

Abstract
Background/Objective
An imbalance between energy intake and expenditure leads to obesity. Increasing metabolism and thermogenesis in brown adipose tissue (BAT) can help in overcoming obesity. Here, we investigated the effect of activation of transient receptor potential vanilloid subfamily 1 (TRPV1) in the upregulation of thermogenic proteins in BAT to counter diet-induced obesity.
Subjects/Methods
We investigated the effect of dietary supplementation of capsaicin (TRPV1 agonist) on the expression of metabolically important thermogenic proteins in BAT of wild type and TRPV1−/− mice that received either a normal chow or high fat (± capsaicin; TRPV1 activator) diet by immunoblotting. We measured the metabolic activity, respiratory quotient and BAT lipolysis.
Results
CAP antagonized high fat diet (HFD)-induced obesity without decreasing energy intake in mice. HFD suppressed TRPV1 expression and activity in BAT and CAP countered this effect. HFD feeding caused glucose intolerance, hypercholesterolemia and decreased the plasma concentration of glucagon like peptide-1 and CAP countered these effects. HFD suppressed the expression of metabolically important thermogenic genes, ucp-1, bmp8b, sirtuin 1, pgc-1α and prdm-16 in BAT and CAP prevented this effect. CAP increased the phosphorylation of sirtuin 1 and induced an interaction between PPARγ with PRDM-16. Further, CAP treatment, in vitro, decreased the acetylation of PRDM-16, which was antagonized by inhibition of TRPV1 by capsazepine, chelation of intracellular Ca2+ by cell permeable BAPTA-AM or the inhibition of SIRT-1 by EX 527. Further, CAP supplementation, post HFD, promoted weight loss and enhanced the respiratory exchange ratio. CAP did not have any effect in TRPV1−/− mice.
Conclusions
Our data show that activation of TRPV1 in BAT enhances the expression of SIRT-1, which facilitates the deacetylation and interaction of PPARγ and PRDM-16. These data suggest that TRPV1 activation is a novel strategy to counter diet-induced obesity by enhancing metabolism and energy expenditure.
Keywords: Obesity, PRDM-16, deacetylation, sirtuin 1, brown fat, TRPV1, capsaicin, thermogenes
Introduction
Diet-induced obesity progressively leads to type II diabetes, dyslipidemia, hypertension and cardiovascular diseases. An effective strategy for healthy weight loss employs either caloric restriction or increase energy expenditure. White adipose tissue (WAT) stores excess energy as fat. Brown adipose tissue (BAT) burns fat into energy. BAT activation by endogenous pathways that regulate metabolic activity causes healthy weight loss1. Therefore, activation of BAT thermogenesis is a good strategy to counter obesity. Pharmacological approach to enhance the BAT activity has geared up the quest for novel molecules that enhance metabolism to counter diet-induced obesity.
Increasing thermogenesis and fat metabolism by stimulating the cellular energy sensor, sirtuin-1 (SIRT-1) is effective against diet-induced obesity2, 3. Deacetylation of adipogenic peroxisome proliferator activated receptor gamma (PPARγ) and its coactivator PGC-1α by SIRT-1 has been recognized to underlie the beneficial effects of SIRT-1 activators to stimulate energy expenditure and counter obesity3, 4. Along with SIRT-1, the thermogenic transcription factor PR domain containing zinc finger protein 16 (PRDM-16) regulates brown fat lineage5–8. Therefore, reengineering thermogenic program in BAT, by activating PRDM-16 will be a novel strategy to counter obesity. Although deacetylation of PPARγ promotes browning of WAT and stabilizes PRDM-163, 9, 10 and is a critical factor maintaining BAT differentiation11, whether PRDM-16 could be regulated by SIRT1 remains elusive.
Transient receptor potential (TRP) vanilloid subfamily 1 (TRPV1) protein is implicated in high fat diet (HFD)-induced obesity12, 13. TRPV1 activation in cultured adipocytic cell line has been shown to induce browning phenotype in white adipocytes14. Also, TRPV1 activation prevented obesity by activating central and peripheral mechanisms, which regulate metabolism and thermogenesis15, 16. Compelling evidences suggest that lack of TRPV1 exacerbates obesity and insulin resistance13 and capsinoids activate BAT to decrease body fat in humans17. However, there is lack of knowledge on the effect of TRPV1 in maintenance of BAT activation. This work analyzed TRPV1 expression in BAT and evaluated the effect of TRPV1 activation by CAP on the expression of thermogenic genes/proteins and metabolic activity to counter obesity.
Research Design and Methods
Animals
Adult male TRPV1−/− (B6.129X1-Trpv1tm1Jul/J) mice and their genetically unaltered wild type (WT) counterparts were purchased from Jackson Laboratory, Maine, USA, and bred in the animal house located in the School of Pharmacy as per protocols approved by the IACUC and as per the ethical regulations. Mice were housed in a climate-controlled environment (22.8 ± 2.0 °C, 45–50% humidity) with a 12/12-light/dark cycle with access to designated diet and water ad libitum. Weight gain, food and water intake were monitored weekly. Starting from week 6, mice were housed in groups of four in separate cages and randomly assigned into feeding groups of NCD or HFD (± CAP) until week 38. At the end of 38 weeks, metabolic study was performed and brown fat pads were obtained and used for in vitro experiments.
Subgroups of WT mice were fed NCD (± CAP) for 32 weeks. We performed a dose response for three concentrations of CAP (0.003%, 0.01% and 0.03% w/w in HFD) to determine their effect on weight gain. For experiments performed with mice to determine the effect of CAP supplementation post HFD, we fed WT and TRPV1−/− mice HFD for 16 weeks (week 6 through 22) followed by supplementation of CAP in HFD (0.01% of CAP) from week 22 through 38. The weight gain and food/water intake were monitored weekly and metabolic studies were performed on weeks 16 and 32.
BAT isolation and quantitative RT-PCR measurements
Following euthanization, mouse was placed with dorsal surface up and cut opened along the back all the way to the neck. The intrascapular BAT found right under the skin between the shoulders, seen as two lobes, was dissected. Quantitative PCR was performed as per previously published methods9 using primers described in Supplemental Table I. The scientist who performed these experiments was blinded on the type of tissues processed for these experiments.
Western blotting and coimmunoprecipitation
Isolated BAT were lysed in lysis buffer. Protein concentrations were determined by Bradford method and equal amounts of protein were separated by SDS-PAGE and transferred to nitrocellulose membrane, immunoblotted with specific antibodies (Supplemental Table II) and detected by chemiluminescence method. All procedure were conducted as per previously published procedures9. The scientist who performed these experiments was blinded on the type of tissues processed for these experiments.
Blood glucose and serum triglyceride measurement and intraperitoneal glucose tolerance test
Fasting blood glucose levels were measured in NCD and HFD (± CAP)-fed mice on weeks 8, 16, 24, 32 and 40. To determine glucose tolerance, IPGTT was performed after fasting mice for 14 hours. Mice were injected with glucose (2g/kg) intraperitoneally. Blood glucose was measured, via tail bleeding, with a glucometer at 0, 15, 30, 60 and 120 minutes. Serum cholesterol and triglycerides were measured using commercially available kits (Biovision, USA).
Quantitative Determination of active plasma glucagon-like peptide-1 (GLP-1)
Mice were fed food ad libitum and blood was drawn into EDTA tubes (BD Biosciences, NJ) containing 10 μl of DPP-IV inhibitor (Millipore, MA) at 9 PM. The tubes were inverted, mixed well and centrifuged at 1000g for 10 min. in a refrigerated centrifuge. GLP-1 in plasma samples was determined as per manufacturer’s instruction (Alpco, NH, USA) using TECAN plate reader.
Cell culture
TRPV1 stably expressing HEK293 cells (HEKTRPV1) were cultured in MEM supplemented with 10% FBS and 1% antibiotics (penicillin and streptomycin). These cells were used for siRNA and other in vitro experiments.
