

Abstract
Background
Fungal plant pathogens belonging to the genus Venturia cause damaging scab diseases of members of the Rosaceae. In terms of economic impact, the most important of these are V. inaequalis, which infects apple, and V. pirina, which is a pathogen of European pear. Given that Venturia fungi colonise the sub-cuticular space without penetrating plant cells, it is assumed that effectors that contribute to virulence and determination of host range will be secreted into this plant-pathogen interface. Thus the predicted secretomes of a range of isolates of Venturia with distinct host-ranges were interrogated to reveal putative proteins involved in virulence and pathogenicity.
Results
Genomes of Venturia pirina (one European pear scab isolate) and Venturia inaequalis (three apple scab, and one loquat scab, isolates) were sequenced and the predicted secretomes of each isolate identified. RNA-Seq was conducted on the apple-specific V. inaequalis isolate Vi1 (in vitro and infected apple leaves) to highlight virulence and pathogenicity components of the secretome. Genes encoding over 600 small secreted proteins (candidate effectors) were identified, most of which are novel to Venturia, with expansion of putative effector families a feature of the genus. Numerous genes with similarity to Leptosphaeria maculans AvrLm6 and the Verticillium spp. Ave1 were identified. Candidates for avirulence effectors with cognate resistance genes involved in race-cultivar specificity were identified, as were putative proteins involved in host-species determination. Candidate effectors were found, on average, to be in regions of relatively low gene-density and in closer proximity to repeats (e.g. transposable elements), compared with core eukaryotic genes.
Conclusions
Comparative secretomics has revealed candidate effectors from Venturia fungal plant pathogens that attack pome fruit. Effectors that are putative determinants of host range were identified; both those that may be involved in race-cultivar and host-species specificity. Since many of the effector candidates are in close proximity to repetitive sequences this may point to a possible mechanism for the effector gene family expansion observed and a route to diversification via transposition and repeat-induced point mutation.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-3699-1) contains supplementary material, which is available to authorized users.
Keywords: Venturia inaequalis, Venturia pirina, Apple, Malus x domestica, European pear, Pyrus communis, Secretome, Effector
Background
Plant-pathogen interactions are a fine interplay between prospective host and pathogen, involving the exchange of molecular signals that determine the outcome of the interaction. The plant endeavours to detect invaders by deploying, as a first line of defence, pattern recognition receptors that recognise pathogen-associated molecular patterns (PAMPs) shared by a wide range of non-specialised microbes, with a resulting induction of a low-level defence response termed PAMP-triggered immunity (PTI). In response, pathogens evolve effectors that enhance a pathogen’s ability to cause disease, often by blocking or suppressing PTI [1–4]. Plants in turn evolve resistance (R) proteins that recognise a subset of these effectors either directly or by their actions on plant host targets. Thus the respective effector and R protein complements can contribute to the determination of host-range for any given pathogen.
The genus Venturia belongs to the order Venturiales, which is assigned to the class Dothideomycetes [5]. The Dothideomycetes include many highly destructive plant pathogens, the Venturiales being no exception. Venturia pathogens are relatively host-specific and infect selected members of the family Rosaceae, perhaps the best known and most widely researched of which is V. inaequalis Cooke (Wint.) that causes the economically important disease apple scab [6, 7]. Related species, for example V. pirina, V. nashicola, V. carpophila and V. cerasi, cause scab diseases of other Rosaceae hosts: European pear, Asian pear, peach and cherry respectively [8–11]. Venturia species cause similar symptoms by adopting analogous biotrophic parasitic strategies [8–10]. During infection they penetrate the host cuticle directly and then develop stromata (laterally dividing, pseudoparenchymatous cells) in the cuticle and sub-cuticular space without penetrating host cells. Ultimately conidiophores and conidia differentiate from stromata and erupt through the cuticle resulting in the formation of dark, crusty, dry lesions (scabs) on leaves and fruit. A fundamental question arises as to what determines the host range of these pathogens, especially given that they have similar modes of biotrophic parasitism and infect closely related host species.
A further level of complexity underlies pathogen/host species specificity in scab fungi, for example, certain isolates classified as V. inaequalis on the basis of morphological and molecular criteria, as well as their ability to mate, are unable to infect Malus, but instead infect different Rosaceous hosts, such as Eriobotrya (loquat) and Pyracantha (firethorn) [12, 13]. Whether these isolates should be considered as separate species or formae speciales is still open to debate. Gladieux and associates [13] suggested that there should be no sub-species delineation based on analysis of six nuclear loci. These isolates are therefore very closely related phylogenetically, but have distinct host specificities. Host cultivar specificity has long been demonstrated in isolates of V. inaequalis that infect Malus [14]; 17 gene-for-gene pairings have been identified to date between races of V. inaequalis and cultivars of Malus. Only two scab R gene loci in apple (Rvi6 and Rvi15) have been fully characterised [15–18]; however, no effectors of V. inaequalis have been characterised to date. Work to identify the V. inaequalis effector repertoire that determines cultivar specificity, and most probably host specificity, has been impeded by the lack of a whole genome sequence for this species.
Genome estimates for V. inaequalis range from 38 Mb [19] to 100 Mb [20]. The de novo transcriptome of a single isolate of V. inaequalis has been published [21], with analysis of both in vitro and in planta transcripts. In our study the whole genome sequences of four isolates of V. inaequalis (three physiological races, differing in their ability to infect apple accessions carrying different R genes, and a loquat-infecting isolate) were sequenced using Illumina sequencing technologies. The V. inaequalis genomes were compared to a single V. pirina genome [22]. Given the extracellular in planta niche the Venturia fungi occupy during parasitism it can be assumed that the majority of the factors that enable successful parasitism are first secreted into the sub-cuticular plant-pathogen interface. Therefore the secretomes of these isolates are of utmost relevance and so were compared to identify putative pathogencity or virulence factors that may have a role in host determination, with special emphasis on secreted enzymes and putative proteinaceous effectors.
Results
Whole genome assemblies and gene predictions
A total of 40 Mb (Vi1.2.8.9) to 61 Mb (Vi1.10) was assembled for Malus-infecting V. inaequalis isolates. The assembled genome size for the loquat scab pathogen (ViL) was 62 and 41 Mb for the pear scab pathogen (Vp). Lack of sequencing of mate-pair libraries with long inserts contributed to the Vi1.2.8.9 assembly having the lowest N50 value (~49 kb) among the five isolates, while Vp had the largest N50 (332 kb, Table 1, Additional file 1). A summary of the repeat content masked for each genome assembly is presented in Table 1 and Additional file 2.
Table 1.
The whole genome assemblies of isolates of Venturia inaequalis and V. pirina
| Species | Venturia inaequalis | Venturia pirina | |||
|---|---|---|---|---|---|
| Isolate (race) | Vi1 (race 1) = ICMP13258 = MNH120 | Vi1.10 (race 1,10) = EU-B04d | Vi1.2.8.9 (race 1,2,8,9) = 1639 | ViL = 1389 | Vp = ICMP11032 |
| Host range | Malus × domestica | Malus × domestica | Malus × domestica | Eriobotrya japonica | Pyrus communis |
| Estimated genome coverage | 120x | 90x | 89x | 89x | 74x |
| Package used for assembly | Velvet | ALLPATHS -LG | Velvet | ALLPATHS -LG | Velvet |
| Number of scaffolds | 1012 | 1415 | 1680 | 1040 | 364 |
| Size (Mb) | 55 | 61 | 40 | 62 | 41 |
| N50 | 233760 | 136376 | 48770 | 213378 | 332167 |
| N90 | 16289 | 23736 | 11843 | 37088 | 81548 |
| Repeats in total genome (%) | 4.11% | 33.41% | 0.74% | 35.60% | 7.28% |
| Number of predicted genesa | 13333/12546 | 12234 | 12868 | 12258 | 11960 |
| Percentage completeness: partial (full)b | 99.60 (99.19) | 98.79 (95.16) | 98.39 (94.76) | 97.58 (94.76) | 98.39 (95.16) |
| Percentage completeness: partial (full)c | 99.51 (96.45) | 99.37 (97.01) | 99.37 (96.94) | 99.51 (97.22) | 99.65 (97.01) |
aHybrid with hints/ab initio gene predictions for Vi1
bCEGMA analysis using both partial and full gene sequences [25]
cBUSCO analysis using both partial and full gene sequences [26]
dNote the recent race designation change reported for EU-B04 [187], previously reported as race (1,14) by Bus et al. [14]
Gene models were predicted with AUGUSTUS gene prediction software (Table 1 and Additional file 3) [23, 24]. The number of gene models in the V. inaequalis isolates ranged from 12,234 to 13,333 whereas there were 11,960 identified in V. pirina. To estimate the completeness of the assembled genomes, the assemblies were scanned for the 248 most highly conserved core eukaryotic genes using the Core Eukaryotic Genes Mapping Approach (CEGMA; Table 1 and Additional file 4) [25]. Comparisons of partial or complete predicted translated proteins demonstrated that the V. inaequalis Vi1 genome is the most complete, with only one or two core eukaryotic genes missing respectively. The lowest representation of the 248 core eukaryotic genes in any genome was still high, 235 complete and another seven partial predicted proteins for V. inaequalis ViL, indicating a reasonably complete whole genome sequence for all isolates. In addition, a Benchmarking Universal Single-Copy Orthologs (BUSCO) analysis was also conducted using BUSCO_v1.1b [26]. This analysis indicated that the V. pirina genome is the most complete and of the V. inaequalis genomes Vi1 and ViL are the most complete when partial predicted proteins are considered, although on comparison of complete predicted proteins the ViL genome is the most complete (Table 1 and Additional file 5). Since Vi1 was the most comprehensive V. inaequalis genome in terms of coverage when CEGMA and BUSCO analyses were taken together, and is predicted to have the most complete set of avirulence effectors (due to the avirulent phenotype of this isolate on all but one of the set of resistant apple host differentials following inoculation in glasshouse trials; Vincent Bus, personal communication), this isolate was selected for preliminary expression (RNA-Seq) analyses.
The predicted secretomes of Venturia inaequalis and V. pirina
To predict the secretome of each isolate, gene models were analysed with a pipeline of programmes (Additional file 6). The total number of genes encoding the predicted secretome for each genome is as follows: 1622 in Vi1; 1158 in Vi1.10; 1324 in Vi1.2.8.9; 1131 in ViL and 1139 in Vp. The number of predicted proteins in the Vi1 secretome is greater than that for the other genomes due to use of both ab initio (based on the trained species model) and hybrid (evidence informed) gene predictions in the analysis. Thakur et al. [21] reported a lower predicted secretome number of 946, however this was based on transcriptome data alone and, as such, direct comparison is problematic. Gene ontology analysis through an in-house annotation pipeline (BioView; Ross Crowhurst, Marcus Davy and Cecilia Deng, unpublished) was used to annotate predicted genes in each secretome; however, the majority of the predicted proteins had no gene ontology classification. Hence, for this reason, the Carbohydrate Active Enzyme (CAZyme) Analysis Toolkit (CAT) [27], and similarity searches utilising BLASTp [28] against the NCBI non-redundant (nr) database [29] were used in addition to gene ontology. Where conflicting designations occurred, CAT, then gene ontology took precedence over similarity search designations. The secretome repertoire, in terms of diversity and abundance of protein classes, was similar between all Venturia isolates, with small, secreted, non-enzymatic proteins (SSPs) dominating (Fig. 1). For comparative purposes the secretomes of three plant pathogenic fungi, Parastagonospora nodorum (synonym: Stagonospora nodorum), Cladosporium fulvum and Puccinia graminis f. sp tritici were also identified using the same pipeline of programmes using predicted proteins downloaded from the Joint Genome Institute (JGI; Additional files 6 and 7). The size of the P. graminis f. sp tritici secretome (1820 predicted proteins) was significantly larger than that recorded for C. fulvum and P. nodorum (1170 and 1122 predicted proteins respectively). The size of the Vi1 secretome was similar to that of P. graminis f. sp. tritici, whereas the remaining Venturia secretomes were of a similar size to those of P. nodorum and C. fulvum.
Fig. 1.
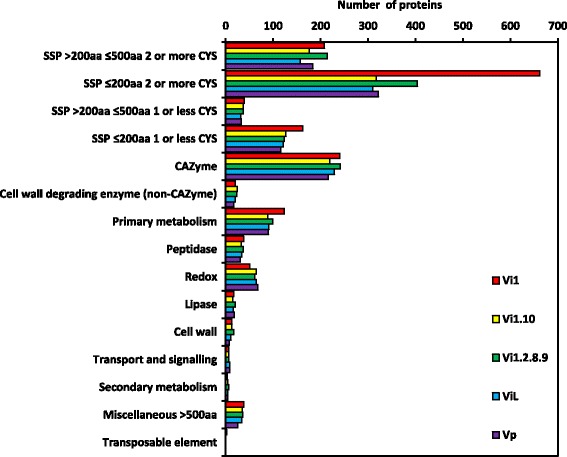
The secretomes of four isolates of V. inaequalis and one isolate of V. pirina. Annotations were based on gene ontology analysis including interrogation of NCBI RefSeq [168], InterPro [169], UniRef [170], ExPASy UniProtKB/Swiss-Prot [171] and ExPASy Prosite [172], CAZyme identification using the CAT server [27] and BLASTp searches against the NCBI non-redundant database [29]. Small secreted proteins (SSPs) include predicted proteins with similarity to known effectors and proteins with no known function, either with or without putative conserved motifs identified by PfamScan [36–38]
OrthoMCL was conducted to assess similarity between the predicted Venturia secretomes (Fig. 2 and Additional file 8) [30]. A total of 898 isolate-specific singletons were identified (310 in Vi1; 37 in Vi1.10; 118 in Vi1.2.8.9; 88 in ViL, and 345 in Vp) but are not included in the OrthoMCL clustering figure (Fig. 2). The Venturia core secretome (those predicted secreted proteins represented in all Venturia isolates) consisted of 514 orthologous clusters, with CAZymes (163) and non-enzymatic SSPs (225) being the predominant classes (Additional file 8). The Venturia pan secretome (the sum of all predicted secreted proteins from all Venturia isolates) totalled 5474 representing 2238 individual proteins (1340 clusters, plus 898 isolate-specific singletons), with many lineage-specific and host-specificity candidates identified, most of which were predicted to encode non-enzymatic SSPs. The majority of these SSPs had no significant similarity to proteins in the public domain. The V. inaequalis-unique (including both apple- and loquat-infecting strains) pan secretome totalled 2033 representing 1156 individual proteins (603 clusters and 553 singletons), with a total of 929 representing 672 individual proteins (207 clusters plus 465 singletons) specific to apple-infecting isolates, with 85 of the clusters (mostly with a single protein from each isolate) found in all three isolates. There were 88 singletons, but no unique clusters specific to the loquat-infecting isolate ViL. The V. pirina-specific secretome totalled 480 representing 387 individual proteins, with 16 unique protein clusters and 345 singletons.
Fig. 2.
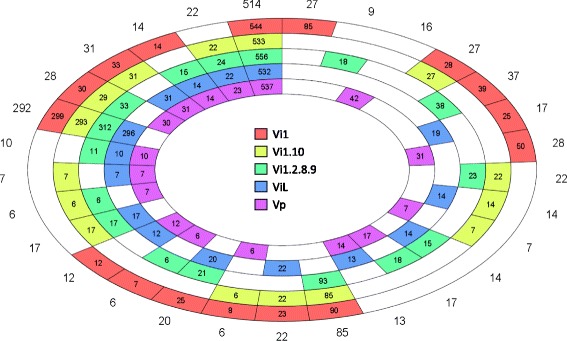
Proteins in the secretomes of four isolates of Venturia inaequalis and one of V. pirina. Similar proteins in each of the secretomes were identified by OrthoMCL-2.0.3 [30] together with the Markov clustering algorithm mcl-09-149 [173]. Similarity levels were calculated based on reciprocal BLASTp similarity searches between the protein sequences with an e value threshold of 1e-10. Figures within the boxes represent the number of proteins in each cluster, whereas figures outside the wheel are the number of clusters in each sector. Those proteins that are singletons within each secretome are not represented
Two sets of RNA-Seq data, from libraries made with RNA harvested from V. inaequalis Vi1 grown in vitro (in cellophane membranes) and Vi1-infected apple seedling leaves (two and seven days post inoculation; dpi), provided evidence of transcription for 93.4% of the Vi1 secretome. The RNA-Seq data were also used to select a smaller subgroup of the Vi1 secretome to analyse in greater detail (sequences are deposited under the BioProject ID PRJNA261633: in vitro: SRR1586226, two dpi in planta: SRR1586224, seven dpi in planta: SRR15862230). These data were used as indicators of involvement in virulence or pathogenicity. Figure 3 shows representative stages of this material (no signs of microbial contamination were evident in the sterile distilled water (SDW)-inoculated apple leaves, as assessed by microscopic evaluation). Two approaches were used: first, overall Vi1 gene expression levels were estimated by analysing fragments per kilobase of exon per million reads mapped (FPKM) values. This enabled a ranking of the genes in terms of overall expression during infection to be calculated at both two and seven dpi and the most highly expressed genes at either time point were selected to give the top 5% data set. Of the 82 genes identified, 48 were expressed at two and seven dpi, 17 at two only and 17 at seven dpi only. In addition, a differential expression analysis detected 268 of the 1622 genes that were up-regulated during growth in planta, compared with in vitro growth (log2 fold change not smaller than two with a false discovery rate of less than 0.05). Thirty-seven genes were in both the top 5% data set and up-regulated during infection gene sets, giving a total of 313 individual genes (which will be referred to henceforth as the V enturia Infection Secretome or ‘VIS’ gene set; Fig. 4).
Fig. 3.
