

Abstract
Fertility in mammals requires appropriate communication within the hypothalamic-pituitary-gonadal axis and the GnRH receptor (GnRHR) is a central conduit for this communication. The GnRHR resides in discrete membrane rafts and raft occupancy is required for signaling by GnRH. The present studies use immunoprecipitation and mass spectrometry to define peptides present within the raft associated with the GnRHR and flotillin-1, a key raft marker. These studies revealed peptides from the F0F1 ATP synthase complex. The catalytic subunits of the F1 domain were validated by immunoprecipitation, flow cytometry, and cell surface biotinylation studies demonstrating that this complex was present at the plasma membrane associated with the GnRHR. The F1 catalytic domain faces the extracellular space and catalyzes ATP synthesis when presented with ADP in normal mouse pituitary explants and a gonadotrope cell line. Steady-state extracellular ATP accumulation was blunted by coadministration of inhibitory factor 1, limiting inorganic phosphate in the media, and by chronic stimulation of the GnRHR. Steady-state extracellular ATP accumulation was enhanced by pharmacological inhibition of ecto-nucleoside triphosphate diphosphohydrolases. Kisspeptin administration induced coincident GnRH and ATP release from the median eminence into the hypophyseal-portal vasculature in ovariectomized sheep. Elevated levels of extracellular ATP augmented GnRH-induced secretion of LH from pituitary cells in primary culture, which was blocked in media containing low inorganic phosphate supporting the importance of extracellular ATP levels to gonadotrope cell function. These studies indicate that gonadotropes have intrinsic ability to metabolize ATP in the extracellular space and extracellular ATP may serve as a modulator of GnRH-induced LH secretion.
The GnRH receptor (GnRHR) is a member of the G protein-coupled receptor (GPCR) superfamily and is a central point of fertility regulation in mammals by integrating hypothalamic secretion of GnRH with gonadotropin production and secretion at the pituitary gonadotrope (1). The GnRHR binds to GnRH resulting in dissociation of the heterotrimeric G protein associated with the receptor and initiation of several intracellular signaling cascades resulting in calcium flux from both extracellular and intracellular pools, the activation of several MAPK pathways including those leading to activation of ERK 1 and 2 and c-Jun N-terminal kinase (JNK) pathways (2–13). Within the GPCR superfamily, the GnRHR is unique in that it lacks an extended cytoplasmic carboxyl terminal tail, which would otherwise be involved in rapid receptor desensitization and internalization, typically via receptor phosphorylation of the carboxyl terminal tail (14–16). Another distinctive feature of the GnRHR is that it is an exclusive and constitutive resident of raft microdomains within the plasma membrane, unlike other GPCRs which partition into or out of raft domains after activation or receptor oligomerization (17–22). Currently, the mechanisms by which the GnRHR and constituents of its signaling network are able to partition into the membrane raft compartment, even in the absence of stimulation, are unknown.
Since the discovery and initial characterization of membrane rafts (reviewed in Refs. 23, 24), these have been associated with numerous physiological processes linked to organization of discrete compartments within the plasma membrane facilitating the activity of signaling molecules including GPCRs. Rafts are thought to be small (10–200 nm), dynamic microdomains within the plasma membrane that are enriched in cholesterol and sphingolipids (25). There are several ways membrane rafts are thought to form in living cells including as a result of the differential miscibility of lipids, which allows lipids with highly acylated side-chains to preferentially associate with each other or with cholesterol, as a result of complex protein-lipid interactions, or protein-protein interactions which “trap” lipids in a given region of the membrane (26–28). It has been hypothesized that membrane rafts serve an organizational role within the plasma membrane acting as scaffolds to compartmentalize or facilitate signal transduction, endocytosis, or to prevent cross talk between the various proteins and lipids that make up the plasma membrane (25). Rafts can be easily, albeit crudely, enriched in low buoyant density fractions using sucrose gradient centrifugation in the presence of a nonionic detergent. The resulting low density detergent-resistant membranes are often taken to represent the membrane raft compartment, although it should be noted that not all entities that copurify with detergent-resistant membranes represent raft microdomains in living cells under physiologic conditions (29).
In the αT3–1 gonadotrope cell line, productive signaling between the GnRHR and the ERK cascade requires that the GnRHR reside in the raft compartment on the plasma membrane (17, 18). The GnRHR-containing raft compartment is marked by the presence of flotillin-1 and flotillin-2, highly conserved membrane raft microdomain-associated proteins (30, 31). Based upon the results of a yeast 2-hybrid screen, Gαq/11 was identified as a binding partner of flotillin-1, suggesting that organization of the flotillin-marked raft compartment may be facilitated by flotillins, serving as a scaffold for other signaling molecules (32). Consistent with these observations, a number of signaling intermediates copurify with the GnRHR in low density membrane fractions including c-Raf kinase, Gαq, calmodulin, 14–3-3 proteins, and ERKs 1 and 2, suggesting that these complexes could facilitate intracellular signaling via GnRHR with rapid kinetics. Moreover, immunoprecipitation (IP) of GnRHR from raft fractions isolated from normal mouse pituitary results in enrichment of ERKs, suggesting that our observations in the αT3–1 cell model are consistent with similar interactions in normal gonadotropes (18). The present studies used flotillin-1 and GnRHR IP approaches and mass spectroscopy (MS) to examine the composition of the raft in gonadotropes and define a more complete set of proteins in an unbiased manner that may contribute to GnRHR action within the raft compartment. Proteomic analysis revealed a cohort of 129 peptides enriched within the raft domain in IPs of flotillin-1 and the GnRHR. Interestingly, a number of proteins from the F0F1 ATP synthase complex are localized within GnRHR-containing rafts and gonadotrope cells are capable of extracellular ATP synthesis and metabolism. To enable its action in vivo, we show that ATP is secreted into the hypophysial portal blood coincident with GnRH. Our studies suggest that extracellular ATP metabolism may reflect a novel regulator of GnRH action on the secretion of LH.
Materials and Methods
Cell culture and reagents
αT3–1 cells, an immortalized mouse gonadotrope cell line (generously provided by Dr Pamela Mellon, University of California, San Diego, CA), were cultured as described previously (33, 34). Briefly, αT3–1 cells were maintained in DMEM containing 2mM glutamine, 100-U penicillin/mL, 100-μg streptomycin/mL and 1× nonessential amino acids and 10% fetal bovine serum. Cells were grown in 5% CO2 in air at 37°C in a humidified environment. Antiflotillin-1 and antiflotillin-2 antibodies were from BD Biosciences. Anti-LHβ antibody was from National Institutes of Health (National Hormone Peptide Program, A. F. Parlow, Torrance, CA; NIDDK). The GnRHR antibody (a gift from Donal Skinner, University of Wyoming) was raised in a rabbit against 20 amino acids of the second extracellular loop (residues 193–212) of the ovine GnRHR (18). ERK2 antibody was purchased from Cell Signaling. Preimmune normal rabbit serum was purchased from Santa Cruz Biotechnology, Inc. Secondary antibodies and Texas Red-streptavidin used for immunohistochemistry were from Jackson ImmunoResearch. Inhibitory factor 1 (IF1) protein was purchased from Novus Biologicals. ARL 67156 was purchased from Tocris Biosciences/R&D Systems. Buserelin (des-GLY10 [D-Ser(t-But)6]-LH-RH ethylamide; referred to as GnRHa) and kisspeptin were obtained from Phoenix Pharmaceuticals Ltd, and all other chemicals were obtained from Sigma. In all experiments, GnRHa was used at 10nM. Glass bottom microwell dishes for confocal studies were obtained from Mat-Tek.
Immunofluorescence in mouse tissues
These animal studies were completed in accordance with the Cornell University Institutional Animal Care and Use Committee. Animals were euthanized by CO2 asphyxiation and whole pituitaries were dissected free and fixed in 4% paraformaldehyde overnight. After fixation, pituitaries were embedded in paraffin and sectioned (5 μm). Pituitary sections were deparaffinized in xylene, and rehydrated through ethanol dilution series to distilled H2O. Epitopes were unmasked by boiling in 10mM citric acid for 10 minutes. Slides were blocked and incubated with an antibody specific for flotillin-1 for 1 hour at room temperature. Slides were then washed with PBS and exposed to an anti-LHβ antibody for 1 hour at room temperature. For visualization, slides were incubated for thirty minutes at 37°C with Texas Red-conjugated antiguinea pig IgG secondary antibodies (1:500) to detect LHβ and Alexa Fluor 488-conjugated mouse IgG to detect flotillin-1 or flotillin-2. Sections were then washed in PBS, stained with 4′,6-diamidino-2-phenylindole (data not shown), mounted, and imaged using a Zeiss LSM 510 confocal microscope.
For some studies, αT3–1 cells were grown in glass-bottom microwell dishes coated with diluted Matrigel (1:100) overnight. The following morning, cells were transiently transfected with GnRHR-GFP for 24 hours using Mirus transfection reagent (Thermo Fisher). Cells were then fixed in 4% paraformaldehyde for 15 minutes at room temperature. After fixation, cells were permeabilized with 0.5% Triton X-100 in PBS for 20 minutes, and then blocked in PBS containing 3% BSA for 1 hour. Cells were then exposed to an antiflotillin-1 antibody (10 μg/mL) in PBS containing 3% BSA for 2 hours. For visualization, cells were incubated with Alexa Fluor 594 antirabbit IgG secondary antibody (2.5 μg/mL) for 1 hour. After immunostaining, cells were stained with 4′,6-diamidino-2-phenylindole and imaged with a Zeiss LSM 710 confocal laser scanning microscope using the appropriate fluorescence filters. Images were analyzed for colocalization within the selected region of interest. Localization of the 2 fluorophores was quantified using ImageJ's colocalization threshold plugin to calculate the Manders (M) and Pearsons coefficients (R). Coefficients closer to 1.0 reflected colocalization of the 2 fluorophores.