SIRT-1 siRNA in HEKTRPV1 cells
HEKTRPV1 cells were grown to 70% confluence. SIRT-1 siRNA treatment was used to deplete SIRT-1 levels in HEKTRPV1 cells. The siRNA against SIRT-1 was purchased from OriGene (SR308256). Scrambled siRNA transfected HEKTRPV1 cells were used as controls. The siRNAs were applied by transfecting cells at a final concentration of 200 nmol/l for 48 hr. using lipofectamine RNAiMAX reagent (Thermofisher.com). Levels of SIRT-1 protein were assessed by western blot and immunoprecipitation method. Western blot analysis was performed with precleared cytosol and immunoprecipitated samples for SIRT-1. Precleared cytosol was also probed for GAPDH as loading control.
Glycerol release from BAT
Isolated BAT were washed in PBS and incubated in pre-warmed DMEM. For basal lipolysis, cut tissue pieces of BAT (approximately 20 mg) were preincubated in 200 μl of DMEM containing 2 % fatty acid free BSA at 37 °C, 5% CO2 and 95% humidity, for 60 min. Basal and glycerol release were determined as pre previously published procedure9.
Metabolic activity measurement
Metabolic activity determined by using the Comprehensive Laboratory Animal Monitoring System9, 18, 19. Mice were individually placed in the metabolic cages with ad libitum access to food and water. After acclimatization for 24 hr., metabolic parameters including the volume of carbon dioxide produced (VCO2), the volume of oxygen consumed (VO2) and the respiratory exchange ratio (RER = VCO2/VO2) were measured.
Statistical analyses
All data are expressed as means ± S.E.M. Comparisons between groups were analyzed using one-way ANOVA and post hoc analyses were done using ANOVA Tukey HSD test or Student t test, whenever appropriate. Samples sizes were set to determine whether the mean value of an outcome variable is one group differed significantly from that in another group. A P value <0.05 was considered as statistically significant.
Drugs and chemicals
HFD was obtained from Research Diets Inc., NJ, USA. All chemicals were from Sigma, USA. Quantitative RT PCR kits were from Qiagen, USA. Sources of all antibodies used for research are given in Supplemental Table II.
Results
CAP antagonizes HFD-induced obesity in the WT mice
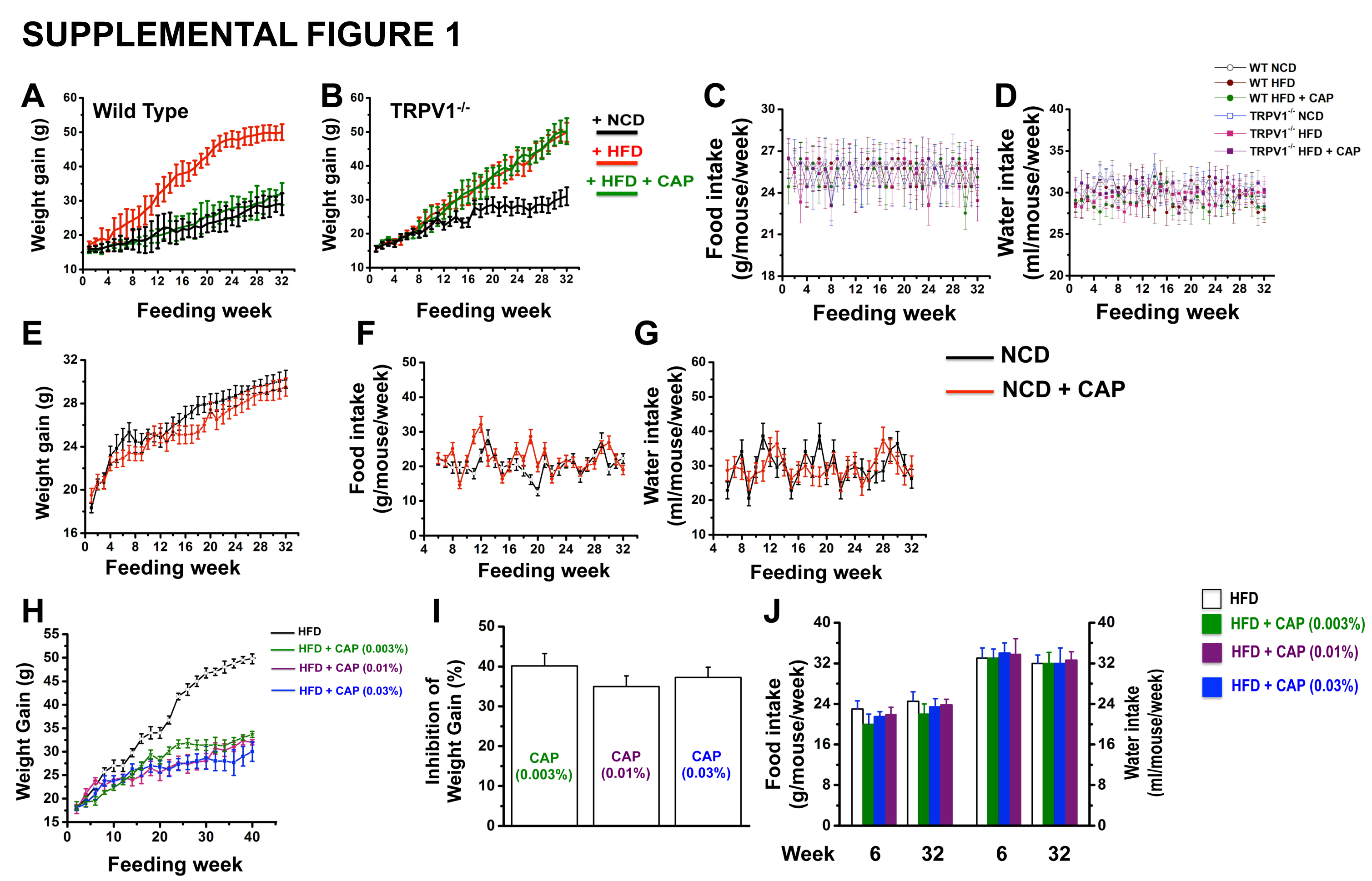
In order to determine the effect of CAP on diet-induced obesity, we fed WT and TRPV1−/− mice NCD, HFD or HFD + CAP (0.01% of the total diet). CAP feeding inhibited weight gain only in the WT but not in TRPV1−/− mice (Supplemental Figure 1A and B). Interestingly, CAP feeding did not alter food and water intake in these mice (Supplemental Figure C and D). We also fed a subgroup of WT mice with either NCD or NCD + CAP. CAP did not alter the weight gain (Supplemental Figure 1E) or food and water intake (Supplemental Figure 1F and G) in these mice. In order to analyze a relationship between dose of CAP in HFD and weight gain inhibition, we fed WT mice HFD [± CAP (0.003%, 0.01% or 0.03%)] for 32 weeks. CAP significantly inhibited HFD-induced weight gain by 35 to 40% (Supplemental Figure 1H and I) without altering diet or water intake (Supplemental Figure 1J).
CAP stimulates metabolic activity
Since CAP significantly inhibited obesity, we evaluated whether CAP increased metabolic activity. Therefore, we determined the RER (respiratory exchange ratio, defined as the ratio of VCO2 to VO2) and ambulatory activity in WT and TRPV1−/− mice-fed NCD or HFD (± CAP) for 10 hr. HFD suppressed RER of WT and TRPV1−/− mice and CAP antagonized this in the WT but not in TRPV1−/− mice (Supplemental Figure 2A, B, C and D). CAP also countered the inhibition of locomotion activity by HFD in the WT but not in TRPV1−/− mice (Supplemental Figure 2C and F).