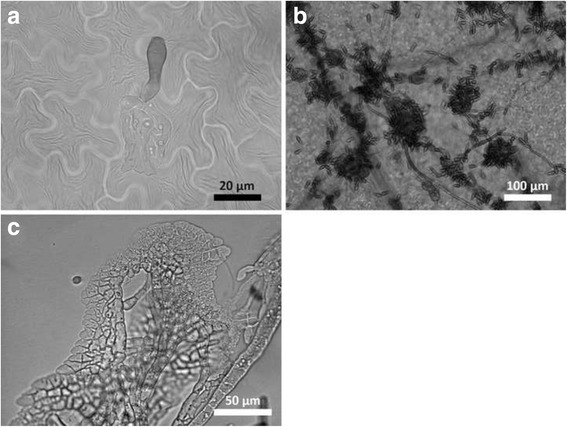
Microscopic evaluation of Venturia inaequalis Vi1 infection of susceptible apple leaves. a Two, and b seven days post inoculation, and c in vitro (in cellophane) showing stromatic growth habit
Fig. 4.
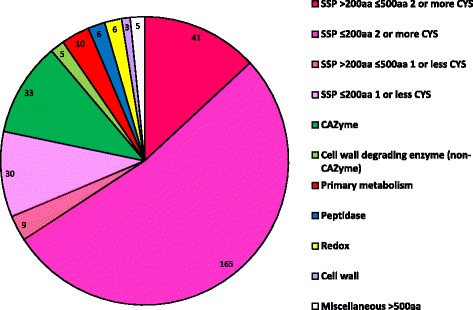
Predicted proteins present in the VIS set from the secretome of Venturia inaequalis isolate Vi1. Annotations were based on gene ontology analysis including interrogation of NCBI RefSeq [168], InterPro [169], UniRef [170], ExPASy UniProtKB/Swiss-Prot [171] and ExPASy Prosite [172], CAZyme identification using the CAT server [27] and BLASTp searches against the NCBI non-redundant database [29]. Small secreted proteins (SSPs) include predicted proteins with similarity to known effectors and proteins with no known function, either with or without putative conserved motifs identified by PfamScan [36–38]. The order of the categories in the legend is the same as that in the chart
Carbohydrate-Active Enzymes (CAZymes) and putative non-CAZyme cell wall degrading enzymes
The CAZyme repertoire (216 to 240 proteins, depending on the isolate) was similar between all Venturia secretomes, with 167 belonging to the core secretome (Additional files 8, 9 and 10). Of these, all isolates have between 16 and 25 proteins with multiple CAZyme domains.
Proteins were identified as being associated with carbohydrate-binding motif (CBM), carbohydrate esterase (CE), glycoside hydrolase (GH), polysaccharide lyase (PL) and glycosyl transferase (GT) domains. Of these, the classes with the greatest number of protein members across each genome were those with: CBM2, CBM1 and CBM13 domains, with specificity for cell wall carbohydrates, and in terms of the latter two, plant cell wall specifically; CE5 domains with specificity for cutin; GH28 with specificity for plant cell wall pectin; GH3 and GH5 with specificity for β-glycans; and GH43 with specificity for pectin and hemicelluloses. Also identified, although not as numerous, were CAZyme domain-containing proteins predicted to interact with fungal cell wall components including: GHs associated with β-glycans, α-glucan, chitin, and CBM domains specific for chitin [31, 32].
The majority of the CAZymes were either present in OrthoMCL clusters that had a single enzyme from each isolate and were thus members of the core secretome set, or in clusters comprised of enzymes from only the four isolates of V. inaequalis, not V. pirina (Additional files 7 and 9).
Similar numbers of predicted proteins (17 to 24) were identified in the secretome sets, which were similar to enzymes that degrade non-carbohydrate cell-wall components, e.g. ligninases (Fig. 1 and Additional file 8).
Peptidases and lipases
Depending on the isolate, 31 to 38 predicted proteins were identified in the Venturia secretomes with similarity to peptidases (Figs. 1 and 4, Additional files 2 and 11). The majority of these were conserved in the core secretome or in isolates of V. inaequalis rather than V. pirina. There were proteins predicted in the secretomes of only the V. inaequalis isolates that were similar to AVR-Pita, the avirulence protein from Magnaporthe with zinc metalloprotease features that binds directly to its cognate R protein [33, 34], albeit with a relatively high similarity score of 8e-16 recorded when the Pathogen Host Interactions database (PHI-base) [35] was interrogated. In addition, a further similar protein was also encoded by the genome of V. pirina but was not predicted to be secreted. There were also between eight and 11 proteins, depending on secretome, with similarity to proteases from Candida and Coccidioides following interrogation of protein sequences against PHI-base. Predicted proteins with similarity to lipases were also identified in each secretome (15 to 20, depending on the secretome).
Primary metabolism and putative proteins involved in redox reactions
Proteins with a wide range of functionality in primary metabolism, e.g. proteins involved in amino acid (aa) or nucleotide metabolism; phosphatases and nitrilases, and unclassified enzymes with an alpha/beta hydrolase fold were identified. Depending on the isolate, there were between 88 and 123 proteins in this category in each secretome, most of which were in the core Venturia secretome. Numerous (51 to 68) proteins with putative functions in redox reactions were identified in the Venturia secretomes.
Putative proteins associated with the cell wall, transport, signalling and secondary metabolism
As expected there were few proteins in the secretomes associated with the cell wall, transport, signalling and secondary metabolism, since these proteins would be eliminated in the secretome prediction pipeline (Additional file 6). The majority of these proteins were members of the core secretome set (Additional file 8).
Small secreted proteins
Proteins classified as SSPs (i.e. mature predicted protein ≤500 aa in length, including proteins with similarity to known effectors, but excluding proteins with similarity to lytic enzymes) made up between 55 and 66% of each secretome (Fig. 1 and Additional file 8). For comparative purposes, three secretomes derived from publicly available genomes (housed at the JGI) were also screened for SSPs using the same pipeline of programmes as those used to analyse the Venturia secretomes. SSPs (less than 500 aa in length) made up 79% of the secretome of P. graminis f. sp. tritici, and 43 and 28% of the secretomes of P. nodorum and C. fulvum respectively. Smaller SSPs less than 200 aa in length predominated in the Venturia and P. graminis f. sp. tritici secretomes, especially those with a predicted signal peptide. In contrast, SSPs of less than 200 aa in length in the predicted SSP repertoires of C. fulvum and P. nodorum were dominated by those predicted to be secreted by a non-classical mechanism (Additional file 7). The majority of genes encoding SSPs in Vi1 were supported with evidence of transcription (95%). Most SSPs are novel to Venturia (i.e. had BLASTp similarity of ≥1e-10 to any non-Venturia proteins in public databases) and most of these novel SSPs were ≤200 aa in length. The vast majority of VIS genes, 245/313, were predicted to encode SSPs (≤500 aa in length), 195 of which were ≤200 aa in length (Fig. 4). Of these smaller SSPs, 165 have two or more cysteines in the mature predicted protein. Larger secreted proteins (>500 aa in length) with no similarity to known proteins were also represented in each of the secretomes (25 to 38 putative proteins; Fig. 1).
Candidate effectors from the SSP set
The secretomes of Venturia have several proteins with a putative role in binding hydrophobic surfaces including between 12 and 15 proteins with hydrophobic surface binding (HsbA) domains (Pfam: PF12296.3) as identified by PfamScan [36–38], with similarity to the Aspergillus oryzae HsbA, Hydrophobic Surface Binding, cell wall galactomannoprotein. Five putative HsbA-like genes were identified amongst the Vi1 VIS gene set. Another gene up-regulated at seven dpi encodes a small predicted mature protein of 120 aa with four cysteines and shares predicted aa sequence identity (e value: 1e-17) to bacterial chaplins, which are membrane-associated proteins known to have similar functions to hydrophobins [39]. Predicted proteins with similarity to bacterial chaplins are also present in Vi1.10, ViL and Vp, but not Vi1.2.8.9.
Two or three proteins with similarity to fungal hydrophobins were identified in the secretomes of V. inaequalis isolates and four in the V. pirina secretome, on the basis of conserved cysteine patterns [40, 41]. None were included in the VIS gene set.
The Venturia secretomes included predicted SSPs with similarity to known effectors from other fungi, including Ecp6 (GenBank: ACF19427.1) from C. fulvum [42], AvrLm6 (CAJ90695.1) from Leptosphaeria maculans [43] and Ave1 (AFB18185.1) from Verticillium spp. [44]. The Venturia candidate effectors were most similar to these effectors in a reciprocal BLAST of the Cladosporium, Leptosphaeria and Verticillium genomes. In addition to the OrthoMCL clustering analysis, further similarity searches within genomes were employed with a more relaxed threshold of 1e-5, to ascertain whether putative paralogues (multigene families) were present. Single orthologues of Ecp6 were identified in each of the Venturia genomes analysed, corroborating the data of Thakur et al. [21] who reported an orthologue of Ecp6 in the transcriptome of a single apple-specific isolate of V. inaequalis. A search of the Pfam database [36] with the Vi1 orthologue of Ecp6 revealed three peptidoglycan-binding, lysin motif (LysM) domains. At the aa level there is 42% identity between Ecp6 and the V. inaequalis orthologues, whereas the V. pirina orthologue has slightly higher identity at 44%. The Vi1 and ViL predicted proteins are identical, whereas that from Vi1.2.8.9 has one aa substitution compared with the Vi1/ViL sequence and that from Vi1.10 has four aa substitutions. The Vi1 Ecp6 orthologue was expressed during growth in planta and in vitro, although expression was not sufficiently high to be in the VIS set.
Orthologues of AvrLm6 [45] and Ave1 are present as large gene families in all of the Venturia genomes examined. The copy number of full length AvrLm6-like genes varied among V. inaequalis isolates (24–29 copies), while the V. pirina isolate had 16 copies. Further characterisation of the Venturia AvrLm6-like gene families is reported in Shiller et al. [45]. Three AvrLm6-like genes are present in the VIS gene set.
Ten full-length Ave1-like genes and seven Ave1-like pseudogenes, were identified in the whole genome sequence of V. inaequalis Vi1, with similar numbers in all other Venturia genomes. The V. inaequalis Ave1-like full length, predicted proteins range in similarity from 37 to 57% identity to V. dahliae Ave1. All but one of the Vi1 Ave1-like proteins have transcript evidence, with expression at all growth stages, including in vitro (on cellophane), with some very highly expressed during both infection time points. Two Ave1-like gene loci are represented in the VIS gene set.
Loci of all 16 of the previously identified V . i naequalis candidate effector (VICE) genes [46] from Vi1 were identified in the Vi1 WGS. However, the majority of the predicted gene models associated with these loci were longer than those identified using ESTs [46]. One VICE gene (VICE 13) did not correspond with any AUGUSTUS gene prediction. Only six VICE predicted proteins were included in the secretome of Vi1 (corresponding to VICE 4, 5, 6, 9, 10 and 11), five of which were in OrthoMCL clusters in the core secretome, with a single predicted protein from each isolate in each cluster. The sixth predicted protein (corresponding to VICE 4) belongs to a cluster with members from only Vi1 (19) and Vp (four). Genes encoding four of these proteins were in the VIS set, including the VICE 4 gene. The remaining nine VICE gene models, assigned by AUGUSTUS, did not encode proteins that were predicted to be secreted. Inspection of these loci revealed that, in the majority of these cases, the N-terminus predicted by AUGUSTUS was upstream of the translation start site predicted by Bowen and associates [46]. Thus the gene predictions at these loci need further validation by sequencing full length cDNAs. Most of the VICE gene transcripts were also detected by Thakur et al. [21], in a single transcriptome of V. inaequalis; however, these authors did not confirm whether these transcripts encoded predicted secreted proteins.
Two cellophane-induced (Cin) genes, Cin1 and Cin3, that encode proteins with internal repeats, have been previously identified as being highly expressed during both in vitro growth in cellophane membranes (10 dpi) and infection of Vi1 on apple (five and 10 dpi) [47]. The RNA-Seq data supported the expression levels reported in the previous study for both of these genes. There were Cin1 orthologues in each of the isolates examined forming a single OrthoMCL cluster, whereas Cin3 orthologues were only in V. inaequalis isolates, again forming a single OrthoMCL cluster. Cin1 was one of the most highly expressed genes in the VIS gene set, with very high expression in vitro as well as at both infection time points.
Two additional genes, which encode smaller cysteine-rich Cin1-like proteins, Cin1L1 and Cin1L2 (106 and 169 aa) [48, 49], are also found in each of the isolates. However, their sequence is more variable since a single cluster with representative proteins from each isolate was not observed. The proteins similar to Cin1L1 from each V. inaequalis isolate clustered together, but that from the V. pirina isolate was classified as a singleton. There are proteins similar to Cin1L2 in each isolate; however, those proteins encoded by the Vi1.10 and Vi1.2.8.9 genomes, although having a signal peptide, were classified as being targeted to the mitochondria by both ProtComp and WoLF PSORT, and the proteins from the remaining isolates formed a single cluster. Cin1L1 and Cin1L2 are in the VIS gene set.
Of those proteins originally classified as SSPs in the Venturia secretomes four shared similarity to proteins in PHI-base [35]: the hydrophobin MHP1 from Magnaporthe oryzae [50]; NPP1 from Hyaloperonospora arabidopsidis [51–53]; GAS1 from Magnaporthe oryzae [54]; BEC1019 from Blumeria graminis f. sp. hordei [55, 56]. None of these proteins were members of the VIS set, although all had evidence of transcription in vitro and in planta (Additional file 12).
SSP families in the VIS set
Of the 195 VIS SSPs ≤200 aa in length in the Vi1 secretome, 64 appear to be single proteins following spectral clustering analysis [57], 121 belong to 38 different families, having between two and 86 members. The families include predicted proteins without in planta up-regulated expression of their corresponding genes and putative proteins that were not predicted by the original gene prediction software (Figs. 5 and 6). The remaining 10 SSPs were not retained in the dataset since, although initially classified as an SSP ≤200 aa in length, they belong to families where the majority of members have a robust annotation to an enzyme based on gene ontology analysis and similarity searches, or are larger than 200 aa in length. Family 4, 6 and 35 are very closely related and as an indication of this these families share members (four are in both family 4 and 6 and one is in both family 6 and 35). A representative sequence logo for family seven is shown in Additional file 13. Although sequence diverse, the families have a putative conserved structure based on conservation of cysteines.
Fig. 5.
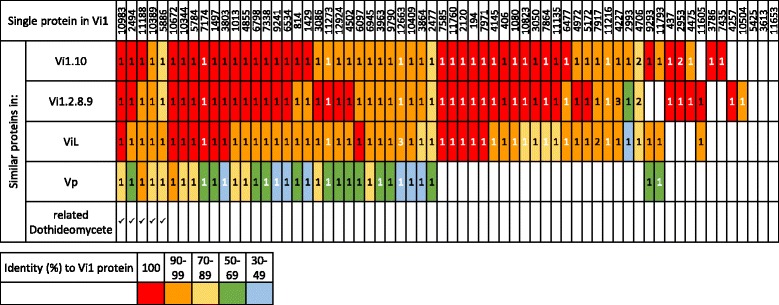
Small secreted proteins (SSPs) in the Venturia inaequalis Vi1 secretome encoded by single genes. Only SSPs ≤200 amino acids in length are included. The number of similar proteins in the Venturia and related Dothideomycete proteomes are indicated by numbers: black indicates a gene predicted by AUGUSTUS, white indicates a putative coding sequence identified by tBLASTn using the protein sequence as query, followed by manual curation. Percentage identity is represented by: red = 100%; orange = 90–99%; yellow = 70–89%; green = 50–69; blue = 30–49%
Fig. 6.
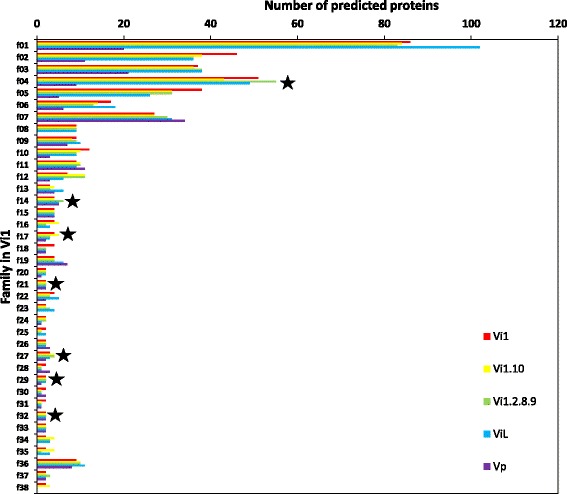
Small secreted protein (SSP) families in four Venturia inaequalis and one V. pirina secretomes. Only SSPs ≤200 amino acids in length are included. Red = proteins in the same family from Vi1; yellow = protein families from Vi1.10; green = families from Vi1.2.8.9; blue = families from ViL; purple = families from Vp. Families f1-f28, and f34-f38 are cysteine rich i.e. two or more cysteines per protein; f29-f33 are those with one or no cysteines. ★ = families with predicted similar proteins in related Dothideomycete genomes
Thirty families of predicted SSPs, and 31 single proteins, have similar proteins present in all of the Venturia isolates analysed. All, except five, of the single proteins (from the Vi1 isolate) are also single proteins in all of the isolates studied. Seven families are unique to and conserved within the isolates of V. inaequalis, whereas there are 20 single proteins in Vi1 with similar proteins present in all V. inaequalis isolates and absent from V. pirina. There is one family, and two single proteins, restricted to isolates that only infect apple (Figs. 5 and 6).
In addition, there are four single SSP proteins, and one family, that are putative candidates for the effector/s that contribute to the race profile of isolate Vi1.2.8.9, i.e. present in Vi1 and Vi1.10, but absent from Vi1.2.8.9 (Table 2 and Additional file 14). There is one family and three single proteins that are candidates for effector AvrRvi10, given their presence (in Vi1 and Vi1.2.8.9) and absence (in Vi1.10) in the Venturia isolates.
Table 2.
