

Abstract
Rationale:
Phosphaturic mesenchymal tumor (PMT) is a new tumor entity of soft tissue and bone tumor recently accepted by the World Health Organization, which typically causes the paraneoplastic syndrome of tumor-induced osteomalacia (TIO). The majority of PMTs follow a benign clinical course and local recurrence occurs in < 10% of cases, malignant PMTs with distant organ metastasis are extremely uncommon.
Patient concerns:
We reported a 41-year-old woman who was diagnosed with PMT 10 years ago with a repeated recurrence and pulmonary metastasis.
Diagnoses:
Based on clinical manifestations, MRI scan, serum biochemical indicators evaluation, followed by histopathological examination, the patient was diagnosed as malignant PMT with pulmonary metastasis.
Interventions:
The patient was treated with calcium, phosphorus, and vitamin D after surgical resection and measured the serum ion concentrations every 3 months.
Outcomes:
The patient had a favorable outcome for 10 months without recurrence.
Lessons:
PMTs lack of characteristic histological morphology, some recurrence cases may appear benign morphologically; the malignant PMTs are easily overlooked. Patients with PMT should be carefully evaluated and monitored, in order to early identify its malignant potential.
Keywords: fibroblast growth factor 23, immunohistochemistry, phosphaturic mesenchymal tumor, tumor-induced osteomalacia
1. Introduction
Phosphaturic mesenchymal tumor (PMT) is a rare mesenchymal tumor, which usually causes paraneoplastic syndrome of tumor-induced osteomalacia (TIO). The main clinical manifestations are hypophosphatemia, decreased serum 1,25-dihydroxy vitamin D3 levels, and increased levels of serum fibroblast growth factor 23 (FGF23) and parathyroid hormone.[1] The majority of PMTs are benign, malignant PMTs are extremely rare, in this study, we reported a case of malignant PMT with repeatedly recurrent and multiple lung metastases.
2. Case description
A 41-year-old Chinese woman felt pain in lower back and left heel after heavy manual labor in 2006, she was diagnosed as lumbar disc herniation in the local hospital without further treatment. The pain exacerbated gradually with myasthenia gravis, walk instability, but without convulsions, paralysis, and other symptoms. Three years later, the pain increased and the patient could not stand, she went to the local hospital for a health check, multiple rib fractures with marked hypophosphatemia (0.23 mmol/L; ref. 0.81–1.52 mmol/L) and found a tumor in right leg. She was diagnosed as TIO, then a directed biopsy confirmed the tumor as a malignant PMT. Her right leg was amputated, and the phosphorus increased significantly 1 week later after the surgery. She was detected the serum level of phosphorus periodical postoperative. Despite undergoing radiotherapy after surgery, 1 year later, the patient was found that the levels of serum phosphorus decreased with inguinal lymph node enlargement. She was underwent a lymph node biopsy, which further confirmed the diagnosis of a PMT with lymph node metastasis. During the scheduled follow-up, she felt pain of her whole body after 3 years of the first surgery, and the phosphorus of serum was decreased (0.27 mmol/L; ref. 0.81–1.52 mmol/L) at the same time, a recurrence mass was found in her right leg above the knee, then it was removed through the surgical operation again. In 2015, 9 years after the first surgery, somatostatin receptor scinigraphy (SRS) showed multiple somatostatin receptor hyperintense lesions in the subcutaneous and middle segments, part of tumor located near the knee joint of the right lower limb. Ultrasound displayed that there were 3 solid hypoechoic lesions located in the medial subcutaneous fat layer of the right femur. The lesions sizes were 2.54∗1.62 cm (Fig. 1A), 1.53∗0.91 cm, and 1.46∗0.83 cm, respectively, with scattered no echo and spot-like color flow signal in hypoechoic visible (Fig. 1B). Computed tomography showed a 6.4∗4.9 cm uneven mass shadow in basal segment of lower lobe of left lung, part of it showed cystic change, the boundary was not clear and the marginal showed false capsule and calcification (Fig. 2A). Enhancement scanning showed the solid part was enhanced obviously (Fig. 2B).
Figure 1.
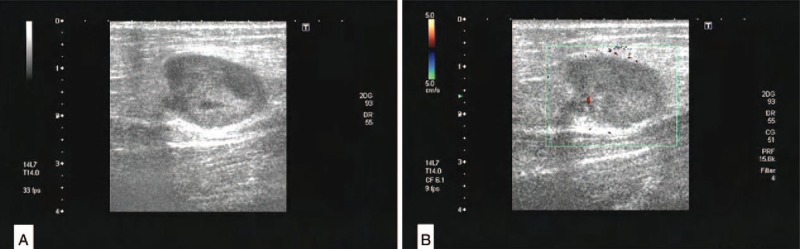
Ultrasound showed the solid hypoechoic in the medial subcutaneous fat layer of the right femur (A), with scattered no echo and spot-like color flow signal in hypoechoic visible (B).