Mammalian BAT expresses TRPV1 protein
Since CAP did not protect TRPV1−/− mice from obesity we hypothesized that TRPV1 is expressed in BAT and CAP activated TRPV1 to mediate its effects. To analyze this, we immunoprecipitated TRPV1 in the interscapular BAT isolated from NCD or HFD (± CAP)-fed WT and TRPV1−/− mice. Lysates of HEK293 cells that stably expressed TRPV1 served as a positive control for this experiment. As shown in Figure 1A, TRPV1 protein was expressed in the lysates of HEKTRPV1 cells and BAT of the NCD-fed WT mice. WT BAT expressed TRPV1 (Figure 1B and C). We also performed quantitative RT-PCR experiments in these samples. The expression level of trpv1 mRNA normalized to 18s is summarized in Figure 1D. HFD-feeding suppressed TRPV1 expression in BAT and dietary CAP prevented this effect. We also determined the effect of HFD on CAP-stimulated Ca2+ influx in primary BAT isolated from NCD or HFD (± CAP)-fed WT mice. As shown in Figure 1E and F, HFD suppressed CAP-stimulated Ca2+ influx while dietary CAP countered this.
Figure 1. HFD suppresses TRPV1 expression in BAT and CAP counters this effect.

A. Immunoblot for TRPV1 in control and TRPV1 expressing HEK293 cells as well as in BAT of NCD-fed WT and TRPV1−/− mice. B. Immunoprecipitation for TRPV1 in the BAT of NCD or HFD (± CAP)-fed WT and TRPV1−/− mice. Relative protein (C) and relative mRNA (D) expression of TRPV1 in the BAT of NCD or HFD (± CAP)-fed WT and TRPV1−/− mice. E. Time courses of basal and CAP (1 μM)-stimulated Ca2+-influx into brown adipocytes isolated from NCD or HFD (± CAP)-fed WT mice. F. Bar graphs represent mean change in fluorescence S.E.M following the addition of CAP in these cells. Numbers in parenthesis indicate the number of cells under each condition. ** represent statistical significance for P<0.05 for n = 8 independent experiments.
CAP increases the expression of PPARs and thermogenes in BAT
Next, we evaluated the effect of CAP on the expression of adipogenic and thermogenic genes, we measured the expression of PPARα and PPARγ, which play a major role in lipid metabolism. Western blots for PPARα and PPARγ (Figure 2A and B) and their mRNA expressions (Figure 2C and D) show that HFD decreased PPARα protein but not its mRNA, while suppressed both protein and mRNA of PPARγ in BAT. We also measured the expression of thermogenic mitochondrial UCP-1 and bone morphogenetic protein 8b (BMP8b) in BAT. Representative western blots (Figure 2E) show the expression of BMP8b and UCP-1 in BAT. The relative mRNA levels of bmp8b and ucp-1 are given in Figure 2G and H. HFD inhibited the expression of both UCP-1 and BMP8b. Dietary CAP prevented the inhibitory effect of HFD on these adipogenic and thermogenic proteins.
Figure 2. CAP increases PPARs and thermogenic BMP8b and UCP-1 in BAT of WT but not TRPV1−/− mice.

A. PPARα and PPARγ expression in BAT obtained from NCD or HFD (± CAP)-fed WT and TRPV1−/− mice. B. Mean PPAR proteins expression intensity ± S.E.M normalized to GAPDH (loading control) for n = 5 independent experiments. The average mRNA levels ± S.E.M of pparα (C) and pparγ (D) normalized to NCD are given for NCD, HFD or HFD + CAP-fed WT and TRPV1−/− mice. E. BMP8b and UCP-1 expression in BAT obtained from NCD or HFD (± CAP)-fed WT and TRPV1−/− mice. F. BMP8b and UCP-1 protein expression mean band intensities ± S.E.M normalized to the loading control GAPDH for 5 independent experiments. The mean mRNA levels ± S.E.M of bmp8b (G) and ucp-1 (H) normalized to NCD are given for NCD, HFD or HFD + CAP-fed WT and TRPV1−/− mice. For quantitative RT-PCR experiments, 18S ribosomal RNA was used as control. $$ and ** represent statistical significance for P<0.05.
CAP counters hyperglycemia and hyperlipidemia and increases plasma GLP-1
Since CAP increased thermogenic proteins’ expression in BAT and enhanced metabolic activity, and BAT controls glucose and lipid metabolism, we measured the glucose tolerance and lipid profiles in NCD or HFD (± CAP)-fed mice. Random blood sugar levels (Supplemental Figure 3A and B) on week 8, 16, 24, 32 and 40 weeks and performed intraperitoneal glucose tolerance test (IPGTT; Supplemental Figure 3C and D) were measured for WT and TRPV1−/− mice. The mean serum cholesterol and triglyceride levels were increased in HFD-fed mice, and dietary CAP reduced these (Supplemental Figure 3E and F). Since CAP countered the glucose intolerance caused by HFD, we determined the effect CAP on plasma GLP-1 levels in the WT and TRPV1−/− mice. The basal GLP-1 levels (pmol/L) for NCD and HFD (± CAP)-fed WT mice were 2.05 ± 0.08, 0.18 ± 0.05 and 2.81 ± 0.09 and that in TRPV1−/− mice were 1.39 ± 0.079, 0.17 ± 0.07 and 0.16 ± 0.89, respectively (Supplemental Figure 3G). CAP restored the inhibition of GLP-1 inhibition caused by HFD in the WT mice.
CAP increases the expression of cox2, foxc2, cidea and dio2 in BAT
Since CAP enhanced the expression of thermogenic UCP-1 and BMP8b in BAT, we measured the expression of other thermogenic genes in the BAT of NCD or HFD (± CAP)-fed mice (Supplemental Figure 4) by quantitative RT-PCR. The relative abundance of mitochondrial protein-cytochrome-C-oxidase subunit-2 [cox2; a protein that regulates mitochondrial functions and is recognized as an important regulator of thermogenesis20, 21], fork-head transcription factor [foxc2; a regulator of metabolism22, 23], cidea, one of the three members of the CIDE (cell-death-inducing DNA-fragmentation-factor-45-like effector) family of proteins, which plays a central role in adaptive thermogenesis24, 25 and dio2 [dioxygenease type 2; regulates brown adipose tissue lipogenesis and adaptive thermogenesis26, 27]. We also determined the expression of bmp8a, the gene duplicate of bmp8b 28, and bmp4 [brown fat adipogenic marker29]. HFD enhanced the expression of bmp8a while suppressed bmp4 and CAP antagonized this.
CAP increases basal and forskolin-stimulated glycerol release from BAT
Since HFD feeding significantly increased serum triglyceride level and CAP countered this, we evaluated whether CAP stimulates lipolysis by increasing fatty acid and glycerol release in BAT. We measured BAT triglyceride levels (Supplemental Figure 5A) and both basal and cyclic AMP-dependent protein kinase A (PKA) stimulated lipolysis30, 31 in BAT of WT and TRPV1−/− mice. We measured forskolin-stimulated glycerol release in the presence of triacsin C, an inhibitor of acyl-CoA synthetase that prevents the reincorporation of hydrolyzed fatty acids into glycerolipids. CAP-stimulated glycerol release under basal as well as forskolin-stimulated conditions (Supplemental Figure 5B and C).
HFD suppresses the expression of PGC-1α, SIRT-1 and PRDM-16 and CAP antagonizes this
PPARγ coactivator 1α (PGC-1α) is an important regulator of energy and lipid metabolism. SIRT-1 is an NAD+ dependent deacetylase that deacetylates and facilitates the interaction of proteins, which regulate thermogenic mechanism in BAT. These proteins are implicated in metabolism and in the pathophysiology of diabetes and obesity. Since CAP increased metabolism and stimulated lipolysis in BAT, we evaluated whether CAP increased the expression of PGC-1α, SIRT-1 and PRDM-16 (a transcription factor that regulates BAT activation) in BAT (Figure 3). CAP countered the inhibitory effect of HFD on PGC-1α, SIRT-1 and PRDM-16 expression in BAT of WT but not TRPV1−/− mice.
Figure 3. CAP increases the expression of pgc-1α and SIRT-1 in BAT of WT but not TRPV1−/− mice.

A. Western blot for PGC-1α and SIRT-1 proteins in BAT. GAPDH was the loading control. B and C. Mean ratio of PGC-1α and SIRT-1 to GAPDH protein expression ± S.E.M in BAT. D, E and F. Relative mRNA abundance mean ± S.E.M of pgc-1α, SIRT-1 and prdm-16 normalized to 18s in BAT for NCD (white), HFD (striped) or HFD + CAP (black)-fed WT and TRPV1−/− mice. ** represent statistical significance for P<0.05 for n = 8 independent experiments.