Small secreted proteins (SSPs) in the Venturia infection secretome (VIS) set and related fungi
| Similar proteins ina: | Families in Vi1 | Single proteins in Vi1 | |||||
|---|---|---|---|---|---|---|---|
| Vi1 | Vi1.10 | Vi1.2.8.9 | ViL | Vp | Related Dothideomycete | ||
| ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | 7 | 5 |
| ✓ | ✓ | ✓ | ✓ | ✓ | 23 | 26 | |
| ✓ | ✓ | ✓ | ✓ | 6 | 20 | ||
| ✓ | ✓ | ✓ | ✓ | 1 | 0 | ||
| ✓ | ✓ | ✓ | ✓ | 0 | 2 | ||
| ✓ | ✓ | ✓ | 0 | 3 | |||
| ✓ | ✓ | ✓ | 0 | 1 | |||
| ✓ | ✓ | 1 | 2 | ||||
| ✓ | ✓ | 0 | 2 | ||||
| ✓ | 0 | 3 | |||||
Both families and single genes encoding small secreted proteins ≤200 amino acids in length are included. Families were analysed by Spectral Clustering followed by manual curation and analysis of non-predicted putative genes
aTicks in columns indicate presence of a similar protein or proteins in that secretome
To ascertain whether any of the 195 SSPs of the VIS set were conserved across the Dothideomycetes, similarity searches (BLASTp) were made against selected Dothideomycete predicted proteomes curated at the JGI (Additional file 15) [58, 59]; this curation includes clustering information pertaining to proteins from each organism. Only SSPs predicted for the Vi1 secretome, which were present in all the Venturia isolates, had putative similar members in related Dothideomycete predicted proteomes (Table 2 and Additional file 14, Figs. 5, 6 and 7).
Fig. 7.
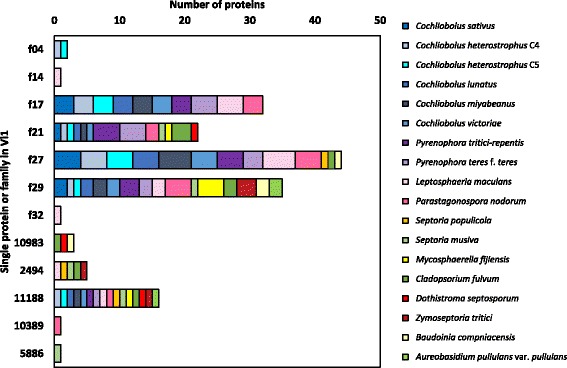
Similar small secreted proteins (SSPs) in Dothideomycete proteomes and the Venturia inaequalis Vi1 secretome. Only SSPs ≤200 amino acids in length are included. Shades of blue, purple and pink = Pleosporales: Cochliobolus sativus, C. heterostrophus C4, C. heterostrophus C5, C. lunatus, C. miyabeanus, C. victoriae, Pyrenophora tritici-repentis, P. teres f. teres, Leptosphaeria maculans, Parastagonospora nodorum; shades of orange, yellow and red = Capnodiales: Septoria populicola (teleomorph Mycosphaerella populicola), S. musiva (teleomorph M. populorum), M. fijiensis, Cladosporium fulvum (syn: Passalora fulva), Dothistroma septosporum, Zymoseptoria tritici, Baudoinia compniacensis; green = Dothideales: Aureobasidium pullulans var. pullulans
Putative host range determinants in the SSP set
In addition to those candidate effectors that are specific to either V. inaequalis, or those isolates able to infect apple described above, SSPs ≤200 aa in length specific to the loquat-infecting isolate of V. inaequalis or to V. pirina were identified by OrthoMCL analysis (Additional file 16). In addition to the 62 candidate effectors specific to ViL, only four of which had any similarity to proteins in the NCBI nr database, 6 were found in both the ViL and Vp secretomes. A further 238 candidate effectors were found to be specific to V. pirina.
Gene density and proximity of genes to transposable elements (or transposable element remnants)
Most genes in the Vi1 genome have intergenic regions between 200 bp and 3 kb (Fig. 8), with a mean intergenic distance of 1677 bp. A subset of core eukaryotic genes [25] had a slightly shorter mean intergenic distance of 1289 bp, whereas the SSP genes in the VIS set (highlighted as pink data points in Fig. 8), display a significantly (P < 0.005) longer average intergenic distance at 2560 bp. Genes located on scaffold ends, hence lacking neighbouring genes, were excluded from this analysis. This excluded 17/440 (3.6%) from the core eukaryotic gene set and 21/245 (8.6%) from the SSPs in the VIS set. The greater proportion of SSP genes located on the ends of scaffolds suggests a bias for association with complex repeat regions that cause problems with sequencing and assembly, resulting in broken genomic scaffolds.
Fig. 8.
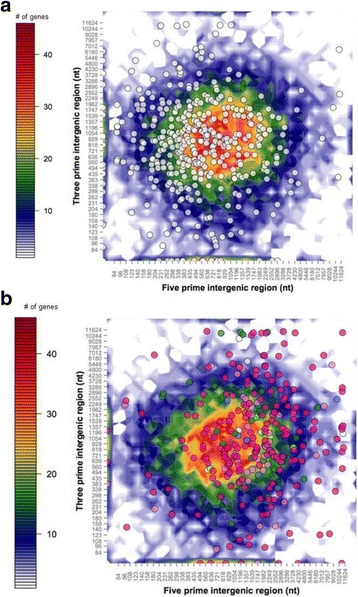
Flanking distance (3′ and 5′) between predicted genes of Venturia inaequalis Vi1. Intergenic distances for all predicted genes are represented in the underlying heatmap, with the number of genes in each bin shown as a colour-coded heat map (on orthogonal projection) generated as in Saunders et al. [185]. Genes were sorted into two-dimensional bins on the basis of the lengths of flanking intergenic distances to neighbouring genes at their 5′ and 3′ ends; overlying this are scatterplots of a 423 Core Eukaryotic Genes (white dots) or b Venturia infection secretome (VIS) gene set, plus AvrLm6- and Ave1-like genes (coloured dots: dark pink = SSPs with two or more cysteines (≤500 amino acids); light pink = SSPs with one or no cysteines (≤500 amino acids); blue = peptidases; dark green = CAZymes; light green = putative cell wall-degrading enzymes (non-CAZyme); white = cell wall associated and miscellaneous proteins >500 amino acids). Note that the axes are not linear. Genes at the scaffold end were excluded from this analysis
Concordantly, SSP genes also appear to be more closely associated with transposable elements (TEs) or TE-like features than core eukaryotic genes and all remaining genes in the Vi1 genome. The mean gene to TE distance was 9024 bp for all genes predicted in the genome (Fig. 9). The core eukaryotic gene set had a significantly (P < 0.005) greater mean distance to the nearest TE of 10,528 bp, whereas the SSPs in the VIS set had a mean distance to the nearest TE (or TE-like features) of only 5401 bp. Twenty-two SSPs in the VIS set were nested within predicted TE-like features. Fourteen of the core eukaryotic genes and six of the VIS set SSP genes were excluded from this analysis as they lacked a neighbouring TE feature on the same scaffold.
Fig. 9.
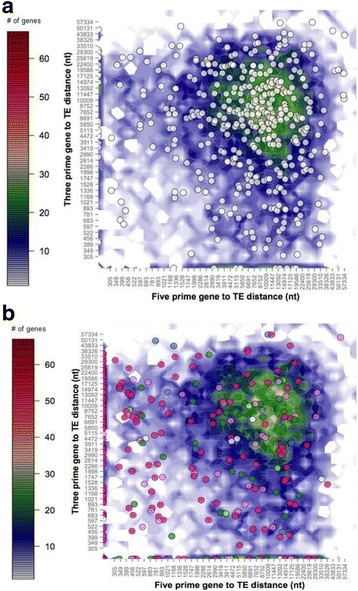
Distance between predicted genes and transposable element (TE)-like features of Venturia inaequalis Vi1. Flanking distances (3′ and 5′) to TE-like features for all predicted genes are represented in the underlying heat map, with the number of genes in each bin shown as a colour-coded heat map on orthogonal projection (generated as in Saunders et al. [185]). Genes were sorted into two dimensional bins on the basis of the lengths of flanking distances. a 423 Core Eukaryotic Genes (white dots) are similar to all genes in that they are not closely associated with TE-like features; whereas b Venturia infection secretome (VIS) gene set, plus AvrLm6- and Ave1-like genes (coloured dots: dark pink = SSPs with two or more cysteines (≤500 amino acid); light pink = SSPs with one or no cysteines or less (≤500 amino acids); blue = enzymes (peptidases/redox/primary metabolism); dark green = CAZymes; light green = putative cell wall-degrading enzymes (non-CAZyme); white = cell wall associated and miscellaneous proteins >500 amino acids in length). Note that the axes are not linear. Genes at the scaffold end were excluded from this analysis
Discussion
Comparisons of whole genome assemblies and gene predictions
This paper reports the first analysis of whole genome sequences from multiple isolates of V. inaequalis (V. pirina being released in 2014) [22]. The sizes of all five Venturia genomes are comparable to those of other Dothideomycetes [60]; however, these may be an underestimate since the majority of the sequencing was carried out on the Illumina platform. Assembling repeat regions using short reads is notoriously difficult [61–64], and hinders genome assembly using de Bruijn graph-based genome assemblers like Velvet and ALLPATHS-LG [62, 64, 65].
In addition, there is an apparent wide range of genome size that can be largely attributed to additional repetitive content, with the assembled genic regions being similar in size for all five Venturia isolates. Whilst variation in genome size between isolates from the same species is not unprecedented; for example, genome size in three isolates of the Dothideomycete Zymoseptoria tritici (synonym Mycosphaerella graminicola) ranged from 31 to 40 Mb as estimated by pulsed field gel electrophoresis [66], this apparent variation in the Venturia genomes may be attributable to the different strategies, and software, used for sequencing. The genomes (Vi1.10 and ViL) assembled using ALLPATHS-LG software, have much larger genome sizes and higher percentages of repeats, than those assembled using Velvet (Vi1, Vi1.2.8.9, and Vp). The major difference between the sequencing of these five isolates is that Vi1.10 and ViL paired-end (PE) libraries were specifically constructed with an insert size of 180 bp so that paired reads overlapped at the end (which is required by ALLPATHS-LG), to produce longer super-reads. For the other three isolates, the PE libraries were constructed with a typical insert size of 400 bp, and paired reads did not overlap and could not be assembled using ALLPATHS-LG. The super-reads generated by ALLPATHS-LG result in repeats being better assembled [67, 68]. As a trial, in addition to ALLPATHS-LG, the Vi1.10 and ViL sequences were also assembled using Velvet. The percentage of repeats in the Velvet scaffolds for each isolate was lower than that for the ALLPATHS-LG assembly, but was still very high compared with the remaining three isolates, with 21.5 and 25% for Vi1.10 and ViL respectively. Additional investigation is required to determine whether the larger genome sizes and higher percentages of repeats in Vi1.10 and ViL are a biological reality.
The number of predicted genes in the Venturia genomes (11,960–13,333) is significantly less than the 24,571 unique fungal genes reported for V. inaequalis by Thakur et al. [21]. Just over half of the reported sequences had no significant similarity to sequences in other species. The Thakur et al. [21] estimate was based on in vitro and in planta transcriptome data only, with multiple gene splice variants likely to account for the higher gene number. Our estimate of predicted gene number is comparable to related Dothideomycete genomes which range from 9739 in S. populicola [60] to 14,127 in C. fulvum [69]. Gene number variation could be due to difference in coverage of the various genomes; however, variation in gene number between isolates is also not unprecedented. For example, Xue and associates [70] found that in the rice blast fungus M. oryzae, gene number varied, with hundreds of isolate-specific genes present in genomes of field isolates. Similar gene number variation was reported in isolates of C. heterostrophus [71].
Venturia pathogens: lifestyle and host determination
Focussing on the secretome of the Venturia pathogens has revealed repertoires of proteins that reflect aspects of their adopted mode of parasitism, with secretion of compounds to adhere to the waxy, water-proof cuticle on leaves and fruit and to enable direct penetration and colonisation of the cuticle and sub-cuticular space. In addition, effectors that are predicted to effect evasion of recognition and suppression of host defence will be secreted to the extracellular plant-pathogen interface, including those that may be directed to be taken up by the host cells.
Attachment and penetration
Several Venturia proteins were identified in the secretomes that may be involved in attachment and penetration of the cuticle. A predicted protein present in all the Dothideomycete genomes analysed has similarity to a M. oryzae fasciclin protein. Fasciclins have been implicated in cell adhesion in diverse organisms including both prokaryotes and eukaryotes [72–75]. The M. oryzae fasciclin has an important role in development and pathogenicity, involved in conidiation and conidial adhesion [76]. A similar role for fasciclins in cell adhesion for all the Venturia isolates analysed is easily envisaged.
Hydrophobins are small, secreted, cysteine-rich, amphipathic proteins that are usually found on the cell walls of fungi [77, 78]. They provide a water-repellent coat and are well characterized for their role in morphogenesis and virulence in plant pathogenic fungi, promoting interactions with hydrophobic surfaces [40, 41, 79, 80]. It is thought that these proteins may also assist in avoiding detection by the host during infection [77]. Hydrophobins are usually highly variable in sequence; however, they have a conserved structure due to conserved cysteine residues. Venturia secretomes have fewer hydrophobin genes (two-to-four) than C. fulvum (11) but have a similar number to D. septosporum (four) [69].
The Venturia secretomes also have a high number of genes (11–14) encoding putative HsbA proteins. HsbA proteins are found in entomopathogenic fungi as well as pathogens of higher animals; with cuticle colonisation being a possible common link. HsbAs are not commonly reported to be encoded by plant pathogen genomes (although genes with low similarity to V. inaequalis HsbA-like genes were identified in genome sequences of members within Colletotrichum, Magnaporthe and Verticillium genera). One sequence with similarity to an HsbA was identified in the genome of the mesophyll apoplast-dwelling C. fulvum and the pine pathogen D. septosporum [69]. HsbAs are not hydrophobins, but they have been reported [81] to have an analagous role in A. oryzae in binding to hydrophobic surfaces and recruiting the CutL1 polyesterase/cutinase to degrade the polyester substrate, poly(butylene succinate-co-adipate) (PBSA). Takahashi and associates [82] also reported that the A. oryzae hydrophobin, RolA, also recruits CutL1, to aid degradation of PBSA surfaces. Thus, A. oryzae appears to use several types of proteins to recruit lytic enzymes to the surface of hydrophobic solid materials and promote their degradation. Proteins similar to the A. oryzae CutL1 polyesterase/cutinase and HsbA were well represented in the VIS set. Extracellular cutinase has been implicated in penetration by V. inaequalis, as cutinase is produced by germinating conidia, and a cutinase inhibitor can prevent penetration [83, 84]. In addition, esterase-like activity has been reported during the germination of conidia and in appressoria [85]. The presence of high numbers of CEs in the Venturia secretomes supports the experimental evidence of enzymatic penetration of the cuticle by V. inaequalis and the inference that V. pirina also may act similarly. We propose that Venturia, like A. oryzae, uses multiple proteins (e.g. hydrophobins and HsbA proteins) to recruit cutinases/esterases to facilitate appressorial adhesion and direct penetration, as well as degradation and digestion of cuticle. This hypothesis fits observations of the cuticular degradation that occurs during colonisation by Venturia fungi potentially drawing nutrition from the cuticle or cuticle precursors [7].
Nutrition
Exploitation of the cuticle may be insufficient to satisfy pathogen nutritional requirements throughout the infection cycle. During biotrophic infection Venturia remains in the cuticle and sub-cuticular space causing relatively little damage to the host. Most damage is due to breaching of the cuticle upon sporulation [6, 7]. At this time the epidermal cells underlying the stroma undergo a progressive depletion of plastids and cytoplasm, accompanied by increasing vacuolation, leading ultimately to cell death. It has been proposed that this damage late in the infection cycle is caused by partial cell wall degradation [86, 87]. Indeed, cellulase and pectinase activities have all been reported for V. inaequalis growing in vitro [6, 88, 89]. However, it has been suggested that the timing of this host cell degradation precludes a significant role for these enzymes in nutrient acquisition [89, 90].
The predominant polysaccharide in apple fruit skin is pectin at 65%, compared with a level of 3% for cellulose [91], with pear fruit also having higher levels of pectin than cellulose [92]. Venturia appears to have tailored its CAZyme repertoire to suit the composition of the host cell wall with pectin-specific CAZymes predominating; the two most numerous classes of CAZymes found in the Venturia secretomes, after those with cutinase activity, are GH28 and GH43 that have pectin as a substrate. A single GH28 enzyme and two putative PLs, that also have pectin as target substrate [31, 93], are up-regulated during infection and are present in the VIS set. Significantly, this lytic activity may therefore not be limited to late in the infection cycle as previously thought, but contribute to nutrition via degrading the surface polysaccharides of the epidermal cells beneath stromata, this damage only becoming evident macroscopically late in infection. In addition, those CAZymes present in the secretomes of the Venturia fungi may also benefit the fungus during saprobic growth and sexual fruiting body development during winter.
Evasion of host defence
The putative proteases present in the secretomes of the Venturia fungi may aid in the evasion of host defense since proteases have been implicated in plant defence avoidance in many plant-pathogen interactions [94]. For example, fungal proteases can target plant chitinases, as in the interaction between Fusarium oxysporum f. sp lycopersici and tomato where both a serine protease and a metalloprotease are required for the inactivation of a chitin-binding domain (CBD)-containing chitinase thus contributing to virulence [95].
As a possible alternative strategy to counteract host chitinases the Venturia strains, in common with many other fungi, all have a homologue of the C. fulvum LysM domain effector, Ecp6. These are likely to either have a similar function to C. fulvum Ecp6 [42, 96] in preventing recognition of chitin fragments capable of initiating a defence response, or to the LysM effectors from Z. tritici that protect hyphae from the action of chitinases, thus preventing the release of chitin-derived PAMPs [97, 98]. In addition, there are single proteins in each secretome similar to the Blumeria effector BEC1019 [55, 56]. The putative Venturia BEC1019 orthologues all have the conserved ETVIC motif that is required for suppression of the hypersensitive response (HR) in barley. A similar function for these proteins during the pathogenicity of Venturia is therefore easily envisaged.
