling.
TAMs have a high impact on cancer development because they facilitate matrix invasion, angiogenesis, and tumor cell mobility. Clinical specimens from ovarian cancer metastasis display strong immunostaining for both CSF-1 and its receptor CSF-1R, in contrast to noninvasive borderline tumors that do not co-express CSF-1 and CSF-1R, and to benign ovarian tissues that express little to no CSF-1/CSF-1R [23]. Blockade of CSF-1R signaling using ribonucleic acid (RNA) interference or pharmacological inhibitors completely inhibits microglial enhancement of invasiveness of several tumor cells. Taken together, the results indicate that CSF-1R is crucial for cancer cell dissemination. To advance human osteosarcoma treatment, it is necessary to understand the molecular mechanisms underlying tumor invasion and metastasis. Overcoming the drug resistance mediated by growth and receptor tyrosine kinases has been of particular interest in lung cancer therapy. In this study, we highlight the importance of CSF-1R in regulating osteosarcoma cell EMT and metastasis, and suggest a new therapeutic option to combat metastatic osteosarcoma cancer cells. The increase in CSF-1R expression in osteosarcoma cells resulted in a highly significant increase in the invasiveness and mobility observed in vitro. In contrast, the CSF knockdown osteosarcoma cell had less invasiveness and mobility. Modulation of CSF-1R levels in osteosarcoma cells is achieved at the EMT protein level and correlated with tumor cell phenotype.
A key observation of our study is that among the many metastasis genes whose expression is altered by CSF-R knockdown, JAG1 is a direct target of CSF-1R. Here, we report a new CSF-1R/JAG1 signaling axis in osteosarcoma cancer cell metastasis. We show that CSF-1R regulates JAG1 expression to regulate osteosarcoma cancer migration and invasion. The JAG signaling pathway is commonly activated and altered in cancer. JAG1 has been implicated in the regulation of multiple processes in cancer, including proliferation, survival, EMT, angiogenesis, metastasis, cancer cell “stemness”, and therapy resistance in several cancer types [24]. Consistent with previous studies, in our osteosarcoma in vitro cancer cell metastatic model system, JAG1 was identified as one gene involved in cancer cell migration and invasion. Further investigation is required to delineate the precise characteristics of different cancer cell lines, and investigation of the CSF-1R/JAG1 axis is needed in our future work on lung cancer. Our study has demonstrated that CSF-1R functions as a potential tumor promoter in human osteosarcoma by regulating the activity of JAG1 and accelerating EMT, invasion, and metastasis (Figure 7). Therefore, CSF-1R protein constitutes a promising therapeutic target for reducing tumor progression and dissemination.
Figure 7.
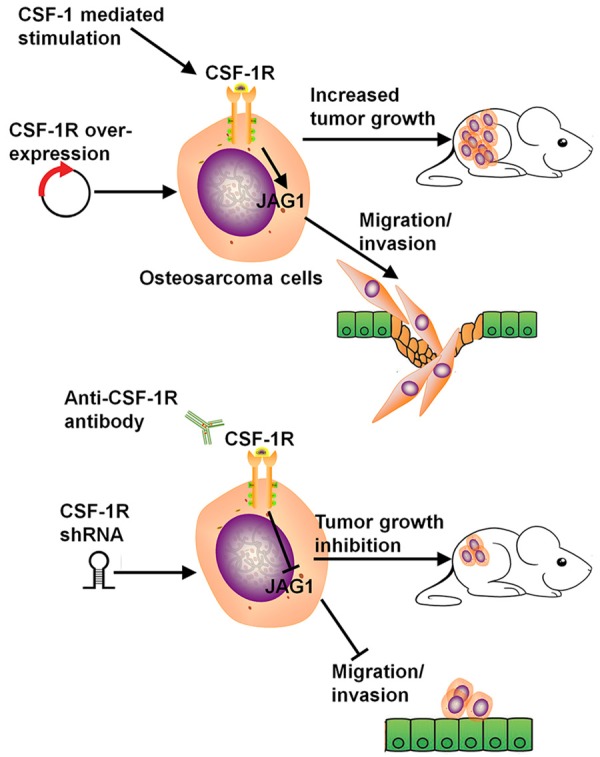
Proposed model: a CSF-1R/JAG1 signaling pathway to regulate osteosarcoma cells metastasis.