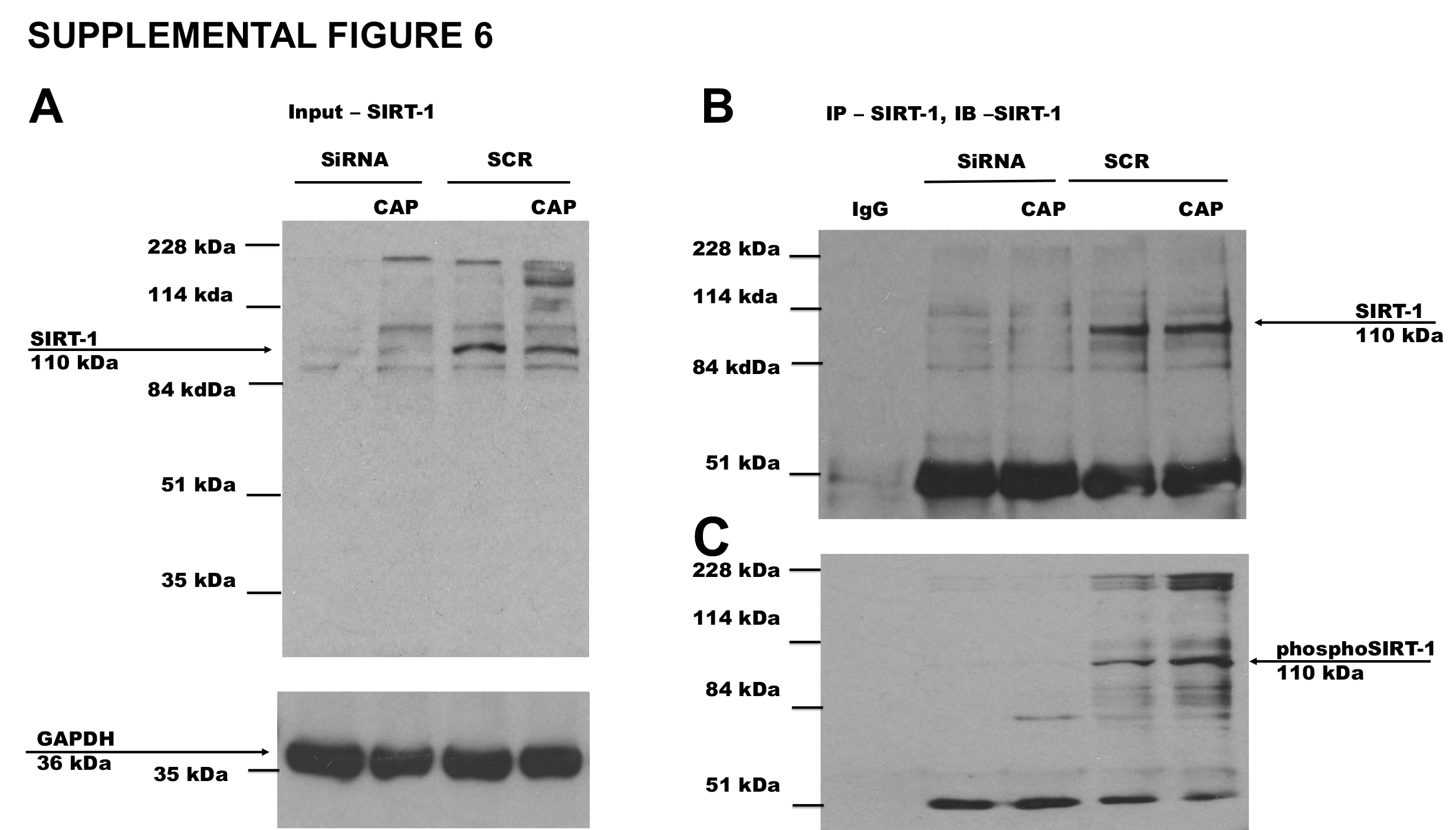
Previous research suggests that CaMKII and 5′-adenosine monophosphate activated kinase (AMPK) phosphorylates and activates SIRT-132. Therefore, we hypothesized that CAP stimulated the phosphorylation of SIRT-1 via CaMKII and AMPK, which is another important metabolic sensor. Consistent to this notion, previous research also suggests that CAP increased AMPK phosphorylation and enhanced the expression of SIRT-1 to suppress matrix metalloproteinase 933. We set to determine the effect of CAP on SIRT-1 and AMPK phosphorylation. Figure 4A shows the SIRT-1 expression in BAT. SIRT-1 immunoprecipitation followed by immunoblotting with phosphoserine antibody is illustrated in Figure 4B. Since cellular protein kinases like CaMKII and AMPK activation can phosphorylate SIRT-1, we measured the phosphorylation of these kinases in BAT. Immunoblots for phospho CaMKII, CaMKII, phospho AMPK and AMPK are shown in Figure 4E and F. HFD suppressed the phosphorylation of CaMKII and AMPK and CAP antagonized this. The phosphorylation and activation of CaMKII and AMPK may enhance the phosphorylation of SIRT-1. If this increases SIRT-1 activity, then SIRT-1 should deacetylate and facilitate the interaction of metabolically important thermogenic proteins in BAT. To address this, we determined the expression, acetylation status and interaction of PRDM-16 and PPARγ in BAT of NCD or HFD (± CAP)-fed WT and TRPV1−/− mice. We also performed experiments in HEKTRPV1 cells after knock-down of SIRT-1 by siRNA. Scrambled siRNA transfected cells were used as controls. Supplemental Figure 6 shows the effect of CAP on SIRT-1 expression and phosphorylation following scrambled or SIRT-1 siRNA transfections in HEKTRPV1 cells under control and CAP (1 μM; 90 min)-stimulated conditions.
Figure 4. HFD suppresses SIRT-1 expression and CAP antagonizes this and enhances SIRT-1 phosphorylation by phosphorylating CaMKII and AMPK.

A. Representative western blot showing SIRT-1 expression in 10% input and β-Actin loading control. B. Representative blot showing SIRT-1 phosphorylation using phospho serine antibody in the SIRT-1 immunoprecipitated sample. C and D. Ratio of SIRT-1 to β-actin and phospho Ser to SIRT-1 in BAT of NCD or HFD (± CAP)-fed WT and TRPV1−/− mice. E and F. Representative western blot shows the expression levels of CaMKII/p-CaMKIIα (Thr286) and AMPK/p-AMPK (Thr172) in 40 μg of BAT lysate with the respective loading controls. G and H. Ratio of phosphoCaMKII to CaMKII and phsphoAMPK to AMPK intensities. ** represent statistical significance for P<0.05 for n = 8 independent experiments.
CAP treatment stimulates PRDM-16 and PPARγ deacetylation
The deacetylation of PPARγ by SIRT-1 activates browning of WAT3, 9. Previous research suggests that PRDM-16 is a transcriptional regulator of BAT development 34 and ablation of PRDM-16 causes metabolic diseases35. These data indicate a critical role of PRDM-16 in metabolism. However, whether SIRT-1 activation deacetylates PRDM-16 to facilitate BAT activation remains unclear. Since HFD suppressed the expression of PRDM-16, we hypothesized that HFD will increase the acetylated PRDM-16 and SIRT-1 activation by CAP will deacetylate it and promote its function in BAT activation. Therefore, we examined this by determining the deacetylation of PRDM-16 by measuring the total and acetylated (lysine acetylation) levels of PRDM-16. As shown in Figure 5A, HFD suppressed the expression of PRDM-16 and CAP prevented this. Also, CAP significantly decreased PRDM-16 acetylation in the BAT of WT mice. The basal expression (NCD-fed condition) of PRDM-16 was significantly lower in TRPV1−/− mice. Also, CAP increased the expression of PPARγ and decreased its acetylation (Figure 5B and C). For these experiments, we immunoprecipitated the acetylated lysine and immunoblot with wither PRDM-16 or PPARγ antibody. The ratio of acetylated lysine to total PRDM-16 or PPARγ is given in Figure 5D and E.