Raft preparations and IP
Detergent-resistant, low-density membrane fractions were prepared essentially as described previously (17, 18). Briefly, αT3–1 cells were grown to approximately 65% confluence. Following the indicated treatments, cells were washed twice in cold PBS, and scraped into PBS with protease inhibitors. Cells were pelleted by centrifugation and then suspended in saline buffer containing 25mM 2-(N-morpholino)ethansulfonic acid (pH 6.5), 130mM NaCl, and protease inhibitors to a final volume of 400 μL. The samples were adjusted to a final concentration of 0.1% Triton X-100 and incubated on ice for 10 minutes. Following gentle homogenization (glass Dounce homogenizer), the samples were mixed with an equal volume of 90% sucrose in MBS, placed in a 5-mL ultracentrifuge tube, and overlaid with a discontinuous gradient of sucrose in MBS consisting of 35% and 5% layers. The gradients were centrifuged at 116 000g in a SW55Ti rotor for 20 hours at 4°C. Low-density detergent-resistant membranes were visible as a band of flocculent material at the 35%–5% interface. Fractions (500 μL) were collected starting from the top of the gradient.
Male B6/129 mice (8–20 wk of age) were euthanized by CO2 asphyxiation. For whole pituitary raft fractionations, pituitaries (n = 10) were collected and suspended in ice cold MBS containing 0.1% Triton X-100, homogenized (glass Dounce homogenizer), and subjected to discontinuous sucrose density centrifugation as described above for preparation of detergent-resistant low-density membranes.
For IP using the GnRHR and flotillin-1 antibodies, raft fractions were initially analyzed to determine the specific fractions that were positive for the GnRHR and flotillin-1. GnRHR and flotillin-1 containing raft fractions were then diluted 1:1 in PBS, containing 0.01% Triton X-100 to reduce sucrose concentrations from the gradient. Diluted raft fractions were precleared with protein A/G agarose beads (Santa Cruz Biotechnology, Inc) for 2 hours at 4°C. Supernatants were then transferred to new tubes and antibodies (GnRHR or flotillin-1) and protein A/G agarose beads (or beads alone) were added and incubated overnight at 4°C. The complexes were then washed 4 times in PBS with 0.01% Triton X-100 followed by the addition of sodium dodecyl sulfate (SDS) loading buffer and boiling. IP studies were carried out on at least 3 independent raft preparations.
Samples used for further analysis by mass spectrometry were immunoprecipitated using the Pierce Crosslink IP kit (Thermo Fisher Scientific) rather than using the above method. Anti-GnRHR or antiflotillin-1 antibodies were cross-linked to protein A/G agarose beads according to the manufacturer's instructions. Samples were precleared using preimmune rabbit serum and the control agarose resin supplied with the kit. Precleared samples were then incubated with antibody-cross-linked protein A/G beads on a rocker overnight at 4°C followed by sample recovery according to the manufacturer's instructions. Eluted sample was then boiled in SDS loading buffer and resolved using SDS-PAGE. Protein concentration in the eluted sample was measured using the Bradford assay.
In-gel digestion and extraction for Nano Liquid Chromatography MS/MS and mass spectrometry data analysis
Preparation of SDS-resolved samples for in-gel digestion, extraction, and subsequent nanoscale liquid chromatography followed by tandem electrospray ionization mass spectrometry (NanoLC MS/MS) and data analysis was performed essentially as described previously (35) with minor modifications. Briefly, whole cell detergent-resistant membrane raft fractions, or immunoprecipitated proteins were resolved by 10% SDS-PAGE, then stained using SYPROruby (Invitrogen). Bands of proteins were then selected and excised manually for further processing before being subjected to in-gel digestion with trypsin. Peptide fragments were recovered and subjected to NanoLC MS/MS. Mascot version 2.3 was used to identify peptides and proteins using the mouse genome as a reference (mouse_refseq_20070725.fasta) and using one decoy database with a 99% confidence interval. All detected peptides were strictly validated by 2 criteria: Mascot expectation value less than 0.01 and Mascot ion score more than 36 for whole floating fraction and more than 25 for the immunoprecipitated samples. The decoy database search in Mascot search engine allows estimation of false discovery rate for detected tryptic peptides, which yielded 1%–3% for each sample submitted. After the additional filters described above were applied, the peptide false discovery rate decreased significantly to 0.8%. In each case, all proteins identified in the false discovery analysis were inferred by a single peptide hit. To be included in our lists we required that each protein be represented by 2 or more unique peptides.
Flow cytometry
αT3–1 cells were suspended in staining buffer (PBS with 0.5% BSA, and 0.09% sodium azide) and incubated with primary antibody to ATP-5A conjugated to fluorescein isothiocyanate (1:200; Abcam) for 30 minutes at 4°C. Cells were washed twice with staining buffer following this incubation and were subjected to flow cytometry analysis to assess the frequency of ATP synthase immunoreactivity in control and GnRH-treated cells. All samples were acquired using a FACSCanto II flow cytometer (BD Bioscience). Raw data were evaluated using FlowJo Flow Cytometry Analysis Software.
Cell surface biotinylation and Western blotting
Cell surface biotinylation studies were carried out as previously described and validated (18) using a commercially available kit (Thermo Scientific) with the following optimization in the protocol. Exposure of the cultured αT3–1 cells to sulfo-NHS-SS-biotin was limited to 5 minutes (rather than 30 min described by the kit) followed by 3 rounds of quenching (rather than a single quench step described by the kit). Our preliminary studies demonstrated this optimization was essential to specifically label cell surface proteins but not intracellular proteins. Specificity of biotin labeling was confirmed by resolving mock and cell surface biotinylated proteins from αT3–1 cells by SDS-PAGE and transferred to nitrocellulose paper by electroblotting. Membranes were blocked in 5% nonfat dry milk in 10mM Tris-HCl (pH 7.5), 150mM NaCl, and 0.05% Tween 20 (TBST) and then incubated with streptavidin-coupled horseradish peroxidase at 1:1000 for 1 hour. Blots were washed 4 times in TBST, and protein bands were visualized using enhanced chemiluminescence according to the manufacturer's instructions (PerkinElmer). Chemiluminescence was detected using a Bio-Rad Chemidoc XRS+ System and pictures analyzed using the Image Lab Software from Bio-Rad. In other studies, cells that were cell surface biotinylated were subsequently used for mitochondrial enrichment/ purification using a commercially available kit (Thermo Scientific). This mitochondrial isolation/purification step was used to demonstrate the specific nature of biotinylation at the cell surface rather than from mitochondria leaking from damaged cells. An aliquot of purified mitochondria from these preparations was retained from each study and used in Western blottings to demonstrate that mitochondrial proteins were present in these preparations, but not biotinylated. For Western blotting studies, cells were washed twice in cold PBS, and scraped into cold radio-IP assay buffer containing 20mM Tris-HCl (pH 8.0), 137mM NaCl, 10% glycerol, 1% Nonidet P-40, 0.1% SDS, 0.5% deoxycholate, 2mM EDTA, 5mM sodium vanadate, 0.2mM phenylmethylsulfonyl fluoride, and 5mM benzamidine. Lysates were cleared by centrifugation and protein concentrations of the lysates were determined by Bradford assay. Protein samples were boiled for 5 minutes in SDS load buffer, resolved by SDS-PAGE, and transferred to polyvinylidene difluoride or nitrocellulose membranes by electroblotting. Membranes were blocked with 5% nonfat dry milk or 1× casein in TBST and then incubated with primary and horseradish peroxidase-conjugated secondary antibodies. Protein bands were visualized and documented as described above.
ATP assays
In all studies examining extracellular and intracellular ATP levels, ADP was administered at a concentration of 10μM. Sample media and cell lysates were centrifuged at 8000g for 6 minutes before ATP assay to pellet and discard cellular debris. The ATP assays were carried out on culture media and cell lysates using a commercially available kit (ATPlite; PerkinElmer) in 96-well microtiter plates per the manufacturer's instructions. All media samples were assayed using identical volumes (100 μL). All cell lysate samples were standardized for protein content using a Bradford assay. Preliminary control studies confirmed the media used in the experiments did not have detectable levels of ATP as measured by this assay. Data are expressed as fold change relative to controls. All studies were carried out in triplicate within a replicate in 3–4 experimental replicates.
Pituitary explant and primary cell culture and LH ELISA
Mature male and ovariectomized (ovx) female mice (>3.0 mo of age; experiments conducted 5–7 d after ovx) were euthanized and pituitaries were collected into ice-cold DMEM containing 10% fetal bovine serum (complete media). For pituitary explant studies, mouse pituitaries were placed into culture as previously described for placental disks (36). Briefly, pituitaries were allowed to equilibrate in media overnight. The explants were then washed in complete media or complete media prepared using DMEM within Pi followed by administration of vehicle or ADP for a period of 1 hour. For primary cultures, pituitaries were digested in collagenase (1.5 mg/mL) with periodic trituration using a sterile Pasteur pipette and then transferred to complete media. The dispersed cells were pelleted and suspended in complete media and aliquoted to 24-well plates pretreated with poly L-lysine at a density of approximately 750 000 cells/well. Primary pituitary cells were incubated in complete media at 37°C in a humidified atmosphere (5% CO2:95% O2) overnight. The following day, the cells were gently washed twice in either in DMEM or DMEM without Pi for 2 hours before ADP administration. The cells were treated with either ADP or vehicle as control for an additional period (30 min), then buserelin was added to the indicated wells and the media were harvested 4 hours later and assayed for LH. The pituitary cells remaining in the wells were lysed to assay for total protein using the Bradford assay. Concentrations of LH were determined using a commercially available ELISA per the manufacturer's instructions (Genway Biotechnology), and LH concentration was standardized by protein content of the specific wells.
Hypophyseal portal vasculature cannulation and GnRH RIA
Procedures carried out in animals in these studies were approved in advance by the Monash University, School of Biomedical Sciences Animal Ethics Committee. Female sheep (adult ewes, n = 3, 3–4 y of age; 50- to 60-kg body weight) were ovx and provided 1-week recovery time from surgery. Hypophyseal portal vasculature and the jugular vein were cannulated as previously described (37). Plasma samples were collected at 10-minute intervals for 1 hour into tubes containing 50-μL aprotonin (1.5 mg/mL), 50mM EDTA, and 5mM bacitracin (Sigma-Aldrich). Following obtaining and initial baseline blood sample, a bolus dose of kisspeptin (50 μg in 1 mL of buffered saline) was administered iv and sample collection continued for the remaining hour. Plasma was collected after centrifugation and stored at −80°C until assayed. Portal plasma samples were assayed for ATP concentrations as described above. Portal plasma concentrations of GnRH and jugular plasma concentrations of LH were analyzed using RIA as previously described (38).