Although single proteins similar to the NPP1 protein from H. arabidopsidis are present in each of the Venturia secretomes [51, 52] the sequence of these from two apple-infecting isolates are truncated. In the remaining secretomes (Vi1, ViL and Vp) the NPP1-similar proteins appear to belong to the type I Nep-1-like protein (NLP) family, with the two conserved cysteines [99]. All three proteins also have two additional C-terminal cysteines, however these are not characteristic of type 2 NLPs [100]. The majority of NLPs induce necrosis in a wide-range of dicotyledonous plants, although whether this necrosis is a direct result of cytotoxicity via dispruption of the plasma membrane or initiation of a defence response remains open to debate [100]. However, the requirement for a functional necrosis-inducing protein in the biotrophic Venturia pathogens is unlikely. Indeed, each of the Venturia proteins has mutations in the critical H residue in the loop region required for necrosis in the NLP from V. dahliae [101], whilst the two V. inaequalis proteins also have a mutation in the heptapeptide domain (GHRHDWE mutated to GHRHEWE) required for necrosis in NLPs from diverse taxa [52, 100, 102]. These mutations may therefore prevent unwanted pathogen-induced necrosis, but the retention of these NLP-like proteins by Venturia may indicate a virulence function that remains to be elucidated.
Venturia predicted proteins with similarity to the Magnaporthe GAS1 protein, required for appressorial penetration and lesion development, may play a similar role in apple and pear scab diseases to that observed in rice. Whether GAS1 is translocated to the host cytoplasm with a role in defence suppression remains equivocal, however; weak fluorescence of fluorescent protein constructs in infectious hyphae in onion epidermal cells was observed [54], as was translocation to rice cytoplasm, albeit under, presumably, the heterologous protomoter from the Magnaporthe gene PWL2 [103]. The Venturia proteins may therefore be translocated to the host cell cytoplasm where they may have a role in re-directing host metabolism during scab disease, however this remains conjecture.
Initiation of host resistance: an avirulence function
The comparative approach undertaken here has enabled preliminary identification of candidates for V. inaequalis avirulence effectors AvrRvi2, AvrRvi8, AvrRvi9 and AvrRvi10, which have an avirulence function and are recognised by cognate R proteins. All candidates have cysteine residues but no recognisable protein motifs, except a single candidate for AvrRvi2, AvrRvi8 or AvrRvi9 that is similar to a bacterial chaplin with a domain of unknown function, that may play a role in the Venturia lifecycle similar to that of hydrophobins. In addition to SSPs that may be involved in race/cultivar specificity on apple within V. inaequalis, further SSPs were identified that are specific to the loquat-specific isolate of V. inaequalis or to V. pirina. The majority of these SSPs were either similar to hypothetical proteins available in the public domain or had no similarity with known proteins. However, of interest is a protein with ankyrin repeats that may be involved in the specificity of V. pirina, since it is dissimilar to proteins found in the other secretomes. Ankyrin repeat-containing proteins are involved in protein-protein interactions and are present in effectors from both prokaryotes and eukaryotes, and as such this protein may play a role in the interaction with European pear [104]. In addition, a single protein specific to the ViL isolate has a SnoaL-like domain. SnoaL-like domains are found in proteins from diverse taxa, including filamentous fungal phytopathogens. For example, the protein PEP2 from Nectria haematococca MPVI has such a domain and contributes to virulence on pea; complementing an isolate of N. haematococca lacking a supernumerary chromosome bearing pathogenicity genes with PEP2 results in a small, but significant, increase in virulence [105]. How these candidate effectors may be contributing to host range determination is unknown. They may be acting as avirulence proteins during attempted infection of a nonhost, with a recognition event triggering resistance, and a concomitant lack of recognition in a compatible host. Indeed, since the hosts are closely related [106], the contribution of R protein-triggered immunity towards nonhost resistance has been postulated to be greater than for host plants that are more distantly related [3]. The variability of the closely related genomes of the Venturiaceae with respect to SSPs reinforces the utility of a comparative genomic approach.
In each of the V. inaequalis secretomes the predicted proteins with similarity to AVR-Pita1 from M. oryzae may play a similar role to that adopted by both AVR-Pita1, and its paralogue AVR-Pita2, in the rice blast interaction [107, 108]. Although the virulence function of AVR-Pita1, a putative zinc metalloprotease, has not been verified it is directly recognised by the cognate R gene product Pi-ta. An avirulence function for the similar proteins in the Venturia pathogens remains to be elucidated.
Effectors with unknown function
In addition to those proteins with similarity to known effectors with roles partially or fully confirmed, the Venturia secretomes comprise a large repertoire of putative effector SSPs with no obvious hints as to function from similarity searches. The paradigm for the role of SSPs is that they are lineage-specific effectors that contribute to the maintenance and evolution of parasitism and that obligate biotrophy is associated with expanded effector repertoires [109, 110]. It is thought that biotrophs require a more extensive and nuanced effector inventory than that required by necrotrophs, that rely more prominently on cell-wall degrading enzymes and toxins [111]. Although this précis of fungal pathogen lifestyles is highly simplistic, it seems to be reflected in the fewer number of SSPs recorded for necrotrophic fungal phytopathogens. For example Parastagonospora nodorum has a suite of only 209 SSPs [60], and a recent analysis of the secretome of Sclerotinia sclerotiorum revealed only 78 effector candidates, albeit using different criteria for selection [112]. This contrasts with the numbers of SSPs reported for the obligate biotrophs: B. graminis (490) [113], although these are up to 400 aa in length; P. graminis f. sp. tritici (540); and Melampsora larici-populina (305) [60]. These studies use different criteria for SSP identification, hampering valid comparsions. In the current study, SSPs from P. nodorum, C. fulvum and P. graminis f. sp. tritici were predicted using the same pipeline as that used for SSP analysis from the Venturia pathogens. The number of SSPs in the Venturia pathogens were between that recorded for the obligate biotroph P. graminis f. sp. tritici and the necrotroph P. nodorum or the facultative biotroph C. fulvum. For those SSPs predicted to be less than 200 aa in length, the proportion predicted to be classically secreted predominated. This profile resembled that of P. graminis f. sp. tritici. Thus overall the repertoire of SSPs from the Venturia pathogens more closely resembles that of an obligate biotroph rather than a necrotroph or facultative biotroph.
Evidence of high expression or up-regulation during infection (compared with in vitro growth) was used to prioritise a smaller set of Venturia SSPs [114]. The smallest of these uncharacterised SSPs were analysed in more detail and of those with similarity to predicted proteins in related Dothideomycete genomes, only four were found in the majority of genomes investigated. These Dothideomycete genomes are from organisms with diverse lifestyles. Up until recently this broad conservation would indicate a core metabolic role, rather than a role in pathogenicity, especially for those proteins also found in the extremophilic sooty mould saprobe, Baudoinia compniacensis, with its compact genome of 21.88 Mb [60]. However, the recent research of Whigham and associates [56] showing that the broadly conserved effector BEC1019 from the pathogen Blumeria, that can suppress HR, may be repurposed to fulfil particular roles in fungi with diverse lifestyles, challenges this view.
A small family of proteins in V. inaequalis with similarity to Bys1 from M. oryzae also has representatives in other diverse Dothideomycetes. The function of the Bys1 domain is unknown, but in Blastomyces dermatitidis the expression of a Bys1-encoding gene is associated with pathogenesis [115, 116], thus a similar role in Dothideomycete pathogens cannot be ruled out.
One of the most highly expressed genes in the VIS gene set, with expression in vitro and in planta, encodes a protein with similarity to the Alternaria alternata major allergen Alt a 1 (GenBank: AAM90320.1). This V. inaequalis protein was identified previously [117] in an analysis of semi-purified secreted proteins that elicited a response from specific resistant apple hosts. Single genes encoding proteins with similarity to the Alt a 1 allergen were identified in all Venturia genomes and a single gene was also identified in some (but not all), related Dothideomycetes (P. nodorum; S. populicola, S. musiva, C. fulvum, Z. tritici). Alt a 1 is well known in clinical settings as a human allergen. It has a structure unique to fungi; however, its role in pathogenesis has not as yet been determined [118].
A large number of SSPs in the VIS set with similar proteins across Dothideomycete spp. have an internal repeat structure. Repeat structures in fungal effectors have been reported previously [104]. For example the SP7 effector from Glomus intraradices, an arbuscular mycorrhizal biotroph, has nine hydrophilic tandem repeats. SP7 re-programmes plant expression to reduce the defence response thus enabling establishment of the fungus within the roots of plants [119]. In addition, a double knock-out mutant of U. maydis lacking the repeat-containing protein effectors, Hum3/Rsp1, has arrested growth in planta, shortly after penetration [120]. The roles of the previously identified repeat-containing proteins, Cin1 and Cin3, which are expressed very highly in the stromata and runner hyphae of Venturia, are yet to be determined [48].
Expanded SSP effector families
Intriguingly, Venturia strains have multiple genes predicted to encode proteins with similarity to AvrLm6 from the Dothideomycete L. maculans and Ave1 from the Sordariomycete Verticillium spp. These effectors are encoded by a single gene (Ave1) in Verticillium spp. [44], or a single gene plus a paralogue (AvrLm6) in L. maculans [121].
The Venturia Ave1-like genes were generally not found in clusters on contigs and both effector families have significant numbers of pseudogenes (lacking a start and/or a stop codon). The role of these Ave1-like proteins is yet to be determined; however, the multiple gene members in the AvrLm6 and Ave1 effector families will present a challenge for functional analyses. AvrLm6-like genes are not found in most of the sequenced Dothideomycete relatives of L. maculans. The same is true for Ave1 from the Sordariomycete, Verticillium. This discontinuous distribution of Ave1 and AvrLm6 suggests that these effectors are either ancient effector ancestors that have been lost or diversified beyond recognition during coevolution and host specialisation, or that there may have been horizontal gene transfer events involving the Ave1- and the AvrLm6-containing species. None of these species share a common host, however, and so mechanisms for gene transfer are not obvious.
Many of these Venturia effector Ave1 and AvrLm6 orthologues were not identified by the secretome prediction pipeline as their N-terminus was incorrectly designated by the automated gene calling software packages. Ave1-like proteins have also been detected by Mass Spectrometry in an analysis of V. pirina proteins expressed in vitro [22]. The presence of a signal peptide, mature N terminus, cleavage site and presence of a conserved intron in the 5′ UTR of the gene, was confirmed for several of the V. pirina Vp Ave1-like genes in this proteogenomic analysis. The conserved intron in the 5′ UTR of Ave1-like genes appears to have caused difficulties in accurate gene prediction for all genes in this family. Rigorous interrogation of the whole set of SSPs, with a size of 200 aa or less, also highlighted the challenges of gene prediction. On initial comparison of gene predictions, via comparison of their encoded proteins, genes encoding SSPs similar to those in the Vi1 secretome were not predicted in the other genomes. To verify this absence, comparison of aa sequence against a six-frame translation of the genomes was undertaken and revealed the presence of identical or near-identical loci. The use of RNA-seq data to inform gene prediction in the Vi1 genome appeared to be more efficient than using a model based upon these data for gene prediction in the other genomes, highlighting the importance of RNA-seq data for individual genomes for accurate gene prediction.
With the exception of the families comprising Ave1- and AvrLm6–like genes, the majority of families of SSP genes in Venturia do not have similar genes in the other Dothideomycete genomes analysed. These families appear to be lineage-specific. Lineage-specific genes are common features of fungal genomes sequenced to date [122]. In the Venturia pathogens many of the lineage-specific SSPs appear to belong to expanded families, for example, eight SSP families with members of less than 200 aa have more than 10 members, and up to 86 members, and are restricted to the Venturia genus. Gene gain and also expansion of these lineage-specific families is obviously associated with host range determination and specificity, and the converse, reduction or loss of gene families can likewise be associated with evolution of virulence on a particular host [122]. Expansion of lineage-specific effector families has recently been reported in B. graminis [123], with the 1350 paralogous copies of AVR k1 and AVR a10, that contribute to the establishment of the haustorium, being an extreme example [124, 125].
Whether the members of the SSP gene families observed in the Venturia genomes are derived from a common ancestor remains equivocal since their overall sequence conservation is low. However, de Guillen et al. [126] identified a family of sequence-unrelated, but structurally conserved effectors (termed MAX-effectors) in Magnaporthe, accounting for 5–10% of the effector repertoire. These effectors have been presumed to have evolved via diversifying selection rather than convergent evolution [126]. In addition, hydrophobins, with their patchy distribution and low level of sequence conservation, contrasting with a highly conserved structure, related to function, appear to evolve by a birth-and-death mechanism and to be phylogenetically related [127, 128]. The families of Venturia SSPs with retention of presumably critical cysteines, in terms of structure and stability, may have also arisen under similar evolutionary constraints and thus also be related phylogenetically. Evolution of protein families involves the duplication of an ancestral gene followed by mutation of the duplicated gene to enable novel functionality to emerge [129, 130]. In the case of effectors, expansion and diversification within a family may enable modification of function to prevent alerting a guarding resistance gene product, or structural modification to avoid a direct recognition event that would otherwise elicit a defense response. Thus having expanded families of effectors may offer an evolutionary selective advantage to a pathogen, enabling recognition of an effector to be overcome with evolution of a novel paralogue and deletion or pseudogenization of the previously recognised effector. The processes involved in how these families expand is still open to debate. The presence of large numbers of presumably structurally related, but sequence diverse, proteins poses problems for the delineation of families. Indeed, three families were delineated in this study that shared common members. Although an unorthodox compromise, these families were deemed to be too diverse to be amalgamated using the thresholds adopted for this analysis, but the members are sufficiently closely related to form a single family if the thresholds were to be slightly relaxed, thereby highlighting the pitfalls of depending on arbitrary thresholds for either separating or grouping proteins.
In many pathogens, effector genes are associated with TEs. Association of TEs is evident with the co-evolution of the B. graminis AVR k1 effector family with a class of LINE-1 retrotransposons [124], the B. graminis AvrPm3a2/f2 effector family with various TEs [123] and the association of miniature impala transposons (mimps) in the promoters of effectors in F. oxysporum [131]. TEs have also been implicated in horizontal gene transfer [132]. The analyses of gene density, and association of effector candidates with repeat elements in Vi1, suggests that effectors and TEs may be similarly associated in Venturia. In addition, most of the AvrLm6- and Ave1-like genes are closely associated with repeats, i.e. TEs and TE-like remnants, as identified by REPET [45]. Oliver [133] outlined the mutagenic potential of transposons and their, at first, seemingly unlikely contribution to the evolution of a successful pathogen: transposons contribute to diversity generation by insertion either in or near a gene, affecting either structure of the resulting encoded protein or the expression pattern, respectively. Transposons may facilitate gene family expansion through capture and translocation of host genes [134], such expansions can become targets for repeat induced point mutation (RIP), with mutations occuring not only in the duplicated transposons but the associated genes [133, 135, 136]. This is an ongoing area of interest and analysis of repeat regions is currently underway in cross progeny of V. inaequalis. Further analyses are required in the Venturia spp. to reveal if TEs and indeed RIP are involved in diversity generation, and expansion of gene families as suggested by this initial analysis.
Conclusions
The comparative analysis of whole secreteomes of multiple races of V. inaequalis with the related scab pathogen, V. pirina, has provided novel insights into the unusual, biotrophic lifestyle niche that these pathogens occupy. It has also yielded significant leads in the hunt for cultivar- and host-specificity determinants of scab fungi. The challenge now will be to prioritise leads, from the expanded arrays of putative effectors, for futher investigation with a mind to delivering durable, scab control.
Methods
Fungal material and culture conditions
All V. inaequalis and V. pirina isolates used in this study (Table 1; Additional file 17) have been reported previously [12, 22, 46, 137, 138] and morphology, pathology as well as DNA sequence comparisons (ribosomal RNA ITS1-5.8-ITS2 and TEF1) have been used to validate the classifications of these isolates as V. inaequalis or V. pirina. All isolates were grown on cellophane (Waugh Rubber Bands, Wellington, New Zealand) [139] overlying potato dextrose agar (PDA) at 20 °C for 18 h to 14 days (16 h light period/day) under white fluorescent lights (4300 K) for the production of conidia for plant inoculation (Vi1 only) and biomass for genomic DNA (gDNA; all isolates) and RNA extraction (Vi1 only). Cellophane was used, as it induces the formation of spores and stromata-like tissues in Venturia spp. similar to those formed during infection [47].
Plant material and infection assays
Four- to six-week-old seedlings originating from open-pollinated Malus x domestica ‘Royal Gala’ (Hawke’s Bay, New Zealand) were used to produce infected plant material. A detached leaf assay was used for generating tissue for RNA sequencing [117], except that 5 μl droplets of conidial suspension (1 × 105 ml−1) were used to cover the entire leaf surface. V. inaequalis Vi1-infected and SDW-inoculated leaves were harvested at two or seven dpi and used for RNA extraction and microscopic evaluation of infection.
gDNA extraction and whole genome sequencing
Extraction of V. inaequalis gDNA was carried out as reported in Kucheryava and associates [47]. Two PE libraries and two mate-pair (MP) libraries (with 5 kb and 10 kb insert sizes) were constructed for Vi1. These libraries were sequenced on the HiSeq2000 platform at the Allan Wilson Centre Genome Service (AWCGS), Massey University, New Zealand, and the Australian Genome Research Facility (AGRF). One plate of a whole genome library was also sequenced by the AWCGS on the Roche 454 Life Sciences Genome Sequencer with read length ranging from 18 to 634 bases. One PE and one MP library were constructed for each of the isolates, Vi1.10 and ViL. The PE libraries were specially built with a fragment size of 180 bp so that read 1 and 2 overlapped at the end. All four libraries were sequenced on the Illumina HiSeq2000 platform at Macrogen, Korea. The Vi.1.2.8.9 PE library was sequenced in one lane on the Illumina HiSeq2000 at the AGRF. Genome sequencing details for Vp were reported in Cooke et al. [22]. The majority of the genomes were sequenced using the Illumina platform, apart from Vi1 which is a hybrid assembly of reads from both Illumina HiSeq and Roche 454 platforms.