Figure 5. CAP increases deacetylation of PRDM-16 and PPARγ by activating SIRT-1.

A. Top panel shows western blot for PRDM-16 in 10% of total protein used for coimmunoprecipitation studies. Middle Panel shows acetylated PRDM-16 in BAT lysates of WT and TRPV1−/− mice-fed NCD or HFD (± CAP). Lysates were immunoprecipitated with acetylated lysine (Ac-K) antibody and immunoblotted for PRDM-16. Bottom panel represents GAPDH, loading control. B. Immunoblotting of 10% total protein used for coimmunoprecipitation with PPARγ and GAPDH. The proteins were resolved using 10% SDS-PAGE gel. C. Immunoprecipitation of samples using Ac-K antibody and immunoblotted with anti-PPARγ antibody. The samples were resolved using 7.5% SDS PAGE gel. D and E show densitometric ratios of acetylated PRDM-16 to total PRDM-16 and acetylated PPARγ to total PPARγ, respectively. F. Top panel shows the immunoblotting of lysates of TRPV1 expressing HEK 293 cells after treatment with CAP, CPZ + CAP, BAPTA-AM + CAP, EX527, EX527 + CAP, Res, and EX527 + Res with SIRT-1 antibody. Middle panel represents phosphorylated SIRT-1 on samples immunoprecipitated with SIRT-1 followed by immunoblotting with phosphoserine and phosphothreonine antibodies. Bottom panel shows β-actin loading control. G. Densitometric ratio of band intensities of phospho SIRT-1 to total SIRT-1. H. Immunoblot of TRPV1 expressing HEK 293 cells lysates after various treatments and probed with anti-PRDM16 and β-actin (loading control) antibodies. I. Immunoblot of acetylated PRDM-16 in TRPV1 expressing HEK 293 cells lysates after immunoprecipitation with AcK and immunoblotting with anti-PRDM-16 antibody. J. The densitometric band intensity ratios between acetylated PRDM-16 to total PRDM-16. (n = 6 independent experiments). K. Mean ratio ± S.E.M of Fura 2-AM fluorescence in TRPV1 expressing HEK 293 cells stimulated with CAP, CAP + CAP, BAPRA-AM + CAP, EX527 + CAP and Res + CAP (n = 88 – 122 cells). ** represent statistical significance for P<0.05.
If CAP mediates BAT activation by enhancing SIRT-1 activation, which deacetylates PRDM-16, then inhibition of TRPV1 should prevent the effect of CAP. To evaluate this we first analyzed the expression and phosphorylation of SIRT-1 in TRPV1 stably expressing HEK293 cells9. We hypothesized that CAP (1 μM)-stimulated Ca2+ influx via TRPV1 activated CaMKII/AMPK-dependent activation/phosphorylation of SIRT-1 and that inhibition of TRPV1 [by capsazepine, CPZ; 10 μM; 10 min. preincubation followed by 90 min. with CAP; 36], preincubation with BAPTA-AM for 1 hr. [cell permeable Ca2+ chelator37; 10 μM) or SIRT-1 inhibition by pretreatment with EX52738 (10 μM; 1 hr.) will prevent the effect of CAP. We also determined the effect of directly activating SIRT-1 with resveratrol39, 40 [Res; 100 μM; 1 hr.]. As summarized in Figure 5F and G, CAP increased SIRT-1 phosphorylation and CPZ, BAPTA-AM or EX527 pretreatment suppressed this effect of CAP. Resveratrol treatment did not increase SIRT-1 phosphorylation. If CAP stimulates SIRT-1 activation/phosphorylation, then it should cause the deacetylation of PRDM-16. We analyzed this and the effect of CAP (± CPZ, BAPTA or EX527) is shown in Figure 5H, I and J. CAP treatment decreased the deacetylation of PRDM-16, while CPZ, BAPTA-AM or EX527 pretreatment prevented this (Figure 5I). Resveratrol (SIRT-1 activator) also decreased PRDM-16 deacetylation. Also, CAP-stimulated intracellular Ca2+ influx was inhibited by CPZ and BAPTA while EX527 or Res had no effect (Figure 5K).
CAP induces PRDM-16-PPARγ interaction
The physical interaction between PPARγ and PRDM-16 is recognized to regulate brown adipose tissue development, fate of lipid storage or induction of thermogenesis program41 and browning of white adipose tissue42 to increase fat metabolism and energy expenditure. Therefore, we hypothesized that HFD suppresses PPARγ and PRDM-16 interaction and CAP antagonized the effects of HFD by facilitating PPARγ and PRDM-16 interaction to stimulate BAT activation. We performed coimmunoprecipitation experiment using specific antibodies against PPARγ and PRDM-16 to determine their interaction in BAT. Figure 6A shows the expression of PPARγ (top panel) and PRDM-16 (bottom panel) in 10% of the total input used for coimmunoprecipitation studies. PPARγ co-precipitated PRDM16 (Figure 6B Top panel) and PRDM16 co-precipitated PPARγ only in BAT of HFD + CAP-fed WT mice (Figure 6B bottom panel).
Figure 6. CAP increases the expression of PRDM-16 and its interaction with PPARγ in BAT of WT but not TRPV1−/− mice.

A. Western blots with anti PPARγ in 10% of total protein used for coimmunoprecipitation studies. B. PPARγ immunoprecipitation shows interaction with PRDM-16 in NCD and CAP fed conditions in WT. C. Western blots for PRDM-16 in 10% of total protein used for coimmunoprecipitation experiment. D. PRDM-16 immunoprecipitation showing its interaction with PPARγ in CAP fed conditions in WT.$$ and ** represent statistical significance for P<0.05 for n = 8 independent experiments.
CAP supplementation, post HFD, promotes weight loss
Since CAP countered HFD-induced weight gain, we evaluated whether CAP can stimulate weight loss in mice post HFD feeding. The mean weight gain ± S.E.M for WT and TRPV1−/− mice is given in Supplemental Figure 7A and B. We also measured the weekly food and water intake in these mice (Supplemental Figure 7C and D). Supplemental Figure 7C, for the HFD + CAP-fed subgroup of WT mice, shows that the food intake decreased from 24.5 ± 2.49 (week 15) to 14.79 ± 0.59 (week 16) and then returned to 20.5 ± 0.95 (week 17) and to 24.3 ± 2.03 thereafter (week 18). CAP supplemented mouse group lost body weight on week 16 and the rate of weight gain was significantly suppressed thereafter compared to HFD-fed group. We did not observe a significant change in water intake in these mice. Supplementation of HFD + CAP did not induce weight loss in TRPV1−/− mice.
CAP supplementation, post HFD, increases metabolic activity
Since CAP supplementation stimulated weight loss and protected mice from further weight gain, we evaluated the metabolic activity and measured the RER, heat output and ambulatory activity in WT and TRPV1−/− mice at 16 weeks, before diet switch (HFD to HFD + CAP), and at the end of 32 weeks. We also measured these parameters for NCD-fed WT and TRPV1−/− mice. CAP supplementation significantly enhanced carbohydrate oxidation, heat production and ambulatory activity in WT but not in TRPV1−/− mice (Supplemental Figure 7E through 7Q).
Discussion
Although TRPV1 protein is predominately expressed in sensory nerve endings, its expression in a wide variety of tissues including liver43, vascular smooth muscle cells 44, neuromuscular junction45, 46, white adipose tissue9, 14, 47 has been recognized recently. We have previously reported that TRPV1 activation caused beiging of inguinal WAT9. This report provides evidence for the expression of TRPV1 protein in BAT and demonstrates that CAP counters diet-induced obesity by activating TRPV1 in BAT. We have delineated a novel mechanism by which CAP-stimulated intracellular Ca2+ influx via TRPV1 activates SIRT-1-dependent deacetylation of PRDM-16.