Statistical analysis
Data are presented as mean ± SEM of a representative example from at least 3 independent experiments. For each experiment, treatments were carried out in triplicate. Results were analyzed for significance by ANOVA. Post hoc comparisons were made using Student's t test using P ≤ .05 as an index of statistical significance.
Results
Flotillins and the GnRHR occupy overlapping expression domains within the plasma membrane
We have previously demonstrated that the GnRHR is an exclusive and constitutive occupant of low buoyant density membrane rafts marked by flotillin-1 (17, 18). In those studies, flotillin-1 and flotillin-2 were used as markers of the raft compartment. Here, we show using immunofluorescence that flotillin-1 and flotillin-2 are expressed ubiquitously in sections of mouse anterior pituitary including within cells of the gonadotrope lineage (marked by costaining for the β-subunit of LH) (Figure 1A). In αT3–1 cells (33), we have previously demonstrated that the GnRHR and ganglioside GM1 (monosialotetrahexosylganglioside; a ganglioside enriched in raft domains) expression domains at the plasma membrane overlap (18). Recent studies from the Hapgood group demonstrate colocalization of flotillin-1 and the GnRHR in the gonadotrope cell line LβT2 (39). Consistent with these results, the plasma membrane expression domain of flotillin-1 overlapped with a GnRHR-GFP fusion protein (Figure 1B) supporting the possibility that flotillin-1 and the GnRHR shared the same sterol-enriched membrane domain. To further confirm that these 2 proteins localized in a common membrane raft, IP studies were carried out using antibodies directed toward flotillin-1 and the GnRHR in low buoyant density raft fractions from αT3–1 cells (Figure 1, C and D). IP studies with antibodies for flotillin-1 and the GnRHR provide direct evidence that the GnRHR and flotillin-1 co-IP from membrane raft fractions.
Figure 1. GnRHR and flotillin-1 localize in membrane rafts.

Studies focused on determining whether the GnRHR and flotillin-1 and flotillin-2 occupied overlapping raft domains. A, Fixed histological section from normal mouse anterior pituitary stained with antibodies for flotillin-1 or flotillin-2 (green) and the β-subunit to LH (red) used to identify the gonadotrope population. Magnification bar (25 μm) is shown in white. B, αT3–1 cells transfected with GnRHR-GFP fusion protein followed by staining for endogenous flotillin-1, a marker for raft domains at the plasma membrane. Localization of the 2 fluorophores was quantified using ImageJ's colocalization threshold plugin to calculate the Manders (M) and Pearsons coefficients (R). Magnification bars (5 μm) are shown in white. C and D, IP of the GnRHR and flotillin-1 (C and D, respectively) show reciprocal enrichment for flotillin-1 and the GnRHR in IPs from membrane rafts. All samples were precleared using preimmune serum. IP of beads alone was used as a negative control, whereas addition of 5% input from the original samples was used as a size marker for the IPs. IPs using a nonspecific antibody (GSK1) were also used as a negative control for flotillin-1 (data not shown). E, Venn diagram depicting the number of unique peptides identified in the whole raft fraction (floating fraction), the GnRHR (IP GnRHR), and flotillin-1 (IP FLOT 1) IPs using mass spectrometry. Individual numbers within each circle reflect the number of peptides detected. For example, 2032 peptides were detected within the floating fraction, only a portion of which overlap with the flotillin-1 and or GnRHR IP. The black arrow identifies the 129 peptides shared from all 3 mass spectrometry approaches.
The GnRHR- and flotillin-1-associated raft proteome
To determine the identity of peptides within the GnRHR/flotillin-1-associated raft compartment, we sought to again use IP approaches from isolated raft fractions along with mass spectrometry (NanoLC MS/MS) to define the raft proteome specifically associated with the GnRHR and flotillin-1. Three distinct protein preparations from αT3–1 cells were analyzed by NanoLC MS/MS: 1) the entire raft fraction marked by the presence of flotillin-1 and the GnRHR within low buoyant density fractions from a discontinuous sucrose gradient; 2) peptides from the flotillin-1 IP from raft fractions analyzed in 1) above; and 3) peptides from the GnRHR IP from raft fractions analyzed in 1) above (Figure 1E). Protein samples were resolved by SDS-PAGE, gels were stained, subjected to in-gel trypsin cleavage, eluted, and analyzed by NanoLC MS/MS. These studies identified 2032 peptides present in the whole raft fraction; 347 peptides present in the flotillin-1 IP, and 243 peptides in the GnRHR IP. Analysis of the peptide composition of each group provided data that 129 peptides were common to all 3 groupings (Figure 1E) and became the focus of our studies.
The complete listing of the 129 shared peptides is presented in Supplemental Table 1. Unexpectedly, a number of the peptides present in this analysis were identified as part of the F0F1 ATP synthase complex and the associated electron transport chain normally found within mitochondrial membranes. Tables 1 and 2 report subsets of peptides linked to the ATP synthase complex and the electron transport chain, respectively. To confirm the presence of the ATP synthase complex within the membrane raft, raft fractions from αT3–1 cells were assayed for the α (ATP5A)- and β (ATP5B)-subunits of the F1 catalytic domain. Both of these catalytic subunits were detectable within low buoyant density fractions from sucrose density gradients isolated from αT3–1 cells (Figure 2A) and whole mouse pituitary (Figure 2B). Flotillin-1 and flotillin-2 were used to mark the raft fractions. Studies in whole mouse pituitary provide important evidence that identification of ATP synthase within membrane rafts was not a sole characteristic of the clonal αT3–1 gonadotrope cell model and suggest that this cell line was an appropriate model system for further study. Interestingly, ATP5A and ATP5B were exclusive and constitutive occupants in these raft fractions consistent with the GnRHR and flotillin-1 (17, 18, 40). To validate these findings, GnRHR IPs from raft fractions were repeated in αT3–1 cells and specifically assayed for the presence of ATP5A and ATP5B (Figure 2C). Both of these ATP synthase catalytic subunits were present in GnRHR IPs from raft fractions. Flotillin-1 was used as a positive control for these experiments.
Table 1.
Peptides from F0/F1 ATP Synthase Complex Associated With GnRH Receptor and Flotillin-1 IPs From αT3–1 Cells
| Accession Number | Protein Identity | Mascot Score | Mass (Da) | % Sequence Coverage | pI | emPAI | % Molar |
|---|---|---|---|---|---|---|---|
| gi 31980648 | ATP synthase, H+ transporting mitochondrial F1 complex, β-subunit | 1676 | 56265 | 52.7 | 5.19 | 2.91 | 1.93 |
| gi 6680748 | ATP synthase, H+ transporting, mitochondrial F1 complex, α-subunit, isoform 1 | 505 | 59830 | 20.1 | 9.22 | 0.62 | 0.41 |
| gi 83715998 | ATP synthase, H+ transporting, mitochondrial F1F0 complex, subunit e | 249 | 8230 | 50.7 | 9.34 | 1.91 | 1.27 |
| gi 21536220 | ATP synthase, H+ transporting, mitochondrial F1 complex, δ-subunit precursor | 229 | 17547 | 13.7 | 5.03 | 0.42 | 0.28 |
| gi 20070412 | ATP synthase, H+ transporting, mitochondrial F1 complex, O subunit | 181 | 23406 | 29.6 | 10 | 1.23 | 0.82 |
| gi 11602916 | ATP synthase, H+ transporting, mitochondrial F1 complex, γ-subunit | 109 | 32945 | 20.5 | 9.06 | 0.33 | 0.22 |
| gi 78214312 | ATP synthase, H+ transporting, mitochondrial F0 complex, subunit b, isoform 1 | 56 | 29044 | 7 | 9.11 | 0.24 | 0.16 |
| gi 13385484 | ATP synthase, H+ transporting, mitochondrial F1 complex, ϵ-subunit | 39 | 5891 | 30.8 | 10.01 | 0.61 | 0.41 |
| gi 21313679 | ATP synthase, H+ transporting, mitochondrial F0 complex, subunit d | 38 | 18795 | 8.7 | 5.52 | 0.39 | 0.26 |
Table 2.
Peptides From Electron Transport Chain Complex Associated With GnRH Receptor and Flotillin-1 IPs From αT3–1 Cells
| Accession Number | Protein Identity | Mascot Score | Mass (Da) | % Sequence Coverage | pI | emPAI | % Molar |
|---|---|---|---|---|---|---|---|
| gi 112181182 | Cytochrome c oxidase, subunit Va | 311 | 16319 | 27.4 | 6.08 | 1.57 | 1.04 |
| gi 54607098 | Succinate dehydrogenase Fp subunit | 254 | 73623 | 3.6 | 7.06 | 0.09 | 0.06 |
| gi 112807195 | Cytochrome c oxidase, subunit Vb | 96 | 14123 | 17.8 | 8.34 | 0.24 | 0.16 |
| gi 21312950 | NADH dehydrogenase (ubiquinone) Fe-S protein 7 | 85 | 24952 | 6.2 | 9.94 | 0.13 | 0.09 |
| gi 33563266 | NADH dehydrogenase (ubiquinone) 1α subcomplex, 4 | 61 | 9321 | 12.2 | 9.52 | 0.37 | 0.25 |
| gi 16716343 | Cytochrome c oxidase, subunit VIc | 46 | 8464 | 9.2 | 10.13 | 0.42 | 0.28 |
| gi 21539585 | Ubiquinol-cytochrome c reductase, complex III subunit VII | 37 | 9762 | 11 | 10.26 | 0.36 | 0.24 |
Figure 2. The catalytic subunits of the F0F1 ATP synthase complex colocalize with flotillins in αT3–1 cells and normal mouse pituitary.

Validation studies focused on determining whether mass spectrometry data were consistent with enrichment of identified peptides in raft fractions from sucrose gradient fractionation. A and B, Sucrose density gradient fractions (1–10) from αT3–1 cells (A) and normal mouse pituitary (B) analyzed by Western blot analyses for ATP5A, ATP5B, and flotillin-1 or flotillin-2. The low and high buoyant density fractions are marked. C, GnRHR IP from αT3–1 cell raft fractions followed by Western blotting (IB) for flotillin-1, ATP5A, and ATP5B.