Figure 2.
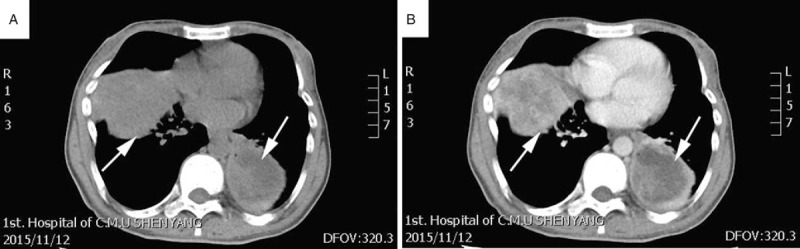
Computed tomography (CT) showed that large nodules in the lungs, with uneven density (arrow, A). The masses exhibited edge strengthening and the internal necrotic area showed no significant enhancement (arrow, B).
Laboratory results showed bone metabolism markers: VitD3 < 3 ng/mL (ref. 11.1–42.9 ng/mL) and osteocalcin 9.48 ng/mL (ref. 11–46 ng/mL) were below the normal range, parathyroid hormone 79.33 pg/mL (ref. 15–65 pg/mL) showed a slightly higher, serum ions: calcium 1.96 mmol/L (ref. 2.17–2.57 mmol/L) and inorganic phosphorus 0.26 mmol/L (ref. 0.81–1.52 mmol/L) were below the normal range. The tubular reabsorption phosphate (TRP, 75.97%), the renal tubular maximum reabsorption of phosphate per liter of GFR (TmP/GFR, 0.5/24 hours), urinary phosphorus (29.38 mmol/L) were within normal range. The patient was carried out with subcutaneous tumor resection and lung tumor puncture biopsy. After 1 week of surgery, the serum phosphorus concentration returned to normal after oral administration of phosphorus.
3. Pathological findings
Histopathological revealed that the tumors were mainly composed of obese spindle cells and osteoclast like giant cells. The spindle cells were arranged in dense bundles or diffuse distribution, the nucleus was oval with homogeneously distributed chromatin, indistinct nucleoli, with frequent mitoses (Fig. 3A). The stromal of tumors were rich in various types of blood vessels, and some areas presented with hemangiopericytoma-like vascular pattern (Fig. 3B), scattered bone islands (Fig. 3 C) and cartilage tissue islands were distributed in tumor stroma, associated with hemorrhage and necrosis (Fig. 3D). Immunohistochemical stainings showed that the spindle tumor cells were perinuclear dot-like positivity expression for FGF23(Fig. 3E), diffuse positive for Vimentin, β-catenin showed spindle tumor cells membrane and cytoplasmic positive, osteoclast like multinucleated giant cells were positive for CD68. The spindle cells were negative for cytokeratin (pan), Bcl-2, S-100, Desmin. The Ki-67 highlighted a high proliferation index (about 40%+) in tumor cells. Similar morphological features and histology were showed in lung biopsy identified the pulmonary metastasis (Fig. 3F). Based on the histopathology findings and clinical features, we confirmed the pathological diagnosis of malignant PMT with pulmonary metastasis. This study was approved by the Institutional Review Board and Committee for Biological and Medical Ethics, China Medical University.
Figure 3.
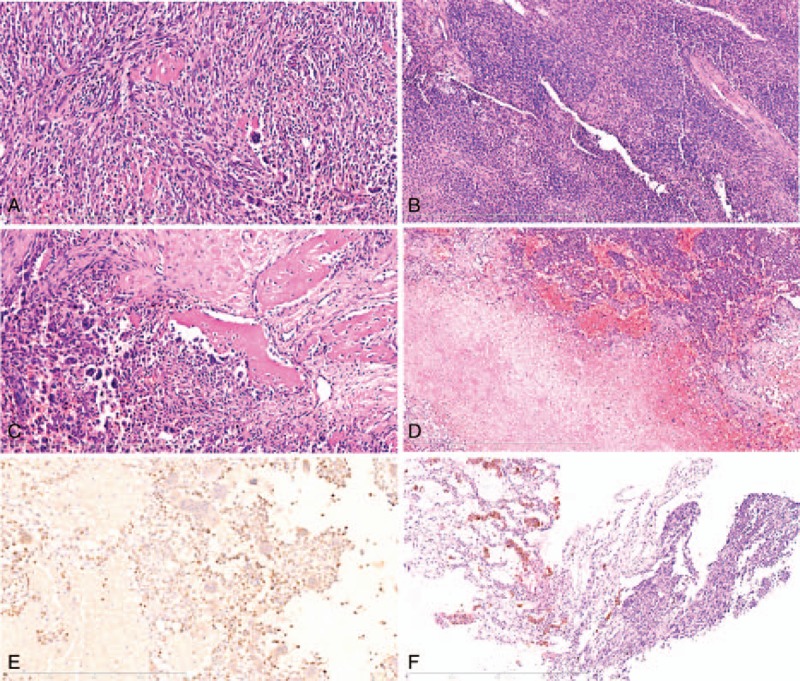
H&E and immunohistochemistry staining. (A) The tumor was mainly composed of obese spindle cells and osteoclast like giant cells (original magnification ×200, scale bar 400 μm). (B) Tumor tissue presented with hemangioma-like structure in some areas (original magnification ×100, scale bar 800 μm). (C) Showed the bone islands (original magnification ×200, scale bar 400 μm). (D) Showed the necrosis and hemorrhage of the tumor (original magnification ×100, scale bar 800 μm). (E) Immunohistochemistry for FGF23 showed a perinuclear dot-like staining pattern. (original magnification ×200, scale bar 400 μm). (F) Showed the lung metastasis (original magnification ×100, scale bar 800 μm).