RNA extraction and transcriptome sequencing
Total RNA was extracted by the method of Chang et al. [140] and concentration quantified using a Nanodrop, ND-1000 Spectrophotometer (NanoDrop Technologies, Rockland, DE). For RNA sequencing cellophane membranes were harvested as follows and combined for a single RNA extraction: three cellophanes 18 h post inoculation (hpi), four cellophanes four dpi, four cellophanes six dpi and one quarter of a cellophane 14 dpi, stripped of aerial hyphae (to enrich for stromatic tissues inside the cellophane sheet). For in planta material three detached leaves inoculated with V. inaequalis conidia (detached leaf assay) were harvested at two and seven dpi and snap frozen in liquid nitrogen. RNA was extracted as above, a single sample being derived from the three leaves. Genomic DNA contamination was excluded by visualisation of RNA on a 0.8% agarose gel and absence of an amplification product using primers specific for glyceraldehyde 3-phosphate dehydrogenase genes from V. inaequalis and apple [47]. The RNA from in vitro and in planta extractions (5 μg), from the two different time points, was sequenced on the Illumina HiSeq2000 platform in PE mode AGRF, giving between 17 and 33 million pairs of reads per sample.
Bioinformatic analyses
Genome assembly
For each Venturia isolate, a quality check (QC) on genome sequencing data was carried out utilising PIQA v1.0 [141], the FASTX toolkit v0.0.13 [142] and FastQC [143]. Reads were trimmed using in-house PERL tools and adaptor sequences were removed using fastq-mcf [144] based on data QC reports. De novo genome assembly was performed using Velvet [61] and SOAPdenovo with a range of Kmer values for Vi1 and Vi1.2.8.9 [145]. For each isolate, the top three assemblies that produced the best statistics in terms of size of contigs and scaffolds, and N50 and N90 metrics, were chosen for futher analysis. CEGMA [25] and BUSCO [26] tests were performed on various genome assemblies to check their completeness; utilising the 248 most conserved core genes for the former [146]. Known Venturia ESTs (ABEA, ABEB, IAAA, MAAB and MAAD libraries deposited as ESTs at NCBI) [46] were aligned to the genome assemblies for quality evaluation i.e. to ensure correct assembly of the corresponding sequences in the genome using a BLASTn similarity threshold of <1e-05. The assembly with the highest percentage completeness measured by the CEGMA analysis and the best-mapping ESTs was selected as the genome to release for the isolate. Isolates Vi1.10, and ViL, were assembled with three genome assemblers, Velvet, SOAPdenovo, and the ALLPATHS-LG programme since their PE libraries were constructed specifically with overlapping pairs and satisfied the requirements for ALLPATHS-LG [62, 64]. As for the other isolates, the assembly with the best statistics was chosen for further improvement. The best genome assembly for each isolate was gapfilled using GapCloser with PE reads (GapCloser in SOAPdenovo version 1.12) [145] then scaffolded using SSPACE with MP reads (SSPACE version 2) [147]. The gapfilling and scaffolding steps were performed iteratively until there was no obvious gap size decrease or reduction in scaffold number.
Gene prediction
The assembled genomes were masked before gene prediction using a customized pipeline which included RepeatMasker-open-3-3-0, RepBase [148], RepeatScout [149], trf [150] and TEClass [151]. Fungal genes downloaded from NCBI (August, 2012) were mapped to the Vi1 genome assembly using Exonerate [152] for similarity based gene prediction. Transcriptome assemblies for in planta and in vitro libraries (see transcriptome analysis section below) together with ESTs from Vi1 grown in vitro and in planta (Vi1-infected susceptible apple leaves) [46] were used to train AUGUSTUS [23, 153] to build a species model file (meta parameter file) for Venturia. Hybrid (evidence-based) gene prediction was performed for Vi1 using this model together with the Vi1 transcript sequences and ESTs as hints in AUGUSTUS. Based on the trained species model, ab initio gene predictions were carried out on the repeat-masked genomes for all the five Venturia isolates, thus there were two sets of gene predictions for the Vi1 genome.
Transcriptome analysis
RNA-Seq data QC was done using FASTX-Toolkit v0.0.13 [142]. Reads were trimmed to 64 nucleotides based on the QC reports (using a median phred score >20 as threshold). For genome-guided transcriptome assembly, trimmed RNA-Seq reads from Vi1 in vitro and two or seven dpi (in planta) were mapped to the reference genome of Vi1 using tophat-1.4.1 [154] and further assembled with cufflinks-1.3.0 [155]. De novo transcriptome assembly was carried out using Inchworm (now evolved to Trinity) [156, 157], and Oases [158] to aid gene prediction (see above). To measure transcriptional support for genes FPKM values were calculated within samples enabling gross ranking of transcriptional activity, with an arbitrary threshold of >1 for evidence of transcription. Differential gene expression tests were carried out between in vitro and in planta samples at different inoculation time points, using rsem-1.2.4 [159] to first map the reads, followed by edgeR v3.2.4 [160] in Bioconductor version 2.12 [161]. Highly differentially expressed genes, with a false discovery rate <0.05 and log2 fold change (LogFC) not smaller than two, were used for prioritisation of genes for analysis.
Identification of the predicted secretomes
Genes encoding putatively secreted proteins were identified in each Venturia genome and gene catalogues from P. nodorum, P. graminis f. sp tritici and C. fulvum downloaded via the Mycocosm portal at the JGI, using a custom software pipeline (Additional file 6). Both the hybrid (based on trancriptome resources used to build the trained species model) and ab initio (based on the trained species model) predictions for Vi1 were used in the secretome discovery pipeline to avoid excluding genes predicted in only one approach; gene predictions in either set that co-localised at the same locus (with a greater than 90% identity and e value of <1e-10) were considered a single prediction. The SignalP 4.0 [162] and the SecretomeP [163] servers were used initially to screen for predicted proteins with a signal peptide or those secreted via a non-classical pathway, respectively. The TMHMM server was used to screen for predicted proteins without a predicted transmembrane domain [164]. Only those predicted proteins, either lacking a transmembrane domain or with a single transmembrane domain with at least 10 aa in the first 60 aa, indicating a probable correspondence with secretion signal, were considered for further analysis. The TargetP server [165] was used to predict cellular location, with those predicted to be extracellular retained. ProtComp v 9.0 [166] and WoLF PSORT servers (with an extracellular score threshold of >17) [167] were used to further analyse the putative location of these predicted proteins. Proteins predicted to be secreted by either method were included to avoid the omission of false negatives.
Annotation of the predicted secretomes
Putative functions of the secretome proteins were initially assigned following gene ontology analysis through an in-house annotation pipeline (BioView; Ross Crowhurst, Marcus Davy and Cecilia Deng, unpublished) that scans public databases including NCBI RefSeq [168], InterPro [169], UniRef [170], ExPASy UniProtKB/Swiss-Prot [171] and ExPASy Prosite [172] and subsequent manual curation on the basis of similarity searches (BLASTp with a similarity threshold of 1e-10) against the NCBI nr database [29]. In addition, identification of the complement of putative carbohydrate-active enzymes (CAZymes) was achieved using the CAT server [27]. Default parameters were used for CAZyme identification, apart from the -threshold which was set to 1e-05.
Venturia genomes were also interrogated (using a combination of automated annotations, BLASTp and tBLASTn, with a similarity threshold of 1e-10), with sequences listed in PHI-base [35] and other effectors (e.g. V. dahliae Ave1, GenBank: AFB18188.1; L. maculans, AvrLm6 GenBank: CAJ90695.1; C. fulvum Ecp6 GenBank: ACF19427.1), as well as sequences annotated as ‘hydrophobin’ and containing the ascomycete-specific hydrophobin motif IPR010636 or conserved eight cysteine pattern, from C. fulvum and D. septosporum (as referred to in [69]), P. nodorum (JGI protein ID 9201, SNOG_03122.3) and L. maculans (JGI protein ID 775, Lema_T007750.1). Conserved domains were identified using Pfamscan [36–38].
Orthologous protein clusters were identified using OrthoMCL version 2.0.3 [30] together with the Markov clustering algorithm mcl [173] to cluster all-vs-all BLASTp (e value <1e-10) results across all isolates.
Small secreted proteins (SSPs)
Small secreted proteins (SSPs) were defined as proteins that were present in the secretome dataset with a length of ≤500 aa (following removal of the secretion signal if identified by SignalP). Enzymes, such as CAZymes, were not included in the SSP set. A more stringent length parameter of 200 aa was also used to identify a more discrete set of candidate effectors. Those SSPs with two or more cysteine residues in the predicted mature protein were also identified. SSPs were screened for repeat content using RADAR [174–176]. The presence of pro-peptide cleavage sites was analysed using ProP 1.0 [177], with those sites with a score ≥0.5 recorded as positive. InterProScan 5 [174, 178] was used to analyse conserved domains in the SSPs.
In addition to the OrthoMCL analysis, the SSPs (≤200 aa in length) were analysed further using Spectral Clustering SCPS 0.9.8 [57] to detect possible clusters of SSPs comprising members with lower sequence similarity than those clustered using the more stringent thresholds of OrthoMCL. The parameters (default) used were those that resulted in the AvrLm6-like proteins forming a cluster with the same members as when clustered manually [45]. The clusters were manually inspected and those members with a similar cysteine pattern retained. In addition, since those SSPs less than 200 aa in length were over-represented in the Vi1 secretome, members of the clusters from Vi1 were used to detect possible gene predictions that were not called automatically by searching each of the Venturia genomes using genBlastG 1.39 [179], then manually curating novel gene models followed by family assignment based on cysteine patterns and construction of multiple sequence alignments using T-Coffee 11.00.8cbe486 [180, 181]. Sequence logos were constructed using the TeX package, TeXshade [182].
Proteins similar to selected SSPs in related Dothideomycete genomes (Additional file 15) were identified either by BLASTp for singletons or HMMER 3.1b2 [183] for families (thresholds 1e-10), and clustering data pertaining to the predicted Dothideomycete orthologues were obtained from the JGI. Dothideomycete genomes were selected for comparison based on phylogenetic spread and diversity of lifestyle, including extremophile saprobes, biotrophs, hemibiotrophs and necrotrophs.
Gene density and proximity to transposable elements (or transposable element remnants) in the Vi1 genome
Intergenic distances were calculated for the predicted genes of Vi1. Gene density across the Vi1 genome was approximated from 5′ and 3′ flanking intergenic lengths. Intergenic lengths were binned according to log (length) and plotted as a 2-dimensional heatmap in R [184] using a method adapted from Saunders and associates [185]. The average distances were also calculated; however, genes lacking an immediate neighbour at either the 5′ or 3′ flank, such as those at the ends of genomic scaffolds, were excluded from this analysis. The VIS set, plus AvrLm6- and Ave1-like genes were compared with 440 core eukaryotic genes [25].
The repeat prediction suite REPET 2.2 [186] was used for the detection and annotation of TEs (repeat sequences) for the whole genome sequence of Vi1. A custom Python script was used to calculate the 5′ and 3′ flanking distances between gene annotations and TE genomic features that were identified in the REPET analysis. Flanking distances between genes and repeats were binned according to log (length) and plotted as a 2-dimensional heatmap in R [184] using a method adapted from Saunders and associates [185]. The VIS and core eukaryotic gene gene sets were plotted for comparison. Student’s t-Tests were conducted to determine whether any differences were statistically significant.
Additional files
Statistics for the whole genome assemblies of isolates of Venturia inaequalis and V. pirina. (XLSX 11 kb)
Repeats in Venturia inaequalis and V. pirina genome assemblies. (XLSX 10 kb)
Analysis of gene models from the Venturia inaequalis and V. pirina genomes. (XLSX 10 kb)
Core Eukaryotic Gene Model Analysis (CEGMA) of the Venturia inaequalis and V. pirina genomes. The 248 most highly-conserved core eukaryotic genes were used in the CEGMA to assess genome completeness. (XLSX 10 kb)
Benchmarking Universal Single-Copy Orthologs (BUSCO_v1.1b) analysis of the Venturia inaequalis and V. pirina genomes. BUSCO analysis was used to assess completeness of the Venturia genome assemblies. (XLSX 10 kb)
Pipeline of programmes used to identify the in silico-predicted secretome of each isolate. (DOCX 147 kb)
The predicted secretomes and small, secreted proteins (SSPs) encoded by the genomes of Venturia and representative related pathogens. The genomes for Parastagonospora nodorum, Cladosporium fulvum and Puccinia graminis f. sp. tritici were downloaded via the Mycocosm portal at the Joint Genome Institute (JGI; see details in Additional file 15). These pathogens were representatives of necrotrophic, facultative and obligate biotrophic fungi, respectively. The pipeline outlined in Additional file 6 was employed to identify the secretome and SSPs in each of the genomes. (XLSX 11 kb)
The predicted proteins belonging to OrthoMCL clusters present in the secretomes of five Venturia isolates. The coloured horizontal bars represent proportions of predicted proteins (scale at bottom of figure) that have been annotated according to the key at the top of the figure, that are present in the secretomes indicated by grey boxes to the left of the figure (white boxes indicate lack of similar proteins). S: indicates singleton proteins. Numbers in the boxes are numbers of proteins. The order of the categories in the key is the same as that in the horizontal bars. For example: the core secretome is represented by the top-most horizontal bar; there are similar proteins present in each of the secretomes (5 grey boxes), of which 15% (77 proteins) are classified as SSPs between 200 and 500 amino acids in length with 2 or more cysteines. (PPTX 56 kb)
The CAZyme domains present in proteins encoded by the Venturia secretomes. CAZyme domains were identified using the CAZymes analysis toolkit (CAT) server. Domains: CBM: carbohydrate binding module; CE: carbohydrate esterase; GH: glycoside hydrolase; GT: glycosyltransferase; PL: polysaccharide lyase. The known substrates for the CAZyme domains were taken from Zhao et al. PCW: plant cell wall. FCW: fungal cell wall. CW: cell wall. ESR: energy storage and recovery. PG: protein glycosylation. dpi: days post inoculation. (XLSX 17 kb)
Summary of distribution of similar proteins with CAZyme domains encoded by the Venturia secretomes. CAZyme domains were identified using the CAZymes analysis toolkit (CAT) server. Filled grey boxes in columns indicate presence of a similar protein or proteins in that secretome identified by OrthoMCL clustering. (XLSX 12 kb)
Classification of proteases in the secretomes of the Venturia pathogens. Filled grey boxes in columns indicate presence of similar proteins in that secretome identified by OrthoMCL clustering: aspartate and serine (including those annotated as subtilases) proteases, metalloproteases and unclassified peptidases. Several of the proteases were similar (<e-10) to proteases in the Pathogen-Host Interactions database (PHI-base). (XLSX 15 kb)
Summary of small, secreted proteins (SSPs) similar to proteins present in the Pathogen-Host Interactions database. Filled grey boxes in columns indicate presence of a similar protein(s) in that secretome identified by OrthoMCL clustering. (XLSX 12 kb)
Small, secreted protein (SSP) family 07 members. Sequence logo of members of A. family 07 from Vi1 and B. family 07 from all isolates. (DOCX 221 kb)
Details of the small (≤200 amino acids in length) secreted proteins in the Venturia infection secretome (VIS) gene set. (XLSX 22 kb)
The genomes used in this study (non-Venturia). Sources of the genomes downloaded or accessed (with permission) from the Joint Genome Institute (JGI)/MycoCosm. (XLSX 14 kb)
Details of the lineage-specific small, secreted proteins (SSPs) identified in Venturia inaequalis ViL and V. pirina secretomes. Only those proteins ≤200 amino acids in length are included. (XLSX 18 kb)
Details of isolates of Venturia inaequalis and V. pirina used in this study. (XLSX 12 kb)
Acknowledgements
The authors wish to thank Jared Bennett for technical assistance with DNA preparation; Mark Andersen and Erik Rikkerink for critical reviewing of the manuscript.
Funding
This work was supported by: the New Zealand Foundation for Research, Science and Technology (now Ministry for Business, Innovation and Employment), contract numbers C06X0302 and C06X0207 and PFR Core funding; Victorian Life Sciences Computation Initiative’s (VLSCI) Life Sciences Computation Centre, a collaboration between La Trobe, Melbourne and Monash Universities (an initiative of the Victorian Government, Australia); La Trobe University eResearch grant; JS was supported by an Australian postgraduate award and VLSCI PhD top-up award.
Availability of data and materials
De novo assemblies for the five Venturia isolates have been deposited at DDBJ/EMBL/GenBank under the accession numbers: National Center for Biotechnology Information (NCBI) [29] BioProject ID PRJNA261633 for V. inaequalis and JEMP01000000 for the V. pirina genome. RNA-seq data (V. inaequalis Vi1) are deposited under the same BioProject (ID PRJNA261633): in vitro: SRR1586226, two dpi in planta: SRR1586224, seven dpi in planta: SRR1586223. In addition, the Vi1 and Vp genomes and gene predictions are publicly accessible via the Mycocosm portal at JGI [58, 59].