The ATP synthase catalytic surface within the plasma membrane is oriented toward the extracellular space
We next sought to determine the orientation of the ATP synthase complex within the raft compartment. The F0F1 ATP synthase is a large multiprotein complex reflecting a large globular catalytic domain (3 ATP5A and 3 ATP5B subunits) associated with ancillary proteins linking the catalytic domain to the membrane-associated F0 domain. The so called “c subunit” is a complex of ten subunits anchoring the synthase within the plasma membrane. To determine the specific orientation of the globular catalytic F1 domain, 2 distinct approaches were taken to provide evidence that the presence of the ATP synthase complex was not an artifact of mitochondrial contamination from αT3–1 cells. First, we assessed ATP5A immunoreactivity using fluorescence activated cell sorting (FACS) to determine whether ATP5A immunoreactivity could be detected in nonpermeablized, live αT3–1 cells (Figure 3, A and B). These FACS studies demonstrated a population of αT3–1 cells staining positively for ATP5A at the plasma membrane. Second, we used cell surface biotinylation to determine whether components of the F0F1 ATP synthase complex and the electron transport chain were present on the cell surface of αT3–1 cells (Figure 4). We have previously validated this approach in αT3–1 cells (18). Initial studies examined the distribution of proteins biotinylated within the raft domain compared with nonraft plasma membrane (Figure 4A). αT3–1 cells were cell surface biotinylated and subjected to raft fraction isolation on discontinuous sucrose gradients. In low (raft) and high buoyant density fractions, several proteins were biotinylated endogenously (see mock lanes Figure 4A) and these varied in terms of magnitude of staining of individual bands and in molecular size with the density of the fraction. As anticipated, cell surface biotinylation increased the number of proteins detected by this method in both low and high density fractions; the arrows in Figure 4A depict differences in biotinylated bands reflecting the specific partitioning of proteins to the raft and nonraft fractions of these preparation. As positive controls for the cell surface biotinylation, initial studies identified the GnRHR and the purinergic receptor P2X2 as cell surface proteins in αT3–1 cells (Figure 4B). Using cell surface biotinylated protein isolated from these studies, immunoblots revealed enrichment of ATP5A (Figure 4C) and ATP5B (data not shown) as cell surface proteins. Importantly, cells for which the cell surfaces were biotinylated and then had mitochondria purified from the same did not demonstrate such biotinylation, suggesting that this approach was specific to the plasma membrane and not from mitochondrial leakage. Further, it would seem highly unlikely that, if mitochondria were present in the extracellular space, mitochondrial F0F1 ATP synthase complex proteins would become biotinylated based upon their localization to the inner mitochondrial leaflet. As a positive control, purified mitochondrial lysates were assayed for these peptides which were found to be present, just not biotinylated (Figure 4C). Further, ERK2, a key component of the raft complex containing the GnRHR (18), was not cell surface biotinylated nor was it present within mitochondria, again suggesting this approach was specific to cell surface proteins. The F0 subunit ATP6 and 2 proteins from the electron transport chain (nicotinamide adenine dinucleotide (NADH) dehydrogenase and succinate dehydrogenase) were also specifically detected at the cell surface of αT3–1 cells. These biochemical studies provide strong support for the conclusion that the ATP synthase and electron transport chain complexes are present at the cell surface and that the catalytic activity of the ATP synthase is facing the extracellular space. Consistent with the inner leaflet of the mitochondria, there is a good likelihood that 2 complexes are functionally linked; however, our data identifying these complexes at the plasma membrane do not provide direct evidence of a physical or functional association. These data do support the conclusion that detection of these proteins by this method was not an artifact of mitochondrial contamination from these cells.
Figure 3. ATP5A immunoreactivity at the cell surface of αT3–1 cells.

Studies were carried out to determine the orientation of the F0F1 ATP synthase catalytic domain at the plasma membrane. Live αT3–1 cells were stained with fluorescein isothiocynate-conjugated antibody for ATP5A then subjected to flow cytometry. The left panel depicts unstained cells and the right panel depicts cells stained for ATP5A on the cell surface. FACS gates were set based upon data from the unstained αT3–1 cells.
Figure 4. Cell surface biotinylation studies and orientation of the F0F1 ATP synthase catalytic domain.

To determine the cell surface orientation of the catalytic subunits of the F0F1 ATP synthase complex in αT3–1 cells, cell surface biotinylation studies were carried out and analyzed by Western blot analyses. A, Preliminary studies examined global cell surface biotinylation (CSB) in low and high density membrane fractions. The white and gray arrows depict specific enrichment of biotinylated proteins in low and high density membrane fractions. Molecular size standards (MW) are shown at the left of the panel. B, The GnRHR and the purinergic P2X2 receptor were identified at the plasma membrane in cell surface biotinylated cells but not in mock biotinylated cells as positive controls for this study (see Materials and Methods). C, CSB was carried in aT3–1 cells followed by mitochondrial extraction. Mock and CSB fractions were then analyzed by Western blot analysis for ATP5A (from the F1 complex), ATP6 (from the F0 complex), NADH dehydrogenase and succinate dehydrogenase (both from the electron transport chain or ETC), and ERK2 as a negative control. M, mock biotinylated; CSB, biotinylated samples; mito, enriched mitochondria.
αT3–1 and normal pituitary cells synthesize ATP in the extracellular space
The observation that the F0F1 ATP synthase and the GnRHR raft-localize at the plasma membrane predicted that pituitary gonadotropes would indeed be capable of synthesizing extracellular ATP if provided the appropriate substrates. To test this hypothesis, αT3–1 cells were placed into culture and administered either vehicle control or ADP in the absence or presence of the GnRH agonist buserelin (GnRHa) (Figure 5, A and B). After a 2-hour time course, media were assayed for steady-state extracellular ATP levels, whereas cell lysates were assayed under identical conditions for potential changes in intracellular ATP levels. In these studies, culture media alone contained undetectable levels of ATP (data not shown). After 5–30 minutes in the presence of ADP, αT3–1 cells were capable of synthesizing ATP in the extracellular media (Supplemental Figure 1 and Figure 5A). Steady-state ATP levels diminished over time and were unaffected by coadministration of GnRHa. Importantly, intracellular ATP concentrations remained relatively unchanged during the time course study (Supplemental Figure 1 and Figure 5B). This experimental outcome was also confirmed using normal mouse pituitaries placed in an explant culture system (Figure 5, C and D).
Figure 5. αT3–1 cells and normal pituitary synthesize extracellular ATP independent of GnRH treatment.

Studies examined the hypothesis that, if presented with the appropriate substrate (ADP; 10μM), pituitary gonadotropes would synthesize extracellular ATP. A, Extracellular ATP levels increased with the addition of ADP (open bars; P < .05) in cell culture media containing normal levels of Pi in a time-dependent manner in αT3–1 cells. This effect occurred independent of buserelin (GnRH agonist) coadministration (black bars). Cell lysates from these studies were assayed for intracellular ATP levels which did not change with ADP or buserelin treatment (B). Whole-mouse pituitaries were established in explant culture overnight, washed, and then treated with ADP in the presence or absence of Pi in the media and extracellular (C) and intracellular (D) ATP levels were assayed after 1 hour of treatment. Addition of ADP to the culture media resulted in an increase (P < .05) in extracellular ATP levels that was blunted when media were deficient in Pi. Intracellular ATP levels did not change during the course of this study. Differing letters depicted over bars within a study designate statistical significance (P < .05).
If true, these observations predict that the ATP synthase activity at the plasma membrane could be manipulated pharmacologically. In mitochondria, IF1 has been shown to be an endogenous inhibitor of F0F1 ATP synthase activity, particularly under conditions of mitochondrial stress (41–45). For example, under conditions such as low oxygen tension, IF1 appears to be up-regulated and interacts with the F1 catalytic head (ATP5A and ATP5B) preventing ATP hydrolysis rather than ATP synthesis. Our studies made use of recombinant IF1 in an effort to blunt extracellular ATP production in αT3–1 cells, speculating that like the intracellular role for IF1, extracellular IF1 would interact with and blunt the activity of the F1 catalytic domain. IF1 blunted steady-state ATP levels in this in vitro system in a dose dependent manner (Figure 6A). A second prediction was that limiting extracellular inorganic phosphate (Pi) availability in the culture media would also result in a blunting of ATP synthase activity. Culturing αT3–1 cells (Figure 6B) or pituitary explants (Figure 5, C and D) in the presence of low extracellular Pi markedly blunted the ability of pituitary cells to synthesize ATP when presented with ADP as a substrate. Importantly, IF1 and low extracellular Pi levels did not appreciably alter intracellular ATP levels in these systems (data not shown), suggesting that the actions of these pharmacological interventions were specific to ATP synthase activity at the cell surface.
Figure 6. IF1 and low extracellular Pi inhibit extracellular ATP synthesis in αT3–1 cells.

Studies sought to regulate F0F1 ATP synthase activity at the cell surface in αT3–1 cells. In A, αT3–1 cells were treated with increasing doses of IF1 (0.1 and 1.0 μg/mL) and vehicle (open bars) or ADP (10μM; black bars) for a 1-hour period. Extracellular ATP levels were then assayed. IF1 blunted (P < .05) extracellular ATP synthesis in a dose-dependent manner. In B, similar studies were carried out examining the effects of reduced Pi in the absence or presence of ADP in the culture media. Media deficient in Pi could not support (P < .05) extracellular ATP synthesis in αT3–1 cells compared with media replete with Pi. Differing letters depicted over bars within a study designate statistical significance (P < .05).