Disclosure of conflict of interest
None.
References
- 1.Luanpitpong S, Wang L, Castranova V, Dinu CZ, Issaragrisil S, Chen YC, Rojanasakul Y. Induction of cancer-associated fibroblast-like cells by carbon nanotubes dictates its tumorigenicity. Sci Rep. 2016;6:39558. doi: 10.1038/srep39558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chambers SK. Role of CSF-1 in progression of epithelial ovarian cancer. Future Oncol. 2009;5:1429–1440. doi: 10.2217/fon.09.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Toy EP, Azodi M, Folk NL, Zito CM, Zeiss CJ, Chambers SK. Enhanced ovarian cancer tumorigenesis and metastasis by the macrophage colony-stimulating factor. Neoplasia. 2009;11:136–144. doi: 10.1593/neo.81150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morandi A, Barbetti V, Riverso M, Dello Sbarba P, Rovida E. The colony-stimulating factor-1 (CSF-1) receptor sustains ERK1/2 activation and proliferation in breast cancer cell lines. PLoS One. 2011;6:e27450. doi: 10.1371/journal.pone.0027450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo J, Marcotte PA, McCall JO, Dai Y, Pease LJ, Michaelides MR, Davidsen SK, Glaser KB. Inhibition of phosphorylation of the colonystimulating factor-1 receptor (c-Fms) tyrosine kinase in transfected cells by ABT-869 and other tyrosine kinase inhibitors. Mol Cancer Ther. 2006;5:1007–1013. doi: 10.1158/1535-7163.MCT-05-0359. [DOI] [PubMed] [Google Scholar]
- 6.Sluijter M, van der Sluis TC, van der Velden PA, Versluis M, West BL, van der Burg SH, van Hall T. Inhibition of CSF-1R supports T-cell mediated melanoma therapy. PLoS One. 2014;9:e104230. doi: 10.1371/journal.pone.0104230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lv YF, Yan GN, Meng G, Zhang X, Guo QN. Enhancer of zeste homolog 2 silencing inhibits tumor growth and lung metastasis in osteosarcoma. Sci Rep. 2015;5:12999. doi: 10.1038/srep12999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lv X, Li L, Lv L, Qu X, Jin S, Li K, Deng X, Cheng L, He H, Dong L. HOXD9 promotes epithelial-mesenchymal transition and cancer metastasis by ZEB1 regulation in hepatocellular carcinoma. J Exp Clin Cancer Res. 2015;34:133. doi: 10.1186/s13046-015-0245-3. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 9.Shi J, Li Q, Sheng M, Zheng M, Yu M, Zhang L. The role of TLR4 in M1 macrophage-induced epithelial-mesenchymal transition of peritoneal mesothelial cells. Cell Physiol Biochem. 2016;40:1538–1548. doi: 10.1159/000453204. [DOI] [PubMed] [Google Scholar]
- 10.Kaowinn S, Kim J, Lee J, Shin DH, Kang CD, Kim DK, Lee S, Kang MK, Koh SS, Kim SJ, Chung YH. Cancer upregulated gene 2 induces epithelial-mesenchymal transition of human lung cancer cells via TGF-beta signaling. Oncotarget. 2017;8:5092–5110. doi: 10.18632/oncotarget.13867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Digiacomo G, Ziche M, Dello Sbarba P, Donnini S, Rovida E. Prostaglandin E2 transactivates the colony-stimulating factor-1 receptor and synergizes with colony-stimulating factor-1 in the induction of macrophage migration via the mitogen-activated protein kinase ERK1/2. FASEB J. 2015;29:2545–2554. doi: 10.1096/fj.14-258939. [DOI] [PubMed] [Google Scholar]