Our data clearly demonstrate that CAP feeding enhanced adipogenic and thermogenic proteins in BAT. It is important to note that although TRPV1 expression in other tissues and its activation by CAP may underlie the effect of CAP in preventing obesity, the enhancement of metabolism and energy expenditure by CAP coupled to its ability to cause SIRT-1-dependent deacetylation of PRDM-16 in BAT is suggestive of the significance of TRPV1 activation in BAT in the process.
SIRT-1 is a central energy sensor that deacetylates PPARγ and promotes its interaction with PRDM-16 to trigger the molecular conversion of WAT to beige phenotype3. Here, we report that CAP stimulated SIRT-1-dependent deacetylation of PPARγ as well as PRDM-16 and their resultant interaction in BAT of WT mice. Such an interaction is important for the differentiation of brown preadipocytes to mature adipocytes48. Our data show that inhibition of SIRT-1 by EX527 suppressed SIRT-1 phosphorylation and PRDM-16 deacetylation. These data suggest that CAP mediates its effects primarily by activating SIRT-1/PRDM-16-dependent mechanism to counter obesity. Also, our research has uncovered the essential role of intracellular Ca2+ in causing SIRT-1 phosphorylation and the resultant deacetylation of PRDM-16. That is, preincubation of TRPV1 expressing HEK293 cells with BAPTA-AM suppressed SIRT-1 phosphorylation and PRDM-16 deacetylation (Figure 5H, I and J). Similar effects were observed when cells were pretreated with CPZ, which is a competitive antagonist of TRPV1. Thus, our data provide evidence for a novel TRPV1-dependent signaling mechanism by which CAP activates SIRT-1 (via CaMKII and AMPK) and promotes the deacetylation of PRDM-16 to cause BAT activation.
PRDM-16 regulates brown fat adipogenesis and BAT functions49. It is a co-regulator of PGC-1α 50 and induces UCP-1 expression51. Therefore, stimulating PRDM-16 expression, its deacetylation and stabilization will enhance BAT activation to combat obesity. Consistently, CAP increased the expression of PRDM-16, and facilitated its interaction with PPARγ. Also, CAP increased the expression of UCP-1 and PGC-1α to trigger BAT differentiation, which increase heat production and energy expenditure to combat obesity.
Weight regulation can be mediated by decreasing appetite or absorption of calories. In this study, we show that CAP mediates weight loss by increasing fat metabolism. Under our experimental conditions, we did not observe any decrease in food intake by CAP. Moreover, feeding either 0.003, 0.01 or 0.03% w/w of CAP in HFD showed a robust inhibition in HFD-induced weight gain without altering the energy intake even at the highest concentration of CAP (0.03%). These results indicate that CAP supplementation effectively counters weight gain without modifying diet intake.
Our metabolic study data (supplemental Figures 2 and 7) reveal that CAP feeding enhanced the utilization of carbohydrate rather than fat for energy (RER data) in the WT mice. This can be explained by an enhanced lipolysis in BAT (Supplementary Figure 5B and C) by CAP leading to glycerol production, which is converted to glucose by the liver for energy utilization52. CAP also increased the expression of PPARα, which could play a major role in glycerol metabolism52. The enhanced fat metabolism stimulated by CAP corroborates with the upregulation of expression of ucp-1, bmp4, bmp8b and other thermogenes in BAT by CAP of WT but not TRPV1−/− mice. Moreover, CAP supplementation countered hyperglycemia, hyperlipidemia and impairment of glucose handling in mice. Also, CAP upregulated of plasma GLP-1 level. These data demonstrate a significant contribution of CAP in controlling comorbidities associated with HFD. Even though our data demonstrate that TRPV1 activation by CAP is essential for its effect to suppress HFD-induced hyperglycemia and decrease in GLP-1, it is possible that CAP may mediate these effects by activating pancreatic beta cells and entero-endocrine cells since TRPV1 expression in these cells has been recognized previously53–55. Further studies are, therefore, required to specifically evaluate the mechanisms by which CAP enhanced GLP-1 and restored glucose handling.
One emerging question is that how HFD downregulates TRPV1 expression in BAT and CAP counter-regulates this. Further studies are required to address this. Nonetheless, our data unambiguously show that TRPV1 activation counters diet-induced obesity by possibly stimulating pathway that is regulated by SIRT-1. These data, however, do not neglect the effect activation of TRPV1 expressed in the sensory nerve endings that innervate BAT. This may facilitate thermogenesis via catecholamine-stimulated beta adrenergic receptor expressed in BAT since CAP has been shown to increase adrenaline secretion56. The enhancement of cAMP by CAP57 may trigger a PKA-dependent signaling to activate lipolysis and thermogenesis. Future studies are required to address this alternate mechanism. Also, it is imperative that development of a mouse model, that lacks TRPV1 in BAT or that expresses TRPV1 only in the adipose tissue, will clarify the BAT specific effect of TRPV1. Collectively, this report provides a new insight into the role of TRPV1 activation in the regulation of cellular metabolism by CAP (Figure 7) in BAT.
Figure 7. Mechanism by which TRPV1 activation enhances metabolism and energy expenditure in BAT.

CAP activates TRPV1 expressed on the brown adipose tissue (BAT) cell membrane to stimulate a Ca2+ influx, which activates CAMKII. This activates AMPK phosphorylation and the subsequent phosphorylation and activation of SIRT-1, which deacetylates PPARγ and PRDM-16 and facilitates their stabilization and interaction. Also, SIRT-1 activates PGC-1α, which transcriptionally activates PPARα and subsequently UCP-1. Activation of TRPV1 also enhances BMP8b. All these enhance metabolism, energy expenditure and thermogenesis to counter HFD-induced obesity.
Although this work presents BAT specific effects of CAP, this work does not discount the effect of CAP on TRPV1-dependent pathways that are independent of its effect on BAT. That is, CAP treatment could promote a gamut of signaling mechanisms that modulate the release of neurotransmitters, neuropeptides and hormones to regulate metabolic activity. Given the broad body of knowledge that describe the benefits of BAT activation in the regulation of metabolism and energy expenditure, our data demonstrate that CAP could significantly enhance SIRT-1-dependent thermogenic processes. Nevertheless, our data provide a new insight into the role of TRPV1 activation in mediating SIRT-1-dependent deacetylation of PPARγ and PRDM-16 and their interaction in BAT, which has clinical relevance.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This work was supported by funding from the AHA Southwest Affiliate Faculty Beginning Grant in Aid (15BGIA23250030), a thematic research project grant from the NIH/NIGMS award 8P20 (GM103432-12) and the University of Wyoming Faculty Seed Research Grant to BT and the National Basic Research Program of China (2013CB531205) to ZZ. We thank Dr. Zhaojie Zhang, for his technical assistance with TECAN plate reader. We thank Dr. Kurt Dolence for critically reading through the manuscript.
Abbreviations
- AMPK
5′-adenosine monophosphate activated kinase
- BMP8b
bone morphogenetic protein 8b
- CAP
capsaicin, CPZ, capsazepine
- BAPTA
1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester)
- BAT
brown adipose tissue
- HFD
high fat diet
- NAD+
GLP-1, glucagon like peptide 1
- FSK
forskolin; nicotinamide adenine dinucleotide
- NCD
normal chow diet
- PKA
protein kinase A
- PPAR
peroxisome proliferator activated receptor
- PGC-1α
PPAR gamma 1 coactivator
- PRDM-16
PR domain containing zinc finger protein 16
- RER
respiratory exchange ratio
- CaMKII
Ca2+/calmodulin dependent protein kinase II
- Res
resveratrol
- Ser
Serine
- SIRT-1
sirtuin 1
- siRNA
small interference RNA
- Thr/Threo
Threonine
- TRPV1
transient receptor potential vanilloid subfamily 1
- TRPV1−/−
TRPV1 knockout
- UCP-1
uncoupling protein 1
- Unt
untreated
- WT
wild type
Footnotes
Conflict of Interest
None. The authors declare that there are no conflicts of interests and competing financial interest.