Chronic GnRH treatment reduces ATP synthase activity at the cell surface in αT3–1 cells
Previously, we demonstrated that (acute) coadministration of GnRHa with ADP did not appreciably affect the ability of αT3–1 cells to synthesize extracellular ATP (Figure 5A). The GnRHR is subject to desensitization with chronic GnRH stimulation, but the kinetics of loss of GnRHR from the plasma membrane with chronic GnRH stimulation of the receptor is relatively slow (∼40% GnRHR internalization by 60 min of stimulation) (46). We reasoned that if at least a portion of the GnRHR population shared a raft compartmentalization with the ATP synthase complex, chronic GnRHa administration might result in a loss of ATP synthetic capacity at the cell surface. To examine this hypothesis, αT3–1 cells were pretreated with GnRHa for 0, 2, or 4 hours, then ADP was added to trigger ATP synthesis (Figure 7, A and B). Chronic GnRHa stimulation diminished the ability of αT3–1 cells to synthesize extracellular ATP in a time-dependent manner. Intracellular ATP concentrations from these treated cells remained unaffected by chronic GnRHa administration. FACS analyses substantiated our speculation that chronic GnRHa treatment would reduce detectable levels of the ATP5A subunit at the cell surface using flow cytometry (Figure 7C). These FACS studies revealed a loss in the proportion of cells with ATP5A immunoreactivity at the cell surface that occurred in a timeframe consistent with GnRHR internalization kinetics (46). An alternative possibility is that chronic GnRH stimulation serves to desensitize ATP synthase activity independent of receptor/raft internalization. Future studies will focus on these possibilities.
Figure 7. Chronic GnRHa treatment of αT3–1 cells disrupts F0F1 ATP synthase activity at the cell surface.

Acute GnRHa treatment did not affect ATP synthase activity in αT3–1 cells (Figure 5). These studies examined the effect of chronic GnRHa treatment (2 or 4 h) before administration of ADP (10μM) to promote extracellular ATP synthesis. Chronic GnRHa treatment reduced extracellular (A) but not intracellular (B) ATP synthesis 1 and 2 hours after ADP administration compared with cells not exposed to chronic GnRHa treatment (P < .05). In C, the proportion of ATP5A-expressing cells (ATP5A+) from FACS analyses was reduced (P < .05) in cells pretreated with buserelin (GnRHa) for 2 hours before flow cytometry. Differing letters depicted over bars within a study designate statistical significance (P < .05).
Inhibition of ecto-nucleoside triphosphate diphosphohydrolase (NTPDase) activity enhances extracellular ATP levels in αT3–1 cells
Thus far, we have employed pharmacological means to blunt or down-regulate ATP synthase activity at the plasma membrane. We considered the possibility that NTPDases could potentially influence the levels of ATP in the extracellular space, as shown previously for the gonadotrope (47, 48). Ecto-NTPDases are membrane associated enzymes that hydrolyze ATP to ADP in the extracellular space exemplified by Cluster of Differentiation (CD)39 (48). In the αT3–1 cell model system, CD39 is enriched in the low buoyant density raft fractions along with ATP5A and flotillin-1 (Figure 8A). Pharmacological disruption of ecto-NTPDase activity (including CD39) can be accomplished using ARL 67156, a relatively nonspecific ecto-NTPDase inhibitor (49–52). The latter is specific for NTPDase 1 (CD39), NTPDase 3, and nucleotide pyrophosphatase/phosphodiesterase 1 (NPP1) and had lesser effects on other members of this family of enzymes (52). We pretreated αT3–1 cells with ARL 67156 (100μM) and then applied ADP to promote ATP synthesis, which augmented extracellular ATP levels in a time-dependent manner (Figure 8B). These pharmacological studies suggest that extracellular ATP metabolism (synthesis and degradation) is an important characteristic of gonadotropes and that ecto-NTPDases (represented by CD39) are likely to play an important role in this.
Figure 8. Inhibition of ecto-NTPDases results in increased extracellular ATP accumulation in αT3–1 cells.
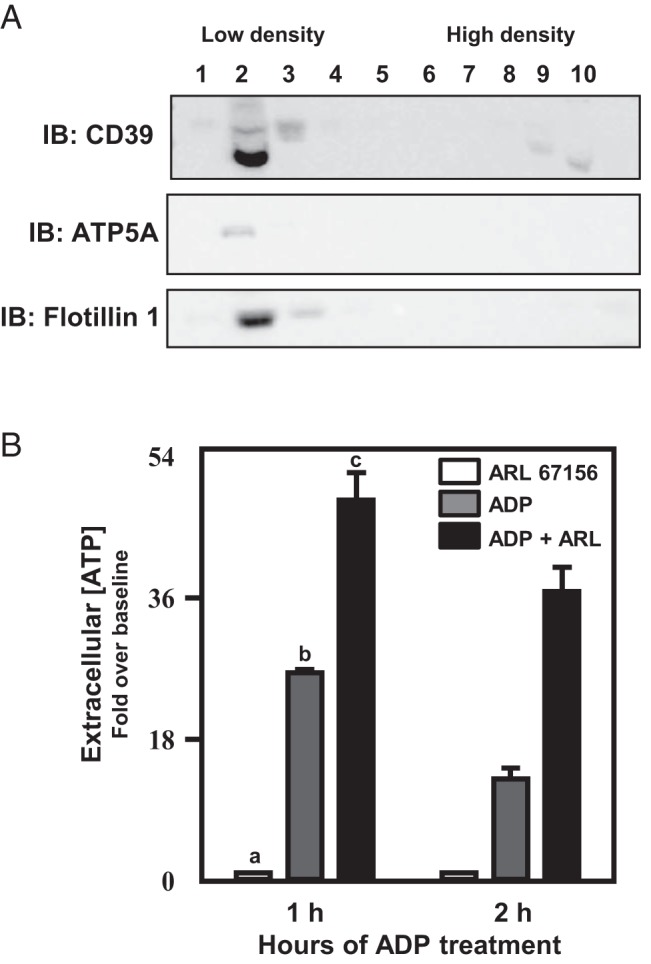
ATP degradation to ADP can occur via extracellular NTPDases such as CD39. In A, Western blot analysis reveals CD39 in the low buoyant density raft fractions coincident with ATP5A and flotillin-1 in αT3–1 cells. Pretreatment of αT3–1 cells with ARL 67156 (ARL; 100μM), an ecto-NTPDase inhibitor, increased extracellular ATP accumulation in the media after 1 and 2 hours of ADP (10μM) administration (B). Differing letters depicted over bars within a study designate statistical significance (P < .05).
The median eminence/hypophyseal portal vasculature is a source of extracellular ATP within the anterior pituitary
Given that F0F1 ATP synthase and ecto-NTPDases metabolic activity at the cell surface of the gonadotrope may only be physiologically relevant with an endogenous source of ATP/ADP, we examined the possibility that ATP is released at the level of the median eminence to provide substrate for these membrane-associated enzymes in the extracellular space within the anterior pituitary. Hypophyseal portal blood collection is a well validated technique for sheep (37). Accordingly, we used this technique in ovx ewes. Baseline plasma samples were collected followed by administration of a bolus dose of kisspeptin (50 μg) to determine whether kisspeptin-stimulated release of GnRH and LH would occur coincident with release of ATP into the portal vasculature. Plasma samples were collected at 10-minute intervals for a period of 1 hour after kisspeptin administration. Figure 9 depicts release of GnRH (portal vasculature), LH (systemic vasculature), and ATP (portal vasculature) from ovx ewes (n = 3) during the study. Kisspeptin administration increased secretion of GnRH from the median eminence and systemic LH concentrations as has been previously shown (53). Coincident with kisspeptin-induced GnRH secretion, ATP concentrations were increased within the portal vasculature, suggesting that the hypothalamus/median eminence may be a key source of extracellular ATP within the pituitary gland.
Figure 9. ATP is released from the median eminence after kisspeptin stimulation in ovx female sheep.
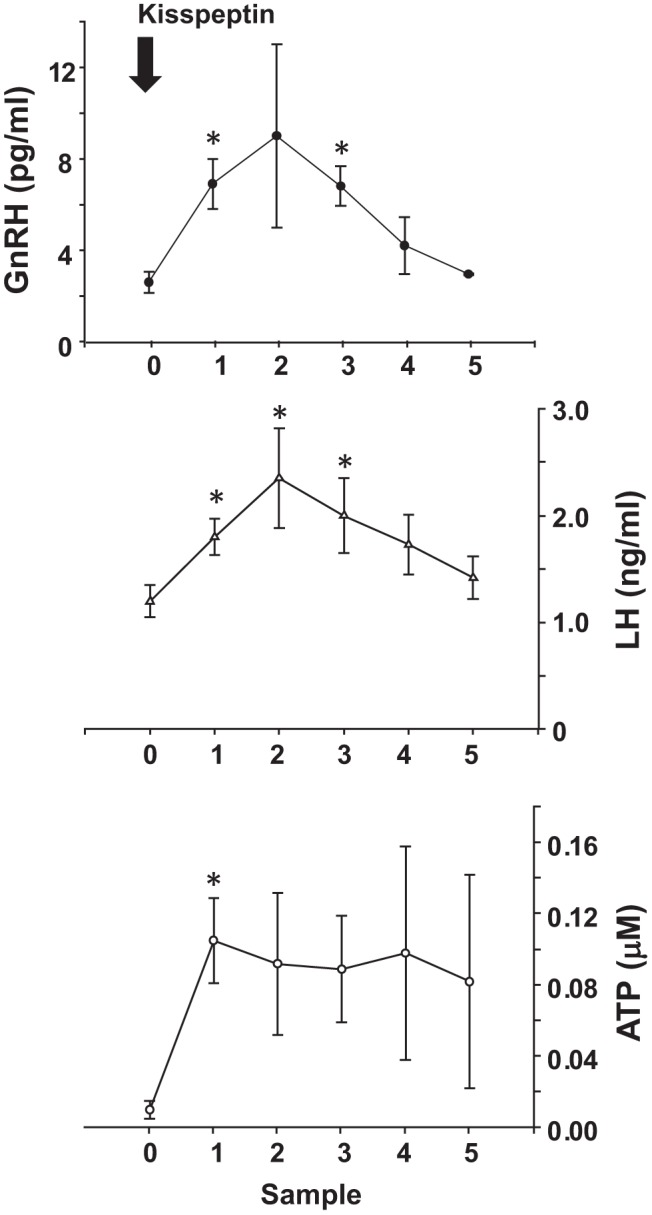
To assess possible sources of ATP/ADP within the extracellular space of the anterior pituitary, adult ovx ewes (n = 3) were outfitted with hypophyseal portal and jugular cannulas. Baseline samples were obtained followed by administration of a bolus dose of kisspeptin to induce GnRH release from the median eminence. Portal plasma samples were then collected at 10-minute intervals for 1 hour and were assayed for GnRH and ATP concentrations. Serial sampling of jugular plasma was assayed for LH concentration; *, data points that differ from the baseline sample (P < .05).