Authors’ contributions
CHD: Genome assemblies for Vi isolates; Repeat elements identification and repeat masking of genomes; Gene prediction and annotation; Comparative genomics of fungal species; Transcriptome assemblies; Gene differential expression profiling; Genome visualization and consortium web site maintenance; Deposition of genome assemblies to NCBI; Deployment of genomes and gene structures to JGI/MycoCosm; Experimental design for WGS and RNASeq (with JKB). KMP: conception and experimental design, bioinformatics analyses, writing of manuscript. DABJ: Analysis of families and singletons in the Vi secretomes. CHM: bioinformatic identification of effector candidates, writing of manuscript. JS: identification of the AvrLm6 orthologues and bioinformatics analyses. APT: interrogation of Vi1 and Vp genomes of Ave1 orthologue members, REPET and intergenic and gene to TE flanking distances analysis. AJR: REPET analyses, bioinformatics assistance and support. PK: bioinformatics and validation of Ave1 orthologue members. NEH: bioinformatics and technical support. MDT: sequence analysis, gene mining. JKB: conception and experimental design, DNA and RNA preparation, infection assays, microscopy, bioinformatics analyses, writing of manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
The plant material used in this research was Malus x domestica ‘Royal Gala’ that was open-pollinated. Seeds were sourced from fruit produced in the Plant & Food Research Orchard (Hawke’s Bay, New Zealand). Since the plant material was not collected from a wild source, any permissions/permits were not necessary. ‘Royal Gala’ is one of the most widely grown apple cultivars worldwide and is readily available for purchase from nurseries.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- aa
Amino acid
- AGRF
Australian Genome Research Facility
- AWCGS
Allan Wilson Centre Genome Service
- BUSCO
Benchmarking Universal Single-Copy Orthologs
- CAT
CAZyme Analysis Toolkit
- CAZyme
Carbohydrate Active Enzyme
- CBD
Chitin-binding domain
- CBM
Carbohydrate-binding motif
- CE
Carbohydrate esterase
- CEGMA
Core Eukaryotic Genes Mapping Approach
- Cin
Cellophane-induced
- dpi
Days post inoculation
- FPKM
Fragments per kilobase of exon per million reads mapped
- gDNA
Genomic DNA
- GH
Glycoside hydrolase
- GT
Glycosyl transferase
- hpi
Hours post inoculation
- HR
Hypersensitive response
- HsbA
hydrophobic surface binding
- JGI
Joint Genome Institute
- LysM
Lysin motif
- Mimp
Miniature impala transposon
- MP
Mate-pair
- NLP
Nep-1-like protein
- PAMP
Pathogen-associated molecular pattern
- PBSA
Poly(butylene succinate-co-adipate)
- PDA
Potato dextrose agar
- PE
Paired-end
- PFR
Plant & Food Research Limited
- PL
Polysaccharide lyase
- PTI
PAMP-triggered immunity
- QC
Quality check
- R
Resistance
- RIP
Repeat induced point mutation
- SDW
Sterile distilled water
- SSP
Small, secreted, non-enzymatic protein
- TEs
Transposable elements
- VICE
V. inaequalis candidate effector
- VIS
Venturia Infection Secretome
- VLSCI
Victorian Life Sciences Computation Initiative
Contributor Information
Cecilia H. Deng, Email: Cecilia.Deng@plantandfood.co.nz
Kim M. Plummer, Phone: +61 (3) 9032 7474, Email: K.Plummer@latrobe.edu.au
Darcy A. B. Jones, Email: darcy.a.jones@postgrad.curtin.edu.au
Carl H. Mesarich, Email: C.Mesarich@Massey.ac.nz
Jason Shiller, Email: jason.shiller@inra.fr.
Adam P. Taranto, Email: adam.taranto@anu.edu.au
Andrew J. Robinson, Email: Andrew.Robinson@latrobe.edu.au
Patrick Kastner, Email: P.Kastner@latrobe.edu.au.
Nathan E. Hall, Email: N.Hall@latrobe.edu.au
Matthew D. Templeton, Email: Matt.Templeton@plantandfood.co.nz
Joanna K. Bowen, Phone: +64 (9) 925 7154, Email: Joanna.Bowen@plantandfood.co.nz
References
- 1.Chisholm ST, Coaker G, Day B, Staskawicz BJ. Host-microbe interactions: shaping the evolution of the plant immune response. Cell. 2006;124:803–14. doi: 10.1016/j.cell.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 2.Jones JD, Dangl JL. The plant immune system. Nature. 2006;444:323–9. doi: 10.1038/nature05286. [DOI] [PubMed] [Google Scholar]
- 3.Schulze-Lefert P, Panstruga R. A molecular evolutionary concept connecting nonhost resistance, pathogen host range, and pathogen speciation. Trends Plant Sci. 2011;16:117–25. doi: 10.1016/j.tplants.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 4.Stergiopoulos I, de Wit P. Fungal effector proteins. Annu Rev Phytopathol. 2009;47:233–63. doi: 10.1146/annurev.phyto.112408.132637. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Y, Crous PW, Schoch CL, Bahkali AH, Guo LD, Hyde KD. A molecular, morphological and ecological re-appraisal of Venturiales - a new order of Dothideomycetes. Fungal Divers. 2011;51:249–77. doi: 10.1007/s13225-011-0141-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.MacHardy WE. Apple scab: biology, epidemiology, and management. St. Paul: The American Phytopathological Society Press; 1996. [Google Scholar]
- 7.Bowen JK, Mesarich CH, Bus VGM, Beresford R, Plummer KM, Templeton MD. Venturia inaequalis: the causal agent of apple scab. Mol Plant Pathol. 2011;12:105–22. doi: 10.1111/j.1364-3703.2010.00656.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sivanesan A. Venturia carpophila IMI descriptions of pathogenic fungi and bacteria. 1974. p. 402. [Google Scholar]
- 9.Sivanesan A, Holliday P. Venturia cerasi IMI descriptions of fungi and bacteria. 1981. p. 706. [Google Scholar]
- 10.Sivanesan A, Waller JM. Venturia pirina. IMI descriptions of fungi and bacteria. 1974. p. 404. [Google Scholar]
- 11.Ishii H, Yanase H. Venturia nashicola, the scab fungus of Japanese and Chinese pears: a species distinct from V. pirina. Mycol Res. 2000;104:755–9. doi: 10.1017/S0953756299001720. [DOI] [Google Scholar]
- 12.Le Cam B, Parisi L, Arene L. Evidence for two formae speciales in Venturia inaequalis, responsible for apple and pyracantha scab. Phytopathology. 2002;92:314–20. doi: 10.1094/PHYTO.2002.92.3.314. [DOI] [PubMed] [Google Scholar]
- 13.Gladieux P, Caffier V, Devaux M, Le Cam B. Host-specific differentiation among populations of Venturia inaequalis causing scab on apple, pyracantha and loquat. Fungal Genet Biol. 2009;47:511–21. doi: 10.1016/j.fgb.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 14.Bus VGM, Rikkerink EHA, Caffier V, Durel CE, Plummer KM. Revision of nomenclature of the differential host-pathogen interactions of Venturia inaequalis and Malus. Annu Rev Phytopathol. 2011;49:391–413. doi: 10.1146/annurev-phyto-072910-095339. [DOI] [PubMed] [Google Scholar]
- 15.Belfanti E, Silfverberg-Dilworth E, Tartarini S, Patocchi A, Barbieri M, Zhu J, et al. The HcrVf2 gene from a wild apple confers scab resistance to a transgenic cultivated variety. Proc Natl Acad Sci U S A. 2004;101:886–90. doi: 10.1073/pnas.0304808101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Joshi SG, Schaart JG, Groenwold R, Jacobsen E, Schouten HJ, Krens FA. Functional analysis and expression profiling of HcrVf1 and HcrVf2 for development of scab resistant cisgenic and intragenic apples. Plant Mol Biol. 2011;75:579–91. doi: 10.1007/s11103-011-9749-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schouten HJ, Brinkhuis J, van der Burgh A, Schaart JG, Groenwold R, Broggini GAL, et al. Cloning and functional characterization of the Rvi15 (Vr2) gene for apple scab resistance. Tree Genet Genomes. 2014;10:251–60. doi: 10.1007/s11295-013-0678-9. [DOI] [Google Scholar]
- 18.Vinatzer BA, Patocchi A, Gianfranceschi L, Tartarini S, Zhang HB, Gessler C, et al. Apple contains receptor-like genes homologous to the Cladosporium fulvum resistance gene family of tomato with a cluster of genes cosegregating with Vf apple scab resistance. Mol Plant Microbe Interact. 2001;14:508–15. doi: 10.1094/MPMI.2001.14.4.508. [DOI] [PubMed] [Google Scholar]
- 19.Celton JM, Christoffels A, Sargent D, Xu X, Rees DJ. Genome-wide SNP identification by high-throughput sequencing and selective mapping allows sequence assembly positioning using a framework genetic linkage map. BMC Biol. 2010;8:155. doi: 10.1186/1741-7007-8-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Broggini GAL, Le Cam B, Parisi L, Wu C, Zhang H-B, Gessler C, et al. Construction of a contig of BAC clones spanning the region of the apple scab avirulence gene AvrVg. Fungal Genet Biol. 2007;44:44–51. doi: 10.1016/j.fgb.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 21.Thakur K, Chawla V, Bhatti S, Swarnkar M, Kaur J, Shankar R, et al. De novo transcriptome sequencing and analysis for Venturia inaequalis, the devastating apple scab pathogen. PLoS One. 2013;8:e53937. doi: 10.1371/journal.pone.0053937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cooke IR, Jones DA, Bowen JK, Deng C, Faou P, Hall NE, et al. Proteogenomic analysis of the Venturia pirina (Pear Scab fungus) secretome reveals potential effectors. J Proteome Res. 2014;13:3635–44. doi: 10.1021/pr500176c. [DOI] [PubMed] [Google Scholar]
- 23.Stanke M, Schöffmann O, Morgenstern B, Waack S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics. 2006;7:62. doi: 10.1186/1471-2105-7-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keller O, Kollmar M, Stanke M, Waack S. A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics. 2011;27:757–63. doi: 10.1093/bioinformatics/btr010. [DOI] [PubMed] [Google Scholar]
- 25.Parra G, Bradnam K, Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23:1061–7. doi: 10.1093/bioinformatics/btm071. [DOI] [PubMed] [Google Scholar]
- 26.Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–12. [DOI] [PubMed]
- 27.Park BH, Karpinets TV, Syed MH, Leuze MR, Uberbacher EC. CAZymes Analysis Toolkit (CAT): web service for searching and analyzing carbohydrate-active enzymes in a newly sequenced organism using CAZy database. Glycobiology. 2010;20:1574–84. doi: 10.1093/glycob/cwq106. [DOI] [PubMed] [Google Scholar]
- 28.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 29.Sayers EW, Barrett T, Benson DA, Bolton E, Bryant SH, Canese K, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2012;40(Database Issue):D13–25. doi: 10.1093/nar/gkr1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li L, Stoeckert CJ, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–89. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. The Carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42(Database Issue):D490–5. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao Z, Liu H, Wang C, Xu J-R. Comparative analysis of fungal genomes reveals different plant cell wall degrading capacity in fungi. BMC Genomics. 2013;14:274. doi: 10.1186/1471-2164-14-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Orbach MJ, Farrall L, Sweigard JA, Chumley FG, Valent B. A telomeric avirulence gene determines efficacy for the rice blast resistance gene Pi-ta. Plant Cell. 2000;12:2019–32. doi: 10.1105/tpc.12.11.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bryan GT, Wu K-S, Farrall L, Jia Y, Hershey HP, McAdams SA, et al. A single amino acid difference distinguishes resistant and susceptible alleles of the rice blast resistance gene Pi-ta. Plant Cell. 2000;12:2033–45. doi: 10.1105/tpc.12.11.2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Urban M, Pant R, Raghunath A, Irvine AG, Pedro H, Hammond-Kosack K. The Pathogen-Host Interactions database: additions and future developments. Nucleic Acids Res. 2015;43(Database Issue):D645–55. doi: 10.1093/nar/gku1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, et al. The Pfam protein families database. Nucleic Acids Res. 2014;42(Database Issue):D222–30. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mistry J, Bateman A, Finn RD. Predicting active site residue annotations in the Pfam database. BMC Bioinformatics. 2007;8:298. doi: 10.1186/1471-2105-8-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li W, Cowley A, Uludag M, Gur T, McWilliam H, Squizzato S, et al. The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res. 2015;43(Web Server Issue):W580–4. doi: 10.1093/nar/gkv279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sunde M, Kwan AHY, Templeton MD, Beever RE, Mackay JP. Structural analysis of hydrophobins. Micron. 2008;39:773–84. doi: 10.1016/j.micron.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 40.Kershaw MJ, Talbot NJ. Hydrophobins and repellents: proteins with fundamental roles in fungal morphogenesis. Fungal Genet Biol. 1998;23:18–33. doi: 10.1006/fgbi.1997.1022. [DOI] [PubMed] [Google Scholar]
- 41.Bayry J, Aimanianda V, Guijarro JI, Sunde M, Latgé J-P. Hydrophobins—unique fungal proteins. PLoS Pathog. 2012;8:e1002700. doi: 10.1371/journal.ppat.1002700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bolton MD, van Esse HP, Vossen JH, de Jonge R, Stergiopoulos I, Stulemeijer IJE, et al. The novel Cladosporium fulvum lysin motif effector Ecp6 is a virulence factor with orthologues in other fungal species. Mol Microbiol. 2008;69:119–36. doi: 10.1111/j.1365-2958.2008.06270.x. [DOI] [PubMed] [Google Scholar]
- 43.Fudal I, Ross S, Gout L, Blaise F, Kuhn ML, Eckert MR, et al. Heterochromatin-like regions as ecological niches for avirulence genes in the Leptosphaeria maculans genome: map-based cloning of AvrLm6. Mol Plant Microbe Interact. 2007;20:459–70. doi: 10.1094/MPMI-20-4-0459. [DOI] [PubMed] [Google Scholar]
- 44.de Jonge R, Peter van Esse H, Maruthachalam K, Bolton M, Santhanam P, Saber M, et al. Tomato immune receptor Ve1 recognizes effector of multiple fungal pathogens uncovered by genome and RNA sequencing. Proc Natl Acad Sci U S A. 2012;109:5110–5. doi: 10.1073/pnas.1119623109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shiller J, Van de Wouw AP, Taranto A, Bowen JK, Dubois D, Robinson A, et al. A large family of AvrLm6-like genes in the apple and pear scab pathogens, Venturia inaequalis and Venturia pirina. Front Plant Sci. 2015;6:980. doi: 10.3389/fpls.2015.00980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bowen JK, Mesarich CH, Rees-George J, Cui W, Fitzgerald A, Win J, et al. Candidate effector gene identification in the ascomycete fungal phytopathogen Venturia inaequalis by expressed sequence tag analysis. Mol Plant Pathol. 2009;10:431–48. doi: 10.1111/j.1364-3703.2009.00543.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kucheryava N, Bowen JK, Sutherland PW, Conolly JJ, Mesarich CH, Rikkerink EHA, et al. Two novel Venturia inaequalis genes induced upon morphogenetic differentiation during infection and in vitro growth on cellophane. Fungal Genet Biol. 2008;45:1329–39. doi: 10.1016/j.fgb.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 48.Mesarich CH, Schmitz M, Tremouihac P, McGillivray DJ, Templeton MD, Dingley AJ. Structure, dynamics and domain organization of the repeat protein Cin1 from the apple scab fungus. Biochim Biophys Acta. 1824;2012:1118–28. doi: 10.1016/j.bbapap.2012.06.015. [DOI] [PubMed] [Google Scholar]
- 49.Mesarich CH. Investigating the structure and function of ViCin1, a novel repeat protein from the apple scab fungus Venturia inaequalis. Auckland: The University of Auckland; 2012. [Google Scholar]
- 50.Kim S, Ahn I-P, Rho H-S, Lee Y-H. MHP1, a Magnaporthe grisea hydrophobin gene, is required for fungal development and plant colonization. Mol Microbiol. 2005;57:1224–37. doi: 10.1111/j.1365-2958.2005.04750.x. [DOI] [PubMed] [Google Scholar]
- 51.Fellbrich G, Romanski A, Varet A, Blume B, Brunner F, Engelhardt S, et al. NPP1, a Phytophthora-associated trigger of plant defense in parsley and Arabidopsis. Plant J. 2002;32:375–90. doi: 10.1046/j.1365-313X.2002.01454.x. [DOI] [PubMed] [Google Scholar]
- 52.Cabral A, Oome S, Sander N, Kufner I, Nurnberger T, Van den Ackerveken G. Nontoxic Nep1-like proteins of the downy mildew pathogen Hyaloperonospora arabidopsidis: repression of necrosis-inducing activity by a surface-exposed region. Mol Plant Microbe Interact. 2012;25:697–708. doi: 10.1094/MPMI-10-11-0269. [DOI] [PubMed] [Google Scholar]
- 53.Oome S, Raaymakers TM, Cabral A, Samwel S, Böhm H, Albert I, et al. Nep1-like proteins from three kingdoms of life act as a microbe-associated molecular pattern in Arabidopsis. Proc Natl Acad Sci U S A. 2014;111:16955–60. doi: 10.1073/pnas.1410031111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xue C, Park G, Choi W, Zheng L, Dean RA, Xu J-R. Two novel fungal virulence genes specifically expressed in appressoria of the rice blast fungus. Plant Cell. 2002;14:2107–19. doi: 10.1105/tpc.003426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pliego C, Nowara D, Bonciani G, Gheorghe DM, Xu R, Surana P, et al. Host-induced gene silencing in barley powdery mildew reveals a class of ribonuclease-like effectors. Mol Plant Microbe Interact. 2013;26:633–42. doi: 10.1094/MPMI-01-13-0005-R. [DOI] [PubMed] [Google Scholar]
- 56.Whigham E, Qi S, Mistry D, Surana P, Xu R, Fuerst GS, et al. Broadly conserved fungal effector BEC1019 suppresses host cell death and enhances pathogen virulence in powdery mildew of barley (Hordeum vulgare L.) Mol Plant Microbe Interact. 2015;28:968–83. doi: 10.1094/MPMI-02-15-0027-FI. [DOI] [PubMed] [Google Scholar]