- 12.Caescu CI, Guo X, Tesfa L, Bhagat TD, Verma A, Zheng D, Stanley ER. Colony stimulating factor-1 receptor signaling networks inhibit mouse macrophage inflammatory responses by induction of microRNA-21. Blood. 2015;125:e1–13. doi: 10.1182/blood-2014-10-608000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Swierczak A, Cook AD, Lenzo JC, Restall CM, Doherty JP, Anderson RL, Hamilton JA. The promotion of breast cancer metastasis caused by inhibition of CSF-1R/CSF-1 signaling is blocked by targeting the G-CSF receptor. Cancer Immunol Res. 2014;2:765–776. doi: 10.1158/2326-6066.CIR-13-0190. [DOI] [PubMed] [Google Scholar]
- 14.Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, Rey-Giraud F, Pradel LP, Feuerhake F, Klaman I, Jones T, Jucknischke U, Scheiblich S, Kaluza K, Gorr IH, Walz A, Abiraj K, Cassier PA, Sica A, Gomez-Roca C, de Visser KE, Italiano A, Le Tourneau C, Delord JP, Levitsky H, Blay JY, Ruttinger D. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. 2014;25:846–859. doi: 10.1016/j.ccr.2014.05.016. [DOI] [PubMed] [Google Scholar]
- 15.Zhang L, Wang Y, Xiao F, Wang S, Xing G, Li Y, Yin X, Lu K, Wei R, Fan J, Chen Y, Li T, Xie P, Yuan L, Song L, Ma L, Ding L, He F, Zhang L. CKIP-1 regulates macrophage proliferation by inhibiting TRAF6-mediated Akt activation. Cell Res. 2014;24:742–761. doi: 10.1038/cr.2014.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barbetti V, Morandi A, Tusa I, Digiacomo G, Riverso M, Marzi I, Cipolleschi MG, Bessi S, Giannini A, Di Leo A, Dello Sbarba P, Rovida E. Chromatin-associated CSF-1R binds to the promoter of proliferation-related genes in breast cancer cells. Oncogene. 2014;33:4359–4364. doi: 10.1038/onc.2013.542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V, Setty M, Leslie CS, Oei Y, Pedraza A, Zhang J, Brennan CW, Sutton JC, Holland EC, Daniel D, Joyce JA. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19:1264–1272. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sampaio NG, Yu W, Cox D, Wyckoff J, Condeelis J, Stanley ER, Pixley FJ. Phosphorylation of CSF-1R Y721 mediates its association with PI3K to regulate macrophage motility and enhancement of tumor cell invasion. J Cell Sci. 2011;124:2021–2031. doi: 10.1242/jcs.075309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flick MB, Sapi E, Perrotta PL, Maher MG, Halaban R, Carter D, Kacinski BM. Recognition of activated CSF-1 receptor in breast carcinomas by a tyrosine 723 phosphospecific antibody. Oncogene. 1997;14:2553–2561. doi: 10.1038/sj.onc.1201092. [DOI] [PubMed] [Google Scholar]
- 20.Kang C, Sun Y, Zhu J, Li W, Zhang A, Kuang T, Xie J, Yang Z. Delivery of nanoparticles for treatment of brain tumor. Curr Drug Metab. 2016;17:745–754. doi: 10.2174/1389200217666160728152939. [DOI] [PubMed] [Google Scholar]
- 21.Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med. 2001;193:727–740. doi: 10.1084/jem.193.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo XK, Sun HP, Shen S, Sun Y, Xie FL, Tao L, Guo QL, Jiang C, You QD. Synthesis and evaluation of gambogic acid derivatives as antitumor agents. Part III. Chem Biodivers. 2013;10:73–85. doi: 10.1002/cbdv.201200126. [DOI] [PubMed] [Google Scholar]
- 23.Ide H, Seligson DB, Memarzadeh S, Xin L, Horvath S, Dubey P, Flick MB, Kacinski BM, Palotie A, Witte ON. Expression of colony-stimulating factor 1 receptor during prostate development and prostate cancer progression. Proc Natl Acad Sci U S A. 2002;99:14404–14409. doi: 10.1073/pnas.222537099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kascinski B. Expression of CSF-1 and its receptor CSF-1R in non-hematopoietic neoplasms. Cancer Treat Res. 2002;107:285–292. doi: 10.1007/978-1-4757-3587-1_13. [DOI] [PubMed] [Google Scholar]