References
- 1.Broeders E, Bouvy ND, van Marken Lichtenbelt WD. Endogenous ways to stimulate brown adipose tissue in humans. Ann Med. 2015;47(2):123–32. doi: 10.3109/07853890.2013.874663. [DOI] [PubMed] [Google Scholar]
- 2.Andrade JM, Frade AC, Guimaraes JB, Freitas KM, Lopes MT, Guimaraes AL, et al. Resveratrol increases brown adipose tissue thermogenesis markers by increasing SIRT1 and energy expenditure and decreasing fat accumulation in adipose tissue of mice fed a standard diet. Eur J Nutr. 2014;53(7):1503–10. doi: 10.1007/s00394-014-0655-6. [DOI] [PubMed] [Google Scholar]
- 3.Qiang L, Wang L, Kon N, Zhao W, Lee S, Zhang Y, et al. Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of Ppargamma. Cell. 2012;150(3):620–32. doi: 10.1016/j.cell.2012.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha} J Biol Chem. 2005;280(16):16456–60. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- 5.Becerril S, Gomez-Ambrosi J, Martin M, Moncada R, Sesma P, Burrell MA, et al. Role of PRDM16 in the activation of brown fat programming. Relevance to the development of obesity. Histology and histopathology. 2013;28(11):1411–25. doi: 10.14670/HH-28.1411. [DOI] [PubMed] [Google Scholar]
- 6.Richard D, Picard F. Brown fat biology and thermogenesis. Front Biosci (Landmark Ed) 2011;16:1233–60. doi: 10.2741/3786. [DOI] [PubMed] [Google Scholar]
- 7.Kajimura S, Seale P, Kubota K, Lunsford E, Frangioni JV, Gygi SP, et al. Initiation of myoblast to brown fat switch by a PRDM16-C/EBP-beta transcriptional complex. Nature. 2009;460(7259):1154–8. doi: 10.1038/nature08262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S, et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature. 2008;454(7207):961–7. doi: 10.1038/nature07182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baskaran P, Krishnan V, Ren J, Thyagarajan B. Capsaicin Induces Browning of White Adipose Tissue and Counters Obesity by activating TRPV1 dependent mechanism. Br J Pharmacol. 2016 doi: 10.1111/bph.13514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ohno H, Shinoda K, Spiegelman BM, Kajimura S. PPARgamma agonists induce a white-to-brown fat conversion through stabilization of PRDM16 protein. Cell Metab. 2012;15(3):395–404. doi: 10.1016/j.cmet.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harms MJ, Ishibashi J, Wang W, Lim HW, Goyama S, Sato T, et al. Prdm16 is required for the maintenance of brown adipocyte identity and function in adult mice. Cell Metab. 2014;19(4):593–604. doi: 10.1016/j.cmet.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Derbenev AV, Zsombok A. Potential therapeutic value of TRPV1 and TRPA1 in diabetes mellitus and obesity. Semin Immunopathol. 2015 doi: 10.1007/s00281-015-0529-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee E, Jung DY, Kim JH, Patel PR, Hu X, Lee Y, et al. Transient receptor potential vanilloid type-1 channel regulates diet-induced obesity, insulin resistance, and leptin resistance. Faseb J. 2015;29(8):3182–92. doi: 10.1096/fj.14-268300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baboota RK, Singh DP, Sarma SM, Kaur J, Sandhir R, Boparai RK, et al. Capsaicin induces “brite” phenotype in differentiating 3T3-L1 preadipocytes. PLoS One. 2014;9(7):e103093. doi: 10.1371/journal.pone.0103093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bargut TC, Mandarim-de-Lacerda CA, Aguila MB. A high-fish-oil diet prevents adiposity and modulates white adipose tissue inflammation pathways in mice. J Nutr Biochem. 2015 doi: 10.1016/j.jnutbio.2015.04.002. [DOI] [PubMed] [Google Scholar]
- 16.de Guglielmo G, Melis M, De Luca MA, Kallupi M, Li HW, Niswender K, et al. PPARgamma activation attenuates opioid consumption and modulates mesolimbic dopamine transmission. Neuropsychopharmacology. 2015;40(4):927–37. doi: 10.1038/npp.2014.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saito M, Yoneshiro T. Capsinoids and related food ingredients activating brown fat thermogenesis and reducing body fat in humans. Current opinion in lipidology. 2013;24(1):71–7. doi: 10.1097/MOL.0b013e32835a4f40. [DOI] [PubMed] [Google Scholar]
- 18.Turdi S, Kandadi MR, Zhao J, Huff AF, Du M, Ren J. Deficiency in AMP-activated protein kinase exaggerates high fat diet-induced cardiac hypertrophy and contractile dysfunction. J Mol Cell Cardiol. 2011;50(4):712–22. doi: 10.1016/j.yjmcc.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ren J. Leptin and hyperleptinemia - from friend to foe for cardiovascular function. J Endocrinol. 2004;181(1):1–10. doi: 10.1677/joe.0.1810001. [DOI] [PubMed] [Google Scholar]
- 20.Muller TD, Lee SJ, Jastroch M, Kabra D, Stemmer K, Aichler M, et al. p62 links beta-adrenergic input to mitochondrial function and thermogenesis. J Clin Invest. 2013;123(1):469–78. doi: 10.1172/JCI64209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alberdi G, Rodriguez VM, Miranda J, Macarulla MT, Churruca I, Portillo MP. Thermogenesis is involved in the body-fat lowering effects of resveratrol in rats. Food Chem. 2013;141(2):1530–5. doi: 10.1016/j.foodchem.2013.03.085. [DOI] [PubMed] [Google Scholar]
- 22.De Matteis R, Lucertini F, Guescini M, Polidori E, Zeppa S, Stocchi V, et al. Exercise as a new physiological stimulus for brown adipose tissue activity. Nutr Metab Cardiovasc Dis. 2013;23(6):582–90. doi: 10.1016/j.numecd.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 23.Toh SY, Gong J, Du G, Li JZ, Yang S, Ye J, et al. Up-regulation of mitochondrial activity and acquirement of brown adipose tissue-like property in the white adipose tissue of fsp27 deficient mice. PLoS One. 2008;3(8):e2890. doi: 10.1371/journal.pone.0002890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chan SC, Lin SC, Li P. Regulation of Cidea protein stability by the ubiquitin-mediated proteasomal degradation pathway. Biochem J. 2007;408(2):259–66. doi: 10.1042/BJ20070690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosell M, Jones MC, Parker MG. Role of nuclear receptor corepressor RIP140 in metabolic syndrome. Biochim Biophys Acta. 2011;1812(8):919–28. doi: 10.1016/j.bbadis.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marsili A, Aguayo-Mazzucato C, Chen T, Kumar A, Chung M, Lunsford EP, et al. Mice with a targeted deletion of the type 2 deiodinase are insulin resistant and susceptible to diet induced obesity. PLoS One. 2011;6(6):e20832. doi: 10.1371/journal.pone.0020832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Christoffolete MA, Linardi CC, de Jesus L, Ebina KN, Carvalho SD, Ribeiro MO, et al. Mice with targeted disruption of the Dio2 gene have cold-induced overexpression of the uncoupling protein 1 gene but fail to increase brown adipose tissue lipogenesis and adaptive thermogenesis. Diabetes. 2004;53(3):577–84. doi: 10.2337/diabetes.53.3.577. [DOI] [PubMed] [Google Scholar]
- 28.Whittle AJ, Carobbio S, Martins L, Slawik M, Hondares E, Vazquez MJ, et al. BMP8B increases brown adipose tissue thermogenesis through both central and peripheral actions. Cell. 2012;149(4):871–85. doi: 10.1016/j.cell.2012.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xue R, Wan Y, Zhang S, Zhang Q, Ye H, Li Y. Role of bone morphogenetic protein 4 in the differentiation of brown fat-like adipocytes. American journal of physiology Endocrinology and metabolism. 2014;306(4):E363–72. doi: 10.1152/ajpendo.00119.2013. [DOI] [PubMed] [Google Scholar]