- 57.Nepusz T, Sasidharan R, Paccanaro A. SCPS: a fast implementation of a spectral method for detecting protein families on a genome-wide scale. BMC Bioinformatics. 2010;11:120. doi: 10.1186/1471-2105-11-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grigoriev IV, Nikitin R, Haridas S, Kuo A, Ohm R, Otillar R, et al. MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res. 2014;42(Database Issue):D699–704. doi: 10.1093/nar/gkt1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grigoriev IV, Nordberg H, Shabalov I, Aerts A, Cantor M, Goodstein D, et al. The genome portal of the Department of Energy Joint Genome Institute. Nucleic Acids Res. 2012;40(Database Issue):D26–32. doi: 10.1093/nar/gkr947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ohm RA, Feau N, Henrissat B, Schoch CL, Horwitz BA, Barry KW, et al. Diverse lifestyles and strategies of plant pathogenesis encoded in the genomes of eighteen Dothideomycetes fungi. PLoS Pathog. 2012;8:e1003037. doi: 10.1371/journal.ppat.1003037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–9. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Butler J, MacCallum I, Kleber M, Shlyakhter IA, Belmonte MK, Lander ES, et al. ALLPATHS: de novo assembly of whole-genome shotgun microreads. Genome Res. 2008;18:810–20. doi: 10.1101/gr.7337908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bradnam KR, Fass JN, Alexandrov A, Baranay P, Bechner M, Birol I, et al. Assemblathon 2: evaluating de novo methods of genome assembly in three vertebrate species. Gigascience. 2013;2:1–31. doi: 10.1186/2047-217X-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.MacCallum I, Przybylski D, Gnerre S, Burton J, Shlyakhter I, Gnirke A, et al. ALLPATHS 2: Small genomes assembled accurately and with high continuity from short paired reads. Genome Biol. 2009;10:R103. doi: 10.1186/gb-2009-10-10-r103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gnerre S, MacCallum I, Przybylski D, Ribeiro FJ, Burton JN, Walker BJ, et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci U S A. 2011;108:1513–8. doi: 10.1073/pnas.1017351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mehrabi R, Taga M, Kema GHJ. Electrophoretic and cytological karyotyping of the foliar wheat pathogen Mycosphaerella graminicola reveals many chromosomes with a large size range. Mycologia. 2007;99:868–76. doi: 10.1080/15572536.2007.11832518. [DOI] [PubMed] [Google Scholar]
- 67.Ruan J, Jiang L, Chong Z, Gong Q, Li H, Li C, et al. Pseudo-Sanger sequencing: massively parallel production of long and near error-free reads using NGS technology. BMC Genomics. 2013;14:711. doi: 10.1186/1471-2164-14-711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang J, Kobert K, Flouri T, Stamatakis A. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics. 2014;30:614–20. doi: 10.1093/bioinformatics/btt593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.de Wit PJGM, van der Burgt A, Ökmen B, Stergiopoulos I, Abd-Elsalam KA, Aerts AL, et al. The genomes of the fungal plant pathogens Cladosporium fulvum and Dothistroma septosporum reveal adaptation to different hosts and lifestyles but also signatures of common ancestry. PLoS Genet. 2012;8:e1003088. doi: 10.1371/journal.pgen.1003088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xue M, Yang J, Li Z, Hu S, Yao N, Dean RA, et al. Comparative analysis of the genomes of two field isolates of the rice blast fungus Magnaporthe oryzae. PLoS Genet. 2012;8:e1002869. doi: 10.1371/journal.pgen.1002869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Condon BJ, Leng Y, Wu D, Bushley KE, Ohm RA, Otillar R, et al. Comparative genome structure, secondary metabolite, and effector coding capacity across Cochliobolus pathogens. PLoS Genet. 2013;9:e1003233. doi: 10.1371/journal.pgen.1003233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Elkins T, Zinn K, McAllister L, HoffMann FM, Goodman CS. Genetic analysis of a Drosophila neural cell adhesion molecule: interaction of fasciclin I and abelson tyrosine kinase mutations. Cell. 1990;60:565–75. doi: 10.1016/0092-8674(90)90660-7. [DOI] [PubMed] [Google Scholar]
- 73.Kawamoto T, Noshiro M, Shen M, Nakamasu K, Hashimoto K, Kawashima-Ohya Y, et al. Structural and phylogenetic analyses of RGD-CAP/βig-h3, a fasciclin-like adhesion protein expressed in chick chondrocytes. Biochim Biophys Acta. 1998;1395:288–92. doi: 10.1016/S0167-4781(97)00172-3. [DOI] [PubMed] [Google Scholar]
- 74.Gaspar Y, Johnson KL, McKenna JA, Bacic A, Schultz CJ. The complex structures of arabinogalactan-proteins and the journey towards understanding function. Plant Mol Biol. 2001;47:161–76. doi: 10.1023/A:1010683432529. [DOI] [PubMed] [Google Scholar]
- 75.Carr M, Bloemink M, Dentten E, Whelan A, Gordon S, Kelly G, et al. Solution structure of the Mycobacterium tuberculosis complex protein MPB70: from tuberculosis pathogenesis to inherited human corneal desease. J Biol Chem. 2003;278:43736–43. doi: 10.1074/jbc.M307235200. [DOI] [PubMed] [Google Scholar]
- 76.Liu T-B, Chen G-Q, Min H, Lin F-C. MoFLP1, encoding a novel fungal fasciclin-like protein, is involved in conidiation and pathogenicity in Magnaporthe oryzae. J Zhejiang Univ Sci B. 2009;10:434–44. doi: 10.1631/jzus.B0920017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Templeton MD, Rikkerink EHA, Beever RE. Small, cysteine-rich proteins and recogntion in fungal-plant interactions. Mol Plant Microbe Interact. 1994;7:320–5. doi: 10.1094/MPMI-7-0320. [DOI] [Google Scholar]
- 78.Wessels JGH. Developmental regulation of fungal cell wall formation. Annu Rev Phytopathol. 1994;32:413–37. doi: 10.1146/annurev.py.32.090194.002213. [DOI] [Google Scholar]
- 79.Wösten HAB. Hydrophobins: multipurpose proteins. Annu Rev Microbiol. 2001;55:625–46. doi: 10.1146/annurev.micro.55.1.625. [DOI] [PubMed] [Google Scholar]
- 80.Bernard M, Latgé J-P. Aspergillus fumigatus cell wall: composition and biosynthesis. Med Mycol. 2001;39:9–17. doi: 10.1080/mmy.39.1.9.17. [DOI] [PubMed] [Google Scholar]
- 81.Ohtaki S, Maeda H, Takahashi T, Yamagata Y, Hasegawa F, Gomi K, et al. Novel hydrophobic surface binding protein, HsbA, produced by Aspergillus oryzae. Appl Environ Microbiol. 2006;72:2407–13. doi: 10.1128/AEM.72.4.2407-2413.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Takahashi T, Maeda H, Yoneda S, Ohtaki S, Yamagata Y, Hasegawa F, et al. The fungal hydrophobin RolA recruits polyesterase and laterally moves on hydrophobic surfaces. Mol Microbiol. 2005;57:1780–96. doi: 10.1111/j.1365-2958.2005.04803.x. [DOI] [PubMed] [Google Scholar]
- 83.Köller W, Parker DM, Becker CM. Role of cutinase in the penetration of apple leaves by Venturia inaequalis. Phytopathology. 1991;81:1375–9. doi: 10.1094/Phyto-81-1375. [DOI] [Google Scholar]
- 84.Köller W, Parker DM. Purification and characterization of cutinase from Venturia inaequalis. Phytopathology. 1989;79:278–83. doi: 10.1094/Phyto-79-278. [DOI] [Google Scholar]
- 85.Nicholson RL, Kuc J, Williams EB. Histochemical demonstration of transitory esterase-activity in Venturia inaequalis. Phytopathology. 1972;62:1242–7. doi: 10.1094/Phyto-62-1242. [DOI] [Google Scholar]
- 86.Nusbaum CJ, Keitt GW. A cytological study of host-parasite relations of Venturia inaequalis on apple leaves. J Agric Res. 1938;56:595–618. [Google Scholar]
- 87.Valsangiacomo C, Gessler C. Role of the cuticular membrane in ontogenic and Vf-resistance of apple leaves against Venturia inaequalis. Phytopathology. 1988;78:1066–9. doi: 10.1094/Phyto-78-1066. [DOI] [Google Scholar]
- 88.Kollar A. Characterization of specific induction, activity, and isozyme polymorphism of extracellular cellulases from Venturia inaequalis detected in vitro and on the host plant. Mol Plant Microbe Interact. 1994;7:603–11. doi: 10.1094/MPMI-7-0603. [DOI] [Google Scholar]
- 89.Valsangiacomo C, Gessler C. Purification and characterization of an exopolygalacturonase produced by Venturia inaequalis, the causal agent of apple scab. Physiol Mol Plant Pathol. 1992;40:63–77. doi: 10.1016/0885-5765(92)90072-4. [DOI] [Google Scholar]
- 90.Kollar A. Characterization of an endopolygalacturonase produced by the apple scab fungus, Venturia inaequalis. Mycol Res. 1998;102:313–9. doi: 10.1017/S0953756297005194. [DOI] [Google Scholar]
- 91.Arnous A, Meyer AS. Quantitative prediction of cell wall polysaccharide composition in grape (Vitis vinifera L.) and apple (Malus domestica) skins from acid hydrolysis monosaccharide profiles. J Agric Food Chem. 2009;57:3611–9. doi: 10.1021/jf900780r. [DOI] [PubMed] [Google Scholar]
- 92.Almeida DPF, Gomes MH, Pintado M. Polysaccharide composition of dietary fiber and cell walls from ‘Rocha’ Pear at two ripening stages. Acta Hortic (ISHS) 2014;1040:23–9. doi: 10.17660/ActaHortic.2014.1040.1. [DOI] [Google Scholar]
- 93.Yip V, Withers S. Breakdown of oligosaccharides by the process of elimination. Curr Opin Chem Biol. 2006;10:147–55. doi: 10.1016/j.cbpa.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 94.Jashni MK, Mehrabi R, Collemare J, Mesarich CH, de Wit PJGM. The battle in the apoplast: further insights into the roles of proteases and their inhibitors in plant–pathogen interactions. Front Plant Sci. 2015;6:584. doi: 10.3389/fpls.2015.00584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jashni MK, Dols IHM, Iida Y, Boeren S, Beenen HG, Mehrabi R, et al. Synergistic action of a metalloprotease and a serine protease from Fusarium oxysporum f. sp. lycopersici cleaves chitin-binding tomato chitinases, reduces their antifungal activity, and enhances fungal virulence. Mol Plant Microbe Interact. 2015;28:996–1008. doi: 10.1094/MPMI-04-15-0074-R. [DOI] [PubMed] [Google Scholar]
- 96.Sánchez-Vallet A, Saleem-Batcha R, Kombrink A, Hansen G, Valkenburg D-J, Thomma BP, et al. Fungal effector Ecp6 outcompetes host immune receptor for chitin binding through intrachain LysM dimerization. Elife. 2013;2:e00790. doi: 10.7554/eLife.00790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Marshall R, Kombrink A, Motteram J, Loza-Reyes E, Lucas J, Hammond-Kosack KE, et al. Analysis of two in planta expressed LysM effector homologs from the fungus Mycosphaerella graminicola reveals novel functional properties and varying contributions to virulence on wheat. Plant Physiol. 2011;156:756–69. doi: 10.1104/pp.111.176347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lee W-S, Rudd JJ, Hammond-Kosack KE, Kanyuka K. Mycosphaerella graminicola LysM effector-mediated stealth pathogenesis subverts recognition through both CERK1 and CEBiP homologues in wheat. Mol Plant Microbe Interact. 2014;27:236–43. doi: 10.1094/MPMI-07-13-0201-R. [DOI] [PubMed] [Google Scholar]
- 99.Gijzen M, Nürnberger T. Nep1-like proteins from plant pathogens: recruitment and diversification of the NPP1 domain across taxa. Phytochemistry. 2006;67:1800–7. doi: 10.1016/j.phytochem.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 100.Oome S, Van den Ackerveken G. Comparative and functional analysis of the widely occurring family of Nep1-like proteins. Mol Plant Microbe Interact. 2014;27:1081–94. doi: 10.1094/MPMI-04-14-0118-R. [DOI] [PubMed] [Google Scholar]
- 101.Zhou B-J, Jia P-S, Gao F, Guo H-S. Molecular characterization and functional analysis of a necrosis-and ethylene-inducing, protein-encoding gene family from Verticillium dahliae. Mol Plant Microbe Interact. 2012;25:964–75. doi: 10.1094/MPMI-12-11-0319. [DOI] [PubMed] [Google Scholar]
- 102.Ottmann C, Luberacki B, Kufner I, Koch W, Brunner F, Weyand M, et al. A common toxin fold mediates microbial attack and plant defense. Proc Natl Acad Sci U S A. 2009;106:10359–64. doi: 10.1073/pnas.0902362106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Saitoh H, Fujisawa S, Mitsuoka C, Ito A, Hirabuchi A, Ikeda K, et al. Large-scale gene disruption in Magnaporthe oryzae identifies MC69, a secreted protein required for infection by monocot and dicot fungal pathogens. PLoS Pathog. 2012;8:e1002711. doi: 10.1371/journal.ppat.1002711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mesarich CH, Bowen JK, Hamiaux C, Templeton MD. Repeat-containing protein effectors of plant-associated organisms. Front Plant Sci. 2015;6:872. doi: 10.3389/fpls.2015.00872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Han Y, Liu X, Benny U, Kistler HC, VanEtten HD. Genes determining pathogenicity to pea are clustered on a supernumerary chromosome in the fungal plant pathogen Nectria haematococca. Plant J. 2001;25:305–14. doi: 10.1046/j.1365-313x.2001.00969.x. [DOI] [PubMed] [Google Scholar]
- 106.Lo EYY, Donoghue MJ. Expanded phylogenetic and dating analyses of the apples and their relatives (Pyreae, Rosaceae) Mol Phylogenet Evol. 2012;63:230–43. doi: 10.1016/j.ympev.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 107.Dai Y, Winston E, Correll JC, Jia Y. Induction of avirulence by AVR-Pita1 in virulent U.S. field isolates of Magnaporthe oryzae. Crop J. 2014;2:1–9. doi: 10.1016/j.cj.2013.12.002. [DOI] [Google Scholar]
- 108.Khang CH, Park S-Y, Lee Y-H, Valent B, Kang S. Genome organization and evolution of the AVR-Pita avirulence gene family in the Magnaporthe grisea species complex. Mol Plant Microbe Interact. 2008;21:658–70. doi: 10.1094/MPMI-21-5-0658. [DOI] [PubMed] [Google Scholar]
- 109.Duplessis S, Cuomo CA, Lin Y-C, Aerts A, Tisserant E, Veneault-Fourrey C, et al. Obligate biotrophy features unraveled by the genomic analysis of rust fungi. Proc Natl Acad Sci U S A. 2011;108:9166–71. doi: 10.1073/pnas.1019315108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Spanu PD. The genomics of obligate (and nonobligate) biotrophs. Annu Rev Phytopathol. 2012;50:91–109. doi: 10.1146/annurev-phyto-081211-173024. [DOI] [PubMed] [Google Scholar]
- 111.Horbach R, Navarro-Quesada AR, Knogge W, Deising HB. When and how to kill a plant cell: infection strategies of plant pathogenic fungi. J Plant Physiol. 2011;168:51–62. doi: 10.1016/j.jplph.2010.06.014. [DOI] [PubMed] [Google Scholar]
- 112.Guyon K, Balagué C, Roby D, Raffaele S. Secretome analysis reveals effector candidates associated with broad host range necrotrophy in the fungal plant pathogen Sclerotinia sclerotiorum. BMC Genomics. 2014;15:336. doi: 10.1186/1471-2164-15-336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pedersen C, van Themaat E, McGuffin L, Abbott J, Burgis T, Barton G, et al. Structure and evolution of barley powdery mildew effector candidates. BMC Genomics. 2012;13:694. doi: 10.1186/1471-2164-13-694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sperschneider J, Dodds PN, Gardiner DM, Manners JM, Singh KB, Taylor JM. Advances and challenges in computational prediction of effectors from plant pathogenic fungi. PLoS Pathog. 2015;11:e1004806. doi: 10.1371/journal.ppat.1004806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Burg EF, Smith LH. Cloning and characterization of bys1, a temperature-dependent cDNA specific to the yeast phase of the pathogenic dimorphic fungus Blastomyces dermatitidis. Infect Immun. 1994;62:2521–8. doi: 10.1128/iai.62.6.2521-2528.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bono JL, Jaber B, Fisher MA, Abuodeh RO, O’Leary-Jepson E, Scalarone GM, et al. Genetic diversity and transcriptional analysis of the bys1 gene from Blastomyces dermatitidis. Mycopathologia. 2001;152:113–23. doi: 10.1023/A:1013121812329. [DOI] [PubMed] [Google Scholar]
- 117.Win J, Greenwood DR, Plummer KM. Characterisation of a protein from Venturia inaequalis that induces necrosis in Malus carrying the Vm resistance gene. Physiol Mol Plant Pathol. 2003;62:193–202. doi: 10.1016/S0885-5765(03)00061-4. [DOI] [Google Scholar]
- 118.Chruszcz M, Chapman MD, Osinski T, Solberg R, Demas M, Porebski PJ, et al. Alternaria alternata allergen Alt a 1: a unique β-barrel protein dimer found exclusively in fungi. J Allergy Clin Immunol. 2012;1:241–7. doi: 10.1016/j.jaci.2012.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kloppholz S, Kuhn H, Requena N. A secreted fungal effector of Glomus intraradices promotes symbiotic biotrophy. Curr Biol. 2011;21:1204–9. doi: 10.1016/j.cub.2011.06.044. [DOI] [PubMed] [Google Scholar]
- 120.Mueller O, Kahmann R, Aguilar G, Trejo-Aguilar B, Wu A, de Vries RP. The secretome of the maize pathogen Ustilago maydis. Fungal Genet Biol. 2008;45(Suppl 1):S63–70. doi: 10.1016/j.fgb.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 121.Grandaubert J, Lowe RG, Soyer JL, Schoch CL, Van de Wouw AP, Fudal I, et al. Transposable element-assisted evolution and adaptation to host plant within the Leptosphaeria maculans-Leptosphaeria biglobosa species complex of fungal pathogens. BMC Genomics. 2014;15:891. doi: 10.1186/1471-2164-15-891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Raffaele S, Kamoun S. Genome evolution in filamentous plant pathogens: why bigger can be better. Nat Rev Microbiol. 2012;10:417–30. doi: 10.1038/nrmicro2790. [DOI] [PubMed] [Google Scholar]