Extracellular ATP metabolism modulates GnRH-induced secretion of LH
Careful studies thus far provide a novel characterization of the enrichment and function of the F0F1 ATP synthase complex at the plasma membrane associated with membrane rafts. We next sought to more fully understand the possible importance of these findings in terms of GnRH action. Here, we investigated the possibility that extracellular ATP levels may augment (directly or indirectly) GnRH-stimulated secretion of LH using the relative simple approach to withholding Pi acutely from the culture media effectively inhibiting the ability of ADP to be converted to ATP by the F0F1 ATP synthase complex at the plasma membrane. Pituitary cells from ovx adult female mice were enzymatically dispersed and seeded into culture dishes. The following day, cells were carefully washed and a subset of cells was placed into complete media and a second cohort was placed into media deficient in Pi. After an equilibration period, cells were treated with vehicle or ADP (due to the low cell density of these primary cultures, changes in ATP levels were not detectable); then saline or GnRH was applied, and the cell media were collected 4 hours later to assess LH secretion/accumulation in the media. After collection of the media, cells were lysed and protein content was determined for use as standardization for the LH data. In complete media, GnRH induced a robust secretion of LH and this response was augmented by addition of ADP to drive extracellular ATP synthesis; culturing cells in media deficient in Pi reversed this effect (Figure 10). Similar data were obtained using primary pituitary cells from adult male mice (data not shown).
Figure 10. Extracellular ATP modulates GnRH-induced LH secretion in mouse pituitary cells in primary culture.
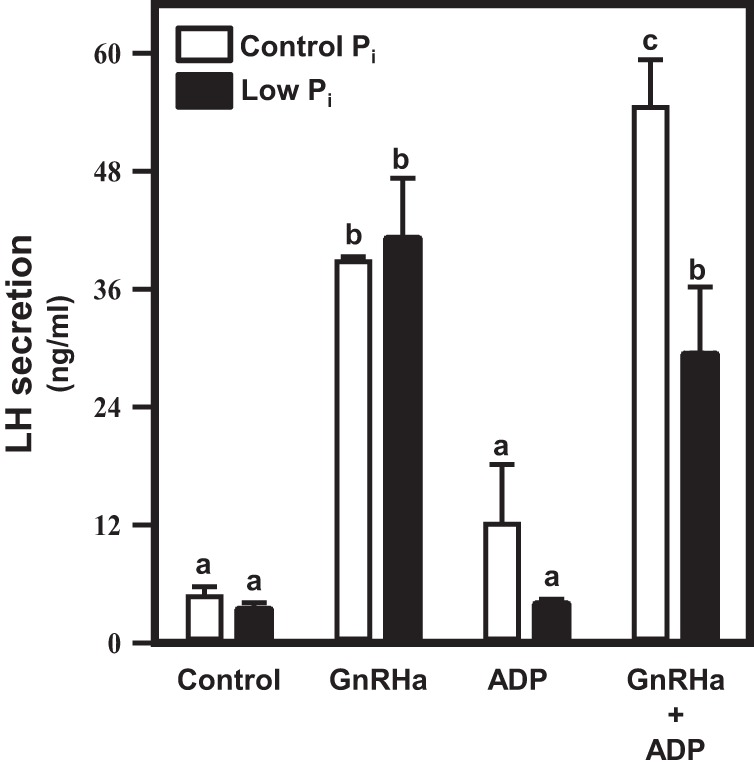
Pituitaries from ovx adult female mice were dispersed into primary culture and allowed to stabilize overnight in serum-containing media. Primary cultures were washed with either DMEM (control) or DMEM without Pi (low Pi). Cells were then treated with vehicle or ADP (10μM) 30 minutes before addition of saline or GnRHa for a period of 4 hours. Media were collected and assayed for LH by ELISA, and cells were lysed and assayed for total protein by Bradford assay to standardize LH secretion. GnRHa induced a marked increase in LH secretion, which was augmented by the addition of ADP (P < .05). Culturing cells in DMEM without Pi reversed this effect.
Discussion
The present studies take advantage of the opportunity to isolate and enrich membrane raft domains and to use IP approaches to reduce protein complexity and study raft-associated peptides using NanoLC MS/MS. Analyses of total raft fractions provide a glimpse of the potential complexity of this membrane compartment, whereas the use of IPs of flotillin-1 and the GnRHR result in more manageable and specific cohorts of peptides associated with the raft domain. A complete listing of the 129 peptides overlapping from each of the groups is presented in Supplemental Table 1. Predictably, a large complement of peptides in this analysis were related to the cytoskeleton in αT3–1 cells including actin, tubulin, actin-related proteins (ARPs) (ARP2/3) and cortactin consistent with recent work from our lab examining the role of ARP2/3 and cortactin in the regulation of GnRH-induced actin reorganization. In those experiments, GnRH-induced cortactin phosphorylation and activation of the actin cytoskeleton was shown to potentially modulate distribution of secretory vesicles and activity of the secretory apparatus (54). We also identified 3 members of the catenin family (Cadherin-associated protein a1 or α-catenin, cadherin-associated protein b1 or β-catenin, and junctional plakoglobin or γ-catenin) (see Supplemental Table 1). Of these, β-catenin has been implicated in regulation of expression of the β-subunit to LH in LβT2 cells, a model for pituitary gonadotropes (55, 56). A precise role for other catenin family members in the GnRH signaling network has yet to be elucidated. The low molecular weight GTP-binding protein Cdc42 was also detected in GnRHR-containing rafts. We have previously identified Cdc42 as a potential upstream activator of GnRH-induced JNK phosphorylation in αT3–1 cells (3). In turn, JNK activation has been linked to control of GnRHR gene promoter activity (57), again underscoring the potential importance of the GnRH-associated raft compartment in the regulation of broad gonadotrope cell function.
Identification of the F0F1 ATP synthase complex along with elements of the electron transport chain in rafts was unexpected (Tables 1 and 2). Our studies used multiple approaches to examine the plasma membrane association with the ATP synthase complex within the raft domain for several reasons. First, the possibility for mitochondrial contamination within the raft preparations could easily explain why the ATP synthase complex was identified using this approach. This is based upon the assumption that the ATP synthase complex was present in the raft compartment within mitochondrial membranes. Our present studies support the conclusion that the identification of the ATP synthase complex was not simply an artifact of the biochemical preparation. If strictly an artifact, it seems unlikely that the ATP synthase complex would coprecipitate with the GnRHR (Figures 1 and 2), a cell surface receptor not known to be expressed in mitochondrial membranes. FACS analysis of live αT3–1 cells and cell-surface biotinylation studies (Figures 3 and 4) support the notion that the ATP synthase complex resides at the plasma membrane likely within the same raft compartment housing the GnRHR and flotillins. FACS analysis, cell surface biotinylation studies and the ability of live cells (including mouse pituitary explants) to synthesize ATP from ADP provides convincing evidence indicating the catalytic face of the complex (ATP5A and ATP5B) faces outwards into the extracellular space. This ATP metabolism appears to occur in the absence of changes in intracellular ATP concentrations in these studies. Further, the ATP synthase catalytic activity is blunted by the endogenous inhibitor of the synthase, IF1 and with media containing low levels of Pi, a necessary substrate in the conversion of ADP to ATP.
One important caveat to our approach using NanoLC MS/MS to examine raft-associated peptides is the inherent difficulty with peptide detection of membrane-associate proteins. Perhaps the most striking example in the present data is the conspicuous absence of the GnRHR and the F0-associated c subunit of the ATP synthase complex in our peptide cohort derived from the spectrometry data. In regard to the former, the strategy for the GnRHR and flotillin-1 IPs was to use antibody cross-linking to enhance our enrichment of GnRHR-associated peptides. This cross-linking may have resulted in an inability to detect the GnRHR using NanoLC MS/MS approaches. In regard to the c subunits of the F0 complex, these subunits are positioned deep within the membrane and serve a central role in the rotational movement of the F1 domain mediating ATP5A and ATP5B catalytic activity and the conversion of ADP to ATP (58). This intimate association with the membrane may have precluded detection using NanoLC MS/MS based upon difficulties with solubility and separation of proteins localizing within membranes (59). Given these caveats, it is highly likely that our proteomics approach may have underestimated the complexity of the raft compartment containing flotillin-1 and the GnRHR. The current studies do not provide definitive evidence that any of the peptides identified by NanoLC MS/MS physically or directly interact with the GnRHR. Our approach was to use specific IPs to recover the complete raft compartment. At present, we can only speculate that enrichment of the raft-associated peptides along with the GnRHR is most likely a consequence of the raft compartment rather than direct interaction with the receptor. Future studies will focus on confirming specific interactions between the GnRHR and a number of the peptides identified in our studies.
Others have identified the ATP synthase complex at the plasma membrane in endothelial cells and various cell lines (60–64). Further, these studies suggest that the presence of the ATP synthase at the plasma membrane may associated with cancer progression and potentially malignant phenotypes. These latter observations underscore the importance of our finding that the ATP synthase complex is present in the low buoyant density/raft domain in normal mouse pituitary consistent with our observations in the αT3–1 cell line (Figure 5). Our studies raise the possibility a more generalized association between key receptor populations like the GnRHR and the ATP synthase complex in low buoyant density membrane compartments that may contribute to extracellular ATP “tone” and modulation of receptor or cellular activities. For example, acute coadministration of ADP/ATP with GnRH had little effect on extracellular ATP synthesis (Figure 5); conversely, chronic GnRH administration resulted in reduced ATP synthase activity which may reflect important direct (receptor desensitization) or indirect actions of GnRH signaling on the ATP synthase at the plasma membrane.