- 30.Mottillo EP, Granneman JG. Intracellular fatty acids suppress beta-adrenergic induction of PKA-targeted gene expression in white adipocytes. Am J Physiol Endocrinol Metab. 2011;301(1):E122–31. doi: 10.1152/ajpendo.00039.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yaney GC, Civelek VN, Richard AM, Dillon JS, Deeney JT, Hamilton JA, et al. Glucagon-like peptide 1 stimulates lipolysis in clonal pancreatic beta-cells (HIT) Diabetes. 2001;50(1):56–62. doi: 10.2337/diabetes.50.1.56. [DOI] [PubMed] [Google Scholar]
- 32.Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458(7241):1056–60. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee GR, Jang SH, Kim CJ, Kim AR, Yoon DJ, Park NH, et al. Capsaicin suppresses the migration of cholangiocarcinoma cells by down-regulating matrix metalloproteinase-9 expression via the AMPK-NF-kappaB signaling pathway. Clin Exp Metastasis. 2014;31(8):897–907. doi: 10.1007/s10585-014-9678-x. [DOI] [PubMed] [Google Scholar]
- 34.Vernochet C, McDonald ME, Farmer SR. Brown adipose tissue: a promising target to combat obesity. Drug News Perspect. 2010;23(7):409–17. doi: 10.1358/dnp.2010.23.7.1487083. [DOI] [PubMed] [Google Scholar]
- 35.Cohen P, Levy JD, Zhang Y, Frontini A, Kolodin DP, Svensson KJ, et al. Ablation of PRDM16 and beige adipose causes metabolic dysfunction and a subcutaneous to visceral fat switch. Cell. 2014;156(1–2):304–16. doi: 10.1016/j.cell.2013.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walpole CS, Bevan S, Bovermann G, Boelsterli JJ, Breckenridge R, Davies JW, et al. The discovery of capsazepine, the first competitive antagonist of the sensory neuron excitants capsaicin and resiniferatoxin. J Med Chem. 1994;37(13):1942–54. doi: 10.1021/jm00039a006. [DOI] [PubMed] [Google Scholar]
- 37.Thyagarajan B, Malli R, Schmidt K, Graier WF, Groschner K. Nitric oxide inhibits capacitative Ca2+ entry by suppression of mitochondrial Ca2+ handling. Br J Pharmacol. 2002;137(6):821–30. doi: 10.1038/sj.bjp.0704949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu GZ, Hou TT, Yuan Y, Hang PZ, Zhao JJ, Sun L, et al. Fenofibrate inhibits atrial metabolic remodelling in atrial fibrillation through PPAR-alpha/sirtuin 1/PGC-1alpha pathway. Br J Pharmacol. 2016;173(6):1095–109. doi: 10.1111/bph.13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jimenez-Gomez Y, Mattison JA, Pearson KJ, Martin-Montalvo A, Palacios HH, Sossong AM, et al. Resveratrol improves adipose insulin signaling and reduces the inflammatory response in adipose tissue of rhesus monkeys on high-fat, high-sugar diet. Cell Metab. 2013;18(4):533–45. doi: 10.1016/j.cmet.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Ligt M, Timmers S, Schrauwen P. Resveratrol and obesity: Can resveratrol relieve metabolic disturbances? Biochim Biophys Acta. 2015;1852(6):1137–44. doi: 10.1016/j.bbadis.2014.11.012. [DOI] [PubMed] [Google Scholar]
- 41.Villanueva CJ, Vergnes L, Wang J, Drew BG, Hong C, Tu Y, et al. Adipose subtype-selective recruitment of TLE3 or Prdm16 by PPARgamma specifies lipid storage versus thermogenic gene programs. Cell Metab. 2013;17(3):423–35. doi: 10.1016/j.cmet.2013.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ohno H, Shinoda K, Spiegelman BM, Kajimura S. PPARgamma agonists induce a white-to-brown fat conversion through stabilization of PRDM16 protein. Cell Metab. 2012;15(3):395–404. doi: 10.1016/j.cmet.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li L, Chen J, Ni Y, Feng X, Zhao Z, Wang P, et al. TRPV1 activation prevents nonalcoholic fatty liver through UCP2 upregulation in mice. Pflugers Arch. 2012;463(5):727–32. doi: 10.1007/s00424-012-1078-y. [DOI] [PubMed] [Google Scholar]
- 44.Toth A, Boczan J, Kedei N, Lizanecz E, Bagi Z, Papp Z, et al. Expression and distribution of vanilloid receptor 1 (TRPV1) in the adult rat brain. Brain Res Mol Brain Res. 2005;135(1–2):162–8. doi: 10.1016/j.molbrainres.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 45.Wong CO, Chen K, Lin YQ, Chao Y, Duraine L, Lu Z, et al. A TRPV channel in Drosophila motor neurons regulates presynaptic resting Ca2+ levels, synapse growth, and synaptic transmission. Neuron. 2014;84(4):764–77. doi: 10.1016/j.neuron.2014.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thyagarajan B, Potian JG, Baskaran P, McArdle JJ. Capsaicin modulates acetylcholine release at the myoneural junction. Eur J Pharmacol. 2014;744:211–9. doi: 10.1016/j.ejphar.2014.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Che H, Yue J, Tse HF, Li GR. Functional TRPV and TRPM channels in human preadipocytes. Pflugers Arch. 2014;466(5):947–59. doi: 10.1007/s00424-013-1355-4. [DOI] [PubMed] [Google Scholar]
- 48.Harms M, Seale P. Brown and beige fat: development, function and therapeutic potential. Nat Med. 2013;19(10):1252–63. doi: 10.1038/nm.3361. [DOI] [PubMed] [Google Scholar]
- 49.Fruhbeck G, Becerril S, Sainz N, Garrastachu P, Garcia-Velloso MJ. BAT: a new target for human obesity? Trends Pharmacol Sci. 2009;30(8):387–96. doi: 10.1016/j.tips.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 50.Seale P, Kajimura S, Yang W, Chin S, Rohas LM, Uldry M, et al. Transcriptional control of brown fat determination by PRDM16. Cell Metab. 2007;6(1):38–54. doi: 10.1016/j.cmet.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Iida S, Chen W, Nakadai T, Ohkuma Y, Roeder RG. PRDM16 enhances nuclear receptor-dependent transcription of the brown fat-specific Ucp1 gene through interactions with Mediator subunit MED1. Genes Dev. 2015;29(3):308–21. doi: 10.1101/gad.252809.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Patsouris D, Mandard S, Voshol PJ, Escher P, Tan NS, Havekes LM, et al. PPARalpha governs glycerol metabolism. J Clin Invest. 2004;114(1):94–103. doi: 10.1172/JCI20468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boesmans W, Owsianik G, Tack J, Voets T, Vanden Berghe P. TRP channels in neurogastroenterology: opportunities for therapeutic intervention. Br J Pharmacol. 2011;162(1):18–37. doi: 10.1111/j.1476-5381.2010.01009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Diaz-Garcia CM, Morales-Lazaro SL, Sanchez-Soto C, Velasco M, Rosenbaum T, Hiriart M. Role for the TRPV1 channel in insulin secretion from pancreatic beta cells. J Membr Biol. 2014;247(6):479–91. doi: 10.1007/s00232-014-9658-8. [DOI] [PubMed] [Google Scholar]
- 55.Fagelskiold AJ, Kannisto K, Bostrom A, Hadrovic B, Farre C, Eweida M, et al. Insulin-secreting INS-1E cells express functional TRPV1 channels. Islets. 2012;4(1):56–63. doi: 10.4161/isl.18915. [DOI] [PubMed] [Google Scholar]
- 56.Watanabe T, Sakurada N, Kobata K. Capsaicin-, resiniferatoxin-, and olvanil-induced adrenaline secretions in rats via the vanilloid receptor. Biosci Biotechnol Biochem. 2001;65(11):2443–7. doi: 10.1271/bbb.65.2443. [DOI] [PubMed] [Google Scholar]
- 57.Xu YP, Zhang JW, Li L, Ye ZY, Zhang Y, Gao X, et al. Complex regulation of capsaicin on intracellular second messengers by calcium dependent and independent mechanisms in primary sensory neurons. Neurosci Lett. 2012;517(1):30–5. doi: 10.1016/j.neulet.2012.04.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.