- 123.Bourras S, McNally KE, Ben-David R, Parlange F, Roffler S, Praz CR, et al. Multiple avirulence loci and allele-specific effector recognition control the Pm3 race-specific resistance of wheat to powdery mildew. Plant Cell. 2015;27:2991–3012. doi: 10.1105/tpc.15.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sacristán S, Vigouroux M, Pedersen C, Skamnioti P, Thordal-Christensen H, Micali C, et al. Coevolution between a family of parasite virulence effectors and a class of LINE-1 retrotransposons. PLoS One. 2009;4:e7463. doi: 10.1371/journal.pone.0007463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Spanu PD, Abbott JC, Amselem J, Burgis TA, Soanes DM, Stüber K, et al. Genome expansion and gene loss in powdery mildew fungi reveal tradeoffs in extreme parasitism. Science. 2010;330:1543–6. doi: 10.1126/science.1194573. [DOI] [PubMed] [Google Scholar]
- 126.de Guillen K, Ortiz-Vallejo D, Gracy J, Fournier E, Kroj T, Padilla A. Structure analysis uncovers a highly diverse but structurally conserved effector family in phytopathogenic fungi. PLoS Pathog. 2015;11:e1005228. doi: 10.1371/journal.ppat.1005228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kubicek CP, Baker S, Gamauf C, Kenerley CM, Druzhinina IS. Purifying selection and birth-and-death evolution in the class II hydrophobin gene families of the ascomycete Trichoderma/Hypocrea. BMC Evol Biol. 2008;8:4. doi: 10.1186/1471-2148-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kwan HY, Winefield RD, Sunde M, Matthews JM, Haverkamp RG, Templeton MD, et al. Structural basis for rodlet assembly in fungal hydrophobins. Proc Natl Acad Sci U S A. 2006;103:3621–6. doi: 10.1073/pnas.0505704103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ohno S. Evolution by gene duplication. London: George Allen & Unwin Ltd; 1970. [Google Scholar]
- 130.He X, Zhang J. Rapid subfunctionalization accompanied by prolonged and substantial neofunctionalization in duplicate gene evolution. Genetics. 2005;169:1157–64. doi: 10.1534/genetics.104.037051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Schmidt S, Houterman P, Schreiver I, Ma L, Amyotte S, Chellappan B, et al. MITEs in the promoters of effector genes allow prediction of novel virulence genes in Fusarium oxysporum. BMC Genomics. 2013;14:119. doi: 10.1186/1471-2164-14-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Schaack S, Gilbert C, Feschotte C. Promiscuous DNA: horizontal transfer of transposable elements and why it matters for eukaryotic evolution. Trends Ecol Evol. 2010;25:537–46. doi: 10.1016/j.tree.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Oliver R. Genomic tillage and the harvest of fungal phytopathogens. New Phytol. 2012;196:1015–23. doi: 10.1111/j.1469-8137.2012.04330.x. [DOI] [PubMed] [Google Scholar]
- 134.Chuma I, Isobe C, Hotta Y, Ibaragi K, Futamata N, Kusaba M, et al. Multiple translocation of the AVR-Pita effector gene among chromosomes of the Rice Blast fungus Magnaporthe oryzae and related species. PLoS Pathog. 2011;7:e1002147. doi: 10.1371/journal.ppat.1002147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Foss EJ, Garrett PW, Kinsey JA, Selker EU. Specificity of repeat-induced point mutation (RIP) in Neurospora: sensitivity of non-Neurospora sequences, a natural diverged tandem duplication, and unique DNA adjacent to a duplicated region. Genetics. 1991;127:711–7. doi: 10.1093/genetics/127.4.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hane JK, Williams AH, Taranto AP, Solomon PS, Oliver RP. Repeat-induced point mutation: a fungal-specific, endogenous mutagenesis process. In: van den Berg MA, Maruthachalam K, editors. Genetic transformation systems in fungi. Berlin: Springer; 2015. pp. 55–68. [Google Scholar]
- 137.Broggini GAL, Bus VGM, Parravicini G, Kumar S, Groenwold R, Gessler C. Genetic mapping of 14 avirulence genes in an EU-B04 × 1639 progeny of Venturia inaequalis. Fungal Genet Biol. 2011;48:166–76. doi: 10.1016/j.fgb.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 138.Stehmann C, Pennycook S, Plummer KM. Molecular identification of a sexual interloper: the pear pathogen, Venturia pirina, has sex on apple. Phytopathology. 2001;91:633–41. doi: 10.1094/PHYTO.2001.91.7.633. [DOI] [PubMed] [Google Scholar]
- 139.Parker DM, Hilber UW, Bodmer M, Smith FD, Yao C, Köller W. Production and transformation of conidia of Venturia inaequalis. Phytopathology. 1995;85:87–91. doi: 10.1094/Phyto-85-87. [DOI] [Google Scholar]
- 140.Chang S, Puryear J, Cairney J. A simple and efficient method for isolating RNA from pine trees. Plant Mol Biol Rep. 1993;11:113–6. doi: 10.1007/BF02670468. [DOI] [Google Scholar]
- 141.Martínez-Alcántara A, Ballesteros E, Feng C, Rojas M, Koshinsky H, Fofanov VY, et al. PIQA: pipeline for Illumina G1 genome analyzer data quality assessment. Bioinformatics. 2009;25:2438–9. doi: 10.1093/bioinformatics/btp429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.FASTX toolkit v0.0.13. http://hannonlab.cshl.edu/fastx_toolkit/. Accessed 21 Aug 2012.
- 143.FastQC. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed 21 Aug 2012.
- 144.fastq-mcf. https://expressionanalysis.github.io/ea-utils/. Accessed 21 Aug 2012.
- 145.Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010;20:265–72. doi: 10.1101/gr.097261.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Genome completeness: a novel approach using core genes. http://korflab.ucdavis.edu/Datasets/genome_completeness/. Accessed 28 Sept 2012.
- 147.Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics. 2011;27:578–9. doi: 10.1093/bioinformatics/btq683. [DOI] [PubMed] [Google Scholar]
- 148.Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110:462–7. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- 149.Price AL, Jones NC, Pevzner PA. De novo identification of repeat families in large genomes. Bioinformatics. 2005;21(Suppl. 1):i351–8. doi: 10.1093/bioinformatics/bti1018. [DOI] [PubMed] [Google Scholar]
- 150.Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–80. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Abrusan G, Grundmann N, DeMeester L, Makalowski W. TEclass: a tool for automated classification of unknown eukaryotic transposable elements. Bioinformatics. 2009;25:1329–33. doi: 10.1093/bioinformatics/btp084. [DOI] [PubMed] [Google Scholar]
- 152.Slater GSC, Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 2005;6:31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Stanke M, Diekhans M, Baertsch R, Haussler D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics. 2008;24:637–44. doi: 10.1093/bioinformatics/btn013. [DOI] [PubMed] [Google Scholar]
- 154.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–11. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–5. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–52. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 2013;8:1494–512. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Schulz MH, Zerbino DR, Vingron M, Birney E. Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics. 2012;28:1086–92. doi: 10.1093/bioinformatics/bts094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Bioconductor version 2.12. http://www.bioconductor.org/. Accessed 29 Sept 2012.
- 162.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–6. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 163.Bendtsen JD, Jensen LJ, Blom N, von Heijne G, Brunak S. Feature-based prediction of non-classical and leaderless protein secretion. Protein Eng Des Sel. 2004;17:349–56. doi: 10.1093/protein/gzh037. [DOI] [PubMed] [Google Scholar]
- 164.The TMHMM server. http://www.cbs.dtu.dk/services/TMHMM/. Accessed 10 Sept 2015.
- 165.Emanuelsson O, Nielsen H, Brunak S, von Heijne G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol. 2000;300:1005–16. doi: 10.1006/jmbi.2000.3903. [DOI] [PubMed] [Google Scholar]
- 166.ProtComp v 9.0. http://www.softberry.com/berry.phtml?topic=protcomppl&group=programs&subgroup=proloc. Accessed 14 Sept 2015.
- 167.Horton P, Park K-J, Obayashi T, Fujita N, Harada H, Adams-Collier CJ, et al. WoLF PSORT: protein localization predictor. Nucleic Acids Res. 2007;35:W585–7. doi: 10.1093/nar/gkm259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Tatusova T, Ciufo S, Fedorov B, O’Neill K, Tolstoy I. RefSeq microbial genomes database: new representation and annotation strategy. Nucleic Acids Res. 2014;42(Database issue):D553–9. doi: 10.1093/nar/gkt1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Mitchell A, Chang H-Y, Daugherty L, Fraser M, Hunter S, Lopez R, et al. The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 2015;43(Database Issue):D213–21. doi: 10.1093/nar/gku1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Suzek BE, Wang Y, Huang H, McGarvey PB, Wu CH, The UniProt Consortium UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics. 2015;31:926–32. doi: 10.1093/bioinformatics/btu739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.The UniProt Consortium UniProt: a hub for protein information. Nucleic Acids Res. 2015;43(Database Issue):D204–12. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Sigrist CJA, de Castro E, Cerutti L, Cuche BA, Hulo N, Bridge A, et al. New and continuing developments at PROSITE. Nucleic Acids Res. 2013;41(Database Issue):D344–7. doi: 10.1093/nar/gks1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.van Dongen S. Graph clustering by flow simulation. The Netherlands: University of Utrecht; 2000. [Google Scholar]
- 174.Goujon M, McWilliam H, Li W, Valentin F, Squizzato S, Paern J, et al. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 2010;38(Web Server Issue):W695–9. doi: 10.1093/nar/gkq313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.McWilliam H, Li W, Uludag M, Squizzato S, Park YM, Buso N, et al. Analysis Tool Web Services from the EMBL-EBI. Nucleic Acids Res. 2013;41(Web Server Issue):W597–600. doi: 10.1093/nar/gkt376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Heger A, Holm L. Rapid automatic detection and alignment of repeats in protein sequences. Proteins. 2000;41:224–37. doi: 10.1002/1097-0134(20001101)41:2<224::AID-PROT70>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 177.Duckert P, Brunak S, Blom N. Prediction of proprotein convertase cleavage sites. Protein Eng Des Sel. 2004;17:107–12. doi: 10.1093/protein/gzh013. [DOI] [PubMed] [Google Scholar]
- 178.Zdobnov EM, Apweiler R. InterProScan - an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17:847–8. doi: 10.1093/bioinformatics/17.9.847. [DOI] [PubMed] [Google Scholar]
- 179.She R, Chu JS-C, Uyar B, Wang J, Wang K, Chen N. genBlastG: using BLAST searches to build homologous gene models. Bioinformatics. 2011;27:2141–3. doi: 10.1093/bioinformatics/btr342. [DOI] [PubMed] [Google Scholar]
- 180.Notredame C, Higgins DG, Heringa J. T-coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–17. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 181.Wallace IM, O’Sullivan O, Higgins DG, Notredame C. M-Coffee: combining multiple sequence alignment methods with T-Coffee. Nucleic Acids Res. 2006;34:1692–9. doi: 10.1093/nar/gkl091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Beitz E. TeXshade: shading and labeling of multiple sequence alignments using LaTeX2e. Bioinformatics. 2000;16:135–9. doi: 10.1093/bioinformatics/16.2.135. [DOI] [PubMed] [Google Scholar]
- 183.Eddy SR. Accelerated Profile HMM Searches. PLoS Comput Biol. 2011;7:e1002195. doi: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.R Core Team . R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2015. [Google Scholar]
- 185.Saunders DGO, Win J, Kamoun S, Raffaele S. Two-dimensional data binning for the analysis of genome architecture in filamentous plant pathogens and other eukaryotes. In: Birch P, Jones J, Bos J, editors. Plant-pathogen interactions: methods and protocols. New York: Springer Science + Business Media; 2014. pp. 29–51. [DOI] [PubMed] [Google Scholar]
- 186.Flutre T, Duprat E, Feuillet C, Quesneville H. Considering transposable element diversification in de novo annotation approaches. PLoS One. 2011;6:e16526. doi: 10.1371/journal.pone.0016526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Caffier V, Patocchi A, Expert P, Bellanger MN, Durel CE, Hilber-Bodmer M, et al. Virulence characterization of Venturia inaequalis reference isolates on the differential set of Malus hosts. Plant Dis. 2015;99:370–5. doi: 10.1094/PDIS-07-14-0708-RE. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Statistics for the whole genome assemblies of isolates of Venturia inaequalis and V. pirina. (XLSX 11 kb)
Repeats in Venturia inaequalis and V. pirina genome assemblies. (XLSX 10 kb)
Analysis of gene models from the Venturia inaequalis and V. pirina genomes. (XLSX 10 kb)
Core Eukaryotic Gene Model Analysis (CEGMA) of the Venturia inaequalis and V. pirina genomes. The 248 most highly-conserved core eukaryotic genes were used in the CEGMA to assess genome completeness. (XLSX 10 kb)
Benchmarking Universal Single-Copy Orthologs (BUSCO_v1.1b) analysis of the Venturia inaequalis and V. pirina genomes. BUSCO analysis was used to assess completeness of the Venturia genome assemblies. (XLSX 10 kb)
Pipeline of programmes used to identify the in silico-predicted secretome of each isolate. (DOCX 147 kb)
The predicted secretomes and small, secreted proteins (SSPs) encoded by the genomes of Venturia and representative related pathogens. The genomes for Parastagonospora nodorum, Cladosporium fulvum and Puccinia graminis f. sp. tritici were downloaded via the Mycocosm portal at the Joint Genome Institute (JGI; see details in Additional file 15). These pathogens were representatives of necrotrophic, facultative and obligate biotrophic fungi, respectively. The pipeline outlined in Additional file 6 was employed to identify the secretome and SSPs in each of the genomes. (XLSX 11 kb)
The predicted proteins belonging to OrthoMCL clusters present in the secretomes of five Venturia isolates. The coloured horizontal bars represent proportions of predicted proteins (scale at bottom of figure) that have been annotated according to the key at the top of the figure, that are present in the secretomes indicated by grey boxes to the left of the figure (white boxes indicate lack of similar proteins). S: indicates singleton proteins. Numbers in the boxes are numbers of proteins. The order of the categories in the key is the same as that in the horizontal bars. For example: the core secretome is represented by the top-most horizontal bar; there are similar proteins present in each of the secretomes (5 grey boxes), of which 15% (77 proteins) are classified as SSPs between 200 and 500 amino acids in length with 2 or more cysteines. (PPTX 56 kb)
The CAZyme domains present in proteins encoded by the Venturia secretomes. CAZyme domains were identified using the CAZymes analysis toolkit (CAT) server. Domains: CBM: carbohydrate binding module; CE: carbohydrate esterase; GH: glycoside hydrolase; GT: glycosyltransferase; PL: polysaccharide lyase. The known substrates for the CAZyme domains were taken from Zhao et al. PCW: plant cell wall. FCW: fungal cell wall. CW: cell wall. ESR: energy storage and recovery. PG: protein glycosylation. dpi: days post inoculation. (XLSX 17 kb)
Summary of distribution of similar proteins with CAZyme domains encoded by the Venturia secretomes. CAZyme domains were identified using the CAZymes analysis toolkit (CAT) server. Filled grey boxes in columns indicate presence of a similar protein or proteins in that secretome identified by OrthoMCL clustering. (XLSX 12 kb)
Classification of proteases in the secretomes of the Venturia pathogens. Filled grey boxes in columns indicate presence of similar proteins in that secretome identified by OrthoMCL clustering: aspartate and serine (including those annotated as subtilases) proteases, metalloproteases and unclassified peptidases. Several of the proteases were similar (<e-10) to proteases in the Pathogen-Host Interactions database (PHI-base). (XLSX 15 kb)
Summary of small, secreted proteins (SSPs) similar to proteins present in the Pathogen-Host Interactions database. Filled grey boxes in columns indicate presence of a similar protein(s) in that secretome identified by OrthoMCL clustering. (XLSX 12 kb)
Small, secreted protein (SSP) family 07 members. Sequence logo of members of A. family 07 from Vi1 and B. family 07 from all isolates. (DOCX 221 kb)
Details of the small (≤200 amino acids in length) secreted proteins in the Venturia infection secretome (VIS) gene set. (XLSX 22 kb)
The genomes used in this study (non-Venturia). Sources of the genomes downloaded or accessed (with permission) from the Joint Genome Institute (JGI)/MycoCosm. (XLSX 14 kb)
Details of the lineage-specific small, secreted proteins (SSPs) identified in Venturia inaequalis ViL and V. pirina secretomes. Only those proteins ≤200 amino acids in length are included. (XLSX 18 kb)
Details of isolates of Venturia inaequalis and V. pirina used in this study. (XLSX 12 kb)
Data Availability Statement
De novo assemblies for the five Venturia isolates have been deposited at DDBJ/EMBL/GenBank under the accession numbers: National Center for Biotechnology Information (NCBI) [29] BioProject ID PRJNA261633 for V. inaequalis and JEMP01000000 for the V. pirina genome. RNA-seq data (V. inaequalis Vi1) are deposited under the same BioProject (ID PRJNA261633): in vitro: SRR1586226, two dpi in planta: SRR1586224, seven dpi in planta: SRR1586223. In addition, the Vi1 and Vp genomes and gene predictions are publicly accessible via the Mycocosm portal at JGI [58, 59].