4. Discussion
In 1987, Weidner first described mesenchymal tumors causing osteomalacia histological morphology and divided it into 4 histological types: PMT, connective tissue subtype (PMTMCT); osteoblastoma-like subtype; nonossifying fibroma-like subtype; and ossifying fibroma-like subtype.[2] Currently, the latter 3 subtypes of PMTMCTs were considered as a series of bone-specific responses. PMTs have been reported only about 250 cases in the literature, they often occur in middle-aged people, occasionally infants and the elderly.[3] It can occur anywhere of soft tissue and bone, the most common in the thigh, followed by the foot, hand, waist, hips, and backcan, but rare in retroperitoneal, visceraland, and mediastinum.[4] About 80% of patients had TIO, clinical manifestations include hypophosphatemia, the serum 1,25(OH)2-vitamin D3 level reduced.[5]
The histological morphology of the PMTs were mainly composed of obese spindle cells and osteoclast like multinucleated giant cells, the spindle cells are the real tumor parenchymal cells. Some cases showed larger staghorn vessels arranged in similar pericytoma or hemangiopericytoma vasculature. The tumor stroma may contain fat components and focal cartilage and bone-like structure, associated with bleeding, microcapsule formation, and mucus change. The matrix may typically exhibited with “grungy” calcification.[6] Due to a morphological of PMTs lack specificity and often showed diversity, they may easily been misdiagnosed as giant cell reparative granuloma, solitary fibrous tumor, chondroblastoma, chondromyxoid fibroma, muscle fiber tumor, spindle cell lipoma, giant cell tumor, osteosarcoma and cutaneous fibrous histiocytoma, and other mesenchymal tumors.[7] To make the tumors more difficult, some recurrence cases may appeared benign morphologically, repeatedly recurrent will result in to malignant transformation. The malignant PMTs were generally high cellularity, more significant cytological atypical or mitotic activity (>5/10 HPF), presented with a sarcomatoid morphology and high Ki67 proliferation index.[1] Uramoto et al[8] reported a case of sublingual PMTMCT with repeated recurrence. Uchihashi et al[9] described 2 cases of PMTs resulting in fatal multiple lung metastases.
Detection of FGF23 and somatostatin receptor 2A expression has subsidiary value in diagnosing a PMT, especially in patients with TIO. FGF23 mRNA expression detected by RT-PCR was present in more than 90% of PMTs with TIO.[10] However, neither FGF23 nor somatostatin receptor 2A expression were specific for confirming the diagnosis of PMT, they were also positive in some aneurysmal bone cyst, osteosarcoma, synovial sarcomas, and hemangiomas.[11] Fibroblast growth factor receptor 1 was a known receptor of FGF23, Lee et al identified fibronectin 1 (FN1)-fibroblast growth factor receptor 1 fusion gene was present in 60% PMTs by next-generation RNA sequencing.[12] And about 6% cases were identified FN1-FGF1 fusion gene.[13] These genetic variants may contributed to PMTs pathology diagnosis.
The prognosis for benign PMTs is excellent after complete surgical resection. For unresectable or incompletely resected tumors, curative intended radiotherapy is a auxiliary therapeutic option, phosphate and active vitamin D supplements are also required.[14] Recent studies have reported that a humanized anti-FGF23 antibody therapy may be useful for TIO patients, octreotide has been found useful for the regulation of phosphate metabolism.[15] This patient was treated with calcium, phosphorus, and vitamin D and measured the serum ion concentrations every 3 months. To date, she was followed up for 10 months from the last operation with no recurrence and no considerable change in serum phosphorus level.