Given the presence of the GnRHR and the ATP synthase within the raft compartment, it will be interesting to examine mechanisms associated with the driving force(s) of the ATP synthase catalytic activity at the plasma membrane. Currently very little is known regarding the specific role of the electron transport chain and the proton gradient at the plasma membrane that typically provides the electrochemical proton driving force that regulates ATP synthase catalytic activity in mitochondria (58). Further, the present studies only begin to address how substrates for the synthase itself are regulated or derived from the median eminence/hypothalamus. Work from other groups suggests that locally derived extracellular ATP (released via pannexins potentially from adrenocorticotropes) may be a source of extracellular ATP in the pituitary (65–66). This posed the interesting possibility of cross talk between cell lineages within the pituitary gland where ADP/ATP may serve a paracrine function via purinergic receptors. The present studies support the additional possibility that substrate for the ATP synthase and ecto-NTPDase is forthcoming from the brain or median eminence, coincident with GnRH secretion into hypophyseal portal blood. Future studies will examine this possibility further in the context of how gonadal steroids may modulate this corelease of GnRH and ATP from the median eminence to affect gonadotrope cell function.
Our studies support the notion that extracellular ADP/ATP levels may be an important modulator of GnRH-induced LH secretion. The emerging model suggests that well-timed changes in extracellular ATP levels may have critical importance to the modulation of GnRH action based on the presence of the ATP synthase along with the GnRHR in the raft compartment. Further, the presence and activity of ecto-NTPDases (such as CD39) are likely critical in extracellular ATP metabolism and homeostasis underscoring the importance of the ATP synthase activity at the plasma membrane to maintain extracellular ATP levels. Currently, little is known regarding how ecto-NTPDases are regulated at the cell surface in gonadotropes or how these enzymes might change throughout the reproductive cycle or pregnancy. The present studies demonstrate that elevated levels of extracellular ATP modulate or augment the ability of GnRH to productively couple with GnRH-induced LH secretion. In its' simplest form, this could be a direct effect of the added ADP/ATP in this culture system; however, given these primary pituitary cultures are a mixed cell population, additional future studies will be needed to resolve the precise mechanism(s) involved. Regulation of hypothalamic contributions to the extracellular ADP/ATP pool along with GnRH may serve as a critical modulator of the sensitivity of the gonadotrope to GnRH at times when extracellular ATP levels are abundant or limiting.
Acknowledgments
We thank Dr Lynn Dong for ongoing technical support in immunohistochemistry, Dr Sheng Zhang and Mr James McCardle for expertise in mass spectrometry, Dr Donal Skinner for generously providing antibody to the GnRHR; and Dr Pamela Mellon for providing αT3–1 cells.
This work was supported by National Institutes of Health/National Institute of Child Health and Human Development Grants R01 HD34772 and R21 HD082780.
Disclosure Summary: The authors have nothing to disclose.
Funding Statement
This work was supported by National Institutes of Health/National Institute of Child Health and Human Development Grants R01 HD34772 and R21 HD082780.
Footnotes
- ARP
- actin-related protein
- CD39
- Cluster of Differentiation 39
- FACS
- fluorescence activated cell sorting
- GnRHR
- GnRH receptor
- GPCR
- G protein-coupled receptor
- IF1
- inhibitory factor 1
- IP
- immunoprecipitation
- JNK
- c-Jun N-terminal kinase
- MS
- mass spectroscopy
- NADH
- nicotinamide adenine dinucleotide
- NTPDase
- nucleoside triphosphate diphosphohydrolase
- ovx
- ovariectomized
- SDS
- sodium dodecyl sulfate
- TBST
- 10mM Tris-HCl (pH 7.5), 150mM NaCl, and 0.05% Tween 20.
References
- 1. Bliss SP, Navratil AM, Xie J, Roberson MS. GnRH signaling, the gonadotrope and endocrine control of fertility. Front Neuroendocrinol. 2010;31:322–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mulvaney JM, Zhang T, Fewtrell C, Roberson MS. Calcium influx through L-type channels is required for selective activation of extracellular signal-regulated kinase by gonadotropin-releasing hormone. J Biol Chem. 1999;274:29796–29804. [DOI] [PubMed] [Google Scholar]
- 3. Mulvaney JM, Roberson MS. Divergent signaling pathways requiring discrete calcium signals mediate concurrent activation of two mitogen-activated protein kinases by gonadotropin-releasing hormone. J Biol Chem. 2000;275:14182–14189. [DOI] [PubMed] [Google Scholar]
- 4. White BR, Duval DL, Mulvaney JM, Roberson MS, Clay CM. Homologous regulation of the gonadotropin-releasing hormone receptor gene is partially mediated by protein kinase C activation of an activator protein-1 element. Mol Endocrinol. 1999;13:566–577. [DOI] [PubMed] [Google Scholar]
- 5. Zhang T, Wolfe MW, Roberson MS. An early growth response protein (Egr) 1 cis-element is required for gonadotropin-releasing hormone-induced mitogen-activated protein kinase phosphatase 2 gene expression. J Biol Chem. 2001;276:45604–45613. [DOI] [PubMed] [Google Scholar]
- 6. Zhang T, Mulvaney JM, Roberson MS. Activation of mitogen-activated protein kinase phosphatase 2 by gonadotropin-releasing hormone. Mol Cell Endocrinol. 2001;172:79–89. [DOI] [PubMed] [Google Scholar]
- 7. Zhang T, Choy M, Jo M, Roberson MS. Structural organization of the rat mitogen-activated protein kinase phosphatase 2 gene. Gene. 2001;273:71–79. [DOI] [PubMed] [Google Scholar]
- 8. Zhang T, Roberson MS. Role of MAP kinase phosphatases in GnRH-dependent activation of MAP kinases. J Mol Endocrinol. 2006;36:41–50. [DOI] [PubMed] [Google Scholar]
- 9. Reiss N, Llevi LN, Shacham S, Harris D, Seger R, Naor Z. Mechanisms of mitogen-activated protein kinase activation by gonadotropin-releasing hormone in the pituitary αT3–1 cell line: differential role of calcium and protein kinase C. Endocrinology. 1997;138:1673–1682. [DOI] [PubMed] [Google Scholar]
- 10. Levi NL, Hanoch T, Benard O, et al. . Stimulation of Jun N-terminal kinase (JNK) by gonadotropin-releasing hormone in pituitary α T3–1 cell line is mediated by protein kinase C, c-Src, and CDC42. Mol Endocrinol. 1998;12:815–824. [DOI] [PubMed] [Google Scholar]
- 11. Dobkin-Bekman M, Naidich M, Pawson AJ, Millar RP, Seger R, Naor Z. Activation of mitogen-activated protein kinase (MAPK) by GnRH is cell-context dependent. Mol Cell Endocrinol. 2006;252:184–190. [DOI] [PubMed] [Google Scholar]
- 12. Bonfil D, Chuderland D, Kraus S, et al. . Extracellular signal-regulated kinase, Jun N-terminal kinase, p38, and c-Src are involved in gonadotropin-releasing hormone-stimulated activity of the glycoprotein hormone follicle-stimulating hormone β-subunit promoter. Endocrinology. 2004;145:2228–2244. [DOI] [PubMed] [Google Scholar]
- 13. Ben-Menahem D, Shraga-Levine Z, Mellon PL, Naor Z. Mechanism of action of gonadotropin-releasing hormone upon gonadotropin α-subunit mRNA levels in the α T3–1 cell line: role of Ca2+ and protein kinase C. Biochem J. 1995;309:325–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sealfon SC, Millar RP. Functional domains of the gonadotropin-releasing hormone receptor. Cell Mol Neurobiol. 1995;15:25–42. [DOI] [PubMed] [Google Scholar]
- 15. Sealfon SC, Millar RP. The gonadotrophin-releasing hormone receptor: structural determinants and regulatory control. Hum Reprod Update. 1995;1:216–230. [DOI] [PubMed] [Google Scholar]
- 16. McArdle CA, Franklin J, Green L, Hislop JN. The gonadotrophin-releasing hormone receptor: signalling, cycling and desensitisation. Arch Physiol Biochem. 2002;110:113–122. [DOI] [PubMed] [Google Scholar]
- 17. Navratil AM, Bliss SP, Berghorn KA, et al. . Constitutive localization of the gonadotropin-releasing hormone (GnRH) receptor to low density membrane microdomains is necessary for GnRH signaling to ERK. J Biol Chem. 2003;278:31593–31602. [DOI] [PubMed] [Google Scholar]
- 18. Bliss SP, Navratil AM, Breed M, Skinner DC, Clay CM, Roberson MS. Signaling complexes associated with the type I gonadotropin-releasing hormone (GnRH) receptor: colocalization of extracellularly regulated kinase 2 and GnRH receptor within membrane rafts. Mol Endocrinol. 2007;21:538–549. [DOI] [PubMed] [Google Scholar]
- 19. Roess DA, Smith SM. Self-association and raft localization of functional luteinizing hormone receptors. Biol Reprod. 2003;69:1765–1770. [DOI] [PubMed] [Google Scholar]
- 20. Lei Y, Hagen GM, Smith SM, Liu J, Barisas G, Roess DA. Constitutively-active human LH receptors are self-associated and located in rafts. Mol Cell Endocrinol. 2007;260–262:65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Steinberg SF. β(2)-Adrenergic receptor signaling complexes in cardiomyocyte caveolae/lipid rafts. J Mol Cell Cardiol. 2004;37:407–415. [DOI] [PubMed] [Google Scholar]
- 22. Sabourin T, Bastien L, Bachvarov DR, Marceau F. Agonist-induced translocation of the kinin B(1) receptor to caveolae-related rafts. Mol Pharmacol. 2002;61:546–553. [DOI] [PubMed] [Google Scholar]
- 23. Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. [DOI] [PubMed] [Google Scholar]
- 24. Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1:31–39. [DOI] [PubMed] [Google Scholar]