Acknowledgements
The authors thank Program of Science and Technology Department of Liaoning Province (No. 201602877) and Natural Science Foundation of Liaoning Province of China (No: L2015598) for the support.
Footnotes
Abbreviations: FGF23 = fibroblast growth factor 23, PMT = phosphaturic mesenchymal tumor, TIO = tumor-induced osteomalacia.
Authorship: SQ designed the study and drafted the manuscript. SQ, LLC, YQ, ZXL, and PY contributed case information and participated in data analysis. QFZ, LMS, and JD participated in the interpretation of staining slides and evaluation of the histology. All authors approved the final version of the manuscript.
Funding/support: This study was supported by Program of Science and Technology Department of Liaoning Province (No.201602877) and Natural Science Foundation of Liaoning Province of China (No:L2015598).
The authors have no funding and conflicts of interest to disclose.
References
- [1].Hautmann AH, Hautmann MG, Kölbl O, et al. Tumor-induced osteomalacia: an up-to-date review. Curr Rheumatol Rep 2015;17:1–6. [DOI] [PubMed] [Google Scholar]
- [2].Noel Weidner MD, Cruz DS. Phosphaturic mesenchymal tumors. A polymorphous group causing osteomalacia or rickets. Cancer 1987;59:1442–54. [DOI] [PubMed] [Google Scholar]
- [3].Chong WH, Molinolo AA, Chen CC, et al. Tumor-induced osteomalacia. Endocr Relat cancer 2011;18:R53–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Jiang Y, Xia W, Xing X, et al. Tumor-induced osteomalacia: An important cause of adult-onset hypophosphatemic osteomalacia in China: Report of 39 cases and review of the literature. J Bone Miner Res 2012;27:1967–75. [DOI] [PubMed] [Google Scholar]
- [5].Siegel HJ, Rock MG, Inwards C, et al. Phosphaturic mesenchymal tumor. Orthopedics 2002;25:1279–81. [DOI] [PubMed] [Google Scholar]
- [6].De Beur SMJ. Tumor-induced osteomalacia. JAMA 2005;294:1260–7. [DOI] [PubMed] [Google Scholar]
- [7].Jimura TT, Sakaguchi K, Aozasa K. Phosphaturic mesenchymal tumor, mixed connective tissue variant (oncogenic osteomalacia). Pathol Int 1996;46:238–41. [DOI] [PubMed] [Google Scholar]
- [8].Uramoto N, Furukawa M, Yoshizaki T. Malignant phosphaturic mesenchymal tumor, mixed connective tissue variant of the tongue. Auris Nasus Larynx 2008;36:104–5. [DOI] [PubMed] [Google Scholar]
- [9].Uchihashi K, Nishijima-Matsunobu A, Matsuyama A, et al. Phosphaturic mesenchymal tumor, nonphosphaturic variant, causing fatal pulmonary metastasis. Hum Pathol 2013;44:2614–8. [DOI] [PubMed] [Google Scholar]
- [10].Bahrami A, Weiss SW, Montgomery E, et al. RT-PCR analysis for FGF23 using paraffin sections in the diagnosis of phosphaturic mesenchymal tumors with and without known tumor induced osteomalacia. Am J Surg Pathol 2009;33:1348–54. [DOI] [PubMed] [Google Scholar]
- [11].Houang M, Clarkson A, Sioson L, et al. Phosphaturic mesenchymal tumors show positive staining for somatostatin receptor 2A (SSTR2A). Hum Pathol 2013;44:2711–8. [DOI] [PubMed] [Google Scholar]
- [12].Jen-Chieh L, Yung-Ming J, Sheng-Yao S, et al. Identification of a novel FN1-FGFR1 genetic fusion as a frequent event in phosphaturic mesenchymal tumor. J Pathol 2014;235:539–45. [DOI] [PubMed] [Google Scholar]
- [13].Lee JC, Su SY, Changou CA, et al. Characterization of FN1–FGFR1 and novel FN1–FGF1 fusion genes in a large series of phosphaturic mesenchymal tumors. Mod Pathol 2016;29:1335–46. [DOI] [PubMed] [Google Scholar]
- [14].Yoshioka K, Nagata RM, Yamaguchi T, et al. Phosphaturic mesenchymal tumor with symptoms related to osteomalacia that appeared one year after tumorectomy. Intern Med 2006;45:1157–60. [DOI] [PubMed] [Google Scholar]
- [15].Folpe AL, Fanburg-Smith JC, Billings SD, et al. Most osteomalacia-associated mesenchymal tumors are a single histopathologic entity: an analysis of 32 cases and a comprehensive review of the literature. Am J Surg Pathol 2004;28:1–30. [DOI] [PubMed] [Google Scholar]