- 25. Owen DM, Magenau A, Williamson D, Gaus K. The lipid raft hypothesis revisited–new insights on raft composition and function from super-resolution fluorescence microscopy. BioEssays. 2012;34:739–747. [DOI] [PubMed] [Google Scholar]
- 26. Kusumi A, Ike H, Nakada C, Murase K, Fujiwara T. Single-molecule tracking of membrane molecules: plasma membrane compartmentalization and dynamic assembly of raft-philic signaling molecules. Semin Immunol. 2005;17:3–21. [DOI] [PubMed] [Google Scholar]
- 27. Chichili GR, Rodgers W. Clustering of membrane raft proteins by the actin cytoskeleton. J Biol Chem. 2007;282:36682–36691. [DOI] [PubMed] [Google Scholar]
- 28. Lillemeier BF, Pfeiffer JR, Surviladze Z, Wilson BS, Davis MM. Plasma membrane-associated proteins are clustered into islands attached to the cytoskeleton. Proc Natl Acad Sci USA. 2006;103:18992–18997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Garner AE, Smith DA, Hooper NM. Visualization of detergent solubilization of membranes: implications for the isolation of rafts. Biophys J. 2008;94:1326–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Galbiati F, Volonte D, Goltz JS, et al. . Identification, sequence and developmental expression of invertebrate flotillins from Drosophila melanogaster. Gene. 1998;210:229–237. [DOI] [PubMed] [Google Scholar]
- 31. Bickel PE, Scherer PE, Schnitzer JE, Oh P, Lisanti MP, Lodish HF. Flotillin and epidermal surface antigen define a new family of caveolae-associated integral membrane proteins. J Biol Chem. 1997;272:13793–13802. [DOI] [PubMed] [Google Scholar]
- 32. Sugawara Y, Nishii H, Takahashi T, et al. . The lipid raft proteins flotillins/reggies interact with Gαq and are involved in Gq-mediated p38 mitogen-activated protein kinase activation through tyrosine kinase. Cell Signal. 2007;19:1301–1308. [DOI] [PubMed] [Google Scholar]
- 33. Windle JJ, Weiner RI, Mellon PL. Cell lines of the pituitary gonadotrope lineage derived by targeted oncogenesis in transgenic mice. Mol Endocrinol. 1990;4:597–603. [DOI] [PubMed] [Google Scholar]
- 34. Horn F, Bilezikjian LM, Perrin MH, et al. . Intracellular responses to gonadotropin-releasing hormone in a clonal cell line of the gonadotrope lineage. Mol Endocrinol. 1991;5:347–355. [DOI] [PubMed] [Google Scholar]
- 35. Asano A, Nelson JL, Zhang S, Travis AJ. Characterization of the proteomes associating with three distinct membrane raft sub-types in murine sperm. Proteomics. 2010;10:3494–3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Berghorn KA, Clark PA, Encarnacion B, et al. . Developmental expression of the homeobox protein Distal-less 3 and its relationship to progesterone production in mouse placenta. J Endocrinol. 2005;186:315–323. [DOI] [PubMed] [Google Scholar]
- 37. Clarke IJ, Brown BW, Tran VV, et al. . Neonatal immunization against gonadotropin-releasing hormone (GnRH) results in diminished GnRH secretion in adulthood. Endocrinology. 1998;139:2007–2014. [DOI] [PubMed] [Google Scholar]
- 38. Jonas HA, Burger HG, Cumming IA, Findlay JK, de Kretser DM. Radioimmunoassay for luteinizing hormone-releasing hormone (LHRH): its application to the measurement of LHRH in ovine and human plasma. Endocrinology. 1975;96:384–393. [DOI] [PubMed] [Google Scholar]
- 39. Wehmeyer L, Du Toit A, Lang DM, Hapgood JP. Lipid raft- and protein kinase C-mediated synergism between glucocorticoid- and gonadotropin-releasing hormone signaling results in decreased cell proliferation. J Biol Chem. 2014;289:10235–10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bliss SP, Navratil AM, Xie J, Miller A, Baccarini M, Roberson MS. ERK signaling, but not c-Raf, is required for gonadotropin-releasing hormone (GnRH)-induced regulation of Nur77 in pituitary gonadotropes. Endocrinology. 2012;153:700–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Campanella M, Seraphim A, Abeti R, Casswell E, Echave P, Duchen MR. IF1, the endogenous regulator of the F(1)F(o) -ATPsynthase, defines mitochondrial volume fraction in HeLa cells by regulating autophagy. Biochim Biophys Acta. 2009;1787:393–401. [DOI] [PubMed] [Google Scholar]
- 42. Cabezón E, Arechaga I, Jonathan P, Butler G, Walker JE. Dimerization of bovine F1-ATPase by binding the inhibitor protein, IF1. J Biol Chem. 2000;275:28353–28355. [DOI] [PubMed] [Google Scholar]
- 43. Tomasetig L, Di Pancrazio F, Harris DA, Mavelli I, Lippe G. Dimerization of F0F1ATP synthase from bovine heart is independent from the binding of the inhibitor protein IF1. Biochim Biophys Acta. 2002;1556:133–141. [DOI] [PubMed] [Google Scholar]
- 44. Cabezón E, Montgomery MG, Leslie AG, Walker JE. The structure of bovine F1-ATPase in complex with its regulatory protein IF1. Nat Struct Biol. 2003;10:744–750. [DOI] [PubMed] [Google Scholar]
- 45. Fujikawa M, Imamura H, Nakamura J, Yoshida M. Assessing actual contribution of IF1, inhibitor of mitochondrial FoF1, to ATP homeostasis, cell growth, mitochondrial morphology, and cell viability. J Biol Chem. 2012;287:18781–18787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hashizume T, Yang WH, Clay CM, Nett TM. Internalization rates of murine and ovine gonadotropin-releasing hormone receptors. Biol Reprod. 2001;64:898–903. [DOI] [PubMed] [Google Scholar]
- 47. He ML, Gonzalez-Iglesias AE, Tomic M, Stojilkovic SS. Release and extracellular metabolism of ATP by ecto-nucleotidase eNTPDase 1–3 in hypothalamic and pituitary cells. Purinergic Signal. 2005;1:135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yegutkin GG, Wieringa B, Robson SC, Jalkanen S. Metabolism of circulating ADP in the bloodstream is mediated via integrated actions of soluble adenylate kinase-1 and NTPDase1/CD39 activities. FASEB J. 2012;26:3875–3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Westfall TD, Kennedy C, Sneddon P. The ecto-ATPase inhibitor ARL 67156 enhances parasympathetic neurotransmission in the guinea-pig urinary bladder. Eur J Pharmacol. 1997;329:169–173. [PubMed] [Google Scholar]
- 50. Ghildyal P, Manchanda R. Effects of cooling and ARL 67156 on synaptic ecto-ATPase activity in guinea pig and mouse vas deferens. Auton Neurosci. 2004;115:28–34. [DOI] [PubMed] [Google Scholar]
- 51. Drakulich DA, Spellmon C, Hexum TD. Effect of the ecto-ATPase inhibitor, ARL 67156, on the bovine chromaffin cell response to ATP. Eur J Pharmacol. 2004;485:137–140. [DOI] [PubMed] [Google Scholar]
- 52. Lévesque SA, Lavoie EG, Lecka J, Bigonnesse F, Sévigny J. Specificity of the ecto-ATPase inhibitor ARL 67156 on human and mouse ectonucleotidases. Br J Pharmacol. 2007;152:141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ezzat A, Pereira A, Clarke IJ. Kisspeptin is a component of the pulse generator for GnRH secretion in female sheep but not the pulse generator. Endocrinology. 2015;156:1828–1837. [DOI] [PubMed] [Google Scholar]
- 54. Navratil AM, Dozier MG, Whitesell JD, Clay CM, Roberson MS. Role of cortactin in dynamic actin remodeling events in gonadotrope cells. Endocrinology. 2014;155:548–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Salisbury TB, Binder AK, Grammer JC, Nilson JH. Maximal activity of the luteinizing hormone β-subunit gene requires β-catenin. Mol Endocrinol. 2007;21:963–971. [DOI] [PubMed] [Google Scholar]
- 56. Salisbury TB, Binder AK, Nilson JH. Welcoming β-catenin to the gonadotropin-releasing hormone transcriptional network in gonadotropes. Mol Endocrinol. 2008;22:1295–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ellsworth BS, White BR, Burns AT, Cherrington BD, Otis AM, Clay CM. c-Jun N-terminal kinase activation of activator protein-1 underlies homologous regulation of the gonadotropin-releasing hormone receptor gene in α T3–1 cells. Endocrinology. 2003;144:839–849. [DOI] [PubMed] [Google Scholar]
- 58. Walker JE. The ATP synthase: the understood, the uncertain and the unknown. Biochem Soc Trans. 2013;41:1–16. [DOI] [PubMed] [Google Scholar]
- 59. Tan S, Tan HT, Chung MC. Membrane proteins and membrane proteomics. Proteomics. 2008;8:3924–3932. [DOI] [PubMed] [Google Scholar]
- 60. Bae TJ, Kim MS, Kim JW, et al. . Lipid raft proteome reveals ATP synthase complex in the cell surface. Proteomics. 2004;4:3536–3548. [DOI] [PubMed] [Google Scholar]
- 61. Kim KB, Lee JW, Lee CS, et al. . Oxidation-reduction respiratory chains and ATP synthase complex are localized in detergent-resistant lipid rafts. Proteomics. 2006;6:2444–2453. [DOI] [PubMed] [Google Scholar]
- 62. Ma Z, Cao M, Liu Y, et al. . Mitochondrial F1Fo-ATP synthase translocates to cell surface in hepatocytes and has high activity in tumor-like acidic and hypoxic environment. Acta Biochim Biophys Sin (Shanghai). 2010;42:530–537. [DOI] [PubMed] [Google Scholar]
- 63. Wang WJ, Ma Z, Liu YW, et al. . A monoclonal antibody (Mc178-Ab) targeted to the ecto-ATP synthase β-subunit-induced cell apoptosis via a mechanism involving the MAPKase and Akt pathways. Clin Exp Med. 2012;12:3–12. [DOI] [PubMed] [Google Scholar]
- 64. Wang WJ, Shi XX, Liu YW, et al. . The mechanism underlying the effects of the cell surface ATP synthase on the regulation of intracellular acidification during acidosis. J Cell Biochem. 2013;114:1695–1703. [DOI] [PubMed] [Google Scholar]
- 65. Li S, Tomi M, Stojilkovic SS. Characterization of novel Pannexin 1 isoforms from rat pituitary cells and their association with ATP-gated P2X channels. Gen Comp Endocrinol. 2011;174:202–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Li S, Bjelobaba I, Yan Z, Kucka M, Tomic M, Stojilkovic SS. Expression and roles of pannexins in ATP release in the pituitary gland. Endocrinology. 2011;152:2342–2352. [DOI] [PMC free article] [PubMed] [Google Scholar]