

Abstract
Glucocorticoid (GC) steroid hormones have well-characterized roles in the regulation of systemic homeostasis, yet less understood is their known role in utero to mature the developing respiratory system in preparation for birth. During late gestation, endogenously produced GCs thin the interstitial tissue of the lung, causing the vasculature and future airspaces to come into close alignment, allowing for efficient gas exchange at birth. More potent synthetic GCs are also used worldwide to reduce the severity of respiratory distress suffered by preterm infants; however, their clinical benefits are somewhat offset by potential detrimental long-term effects on health and development. Here, we review the recent literature studying both global and conditional gene-targeted respiratory mouse models of either GC deficiency or glucocorticoid receptor ablation. Although some discrepancies exist between these transgenic mouse strains, these models have revealed specific roles for GCs in particular tissue compartments of the developing lung and identify the mesenchyme as the critical site for glucocorticoid receptor–mediated lung maturation, particularly for the inhibition of cell proliferation and epithelial cell differentiation. Specific mesenchymal and epithelial cell–expressed gene targets that may potentially mediate the effect of GCs have also been identified in these studies and imply a GC-regulated system of cross talk between compartments during lung development. A better understanding of the specific roles of GCs in specific cell types and compartments of the fetal lung will allow the development of a new generation of selective GC ligands, enabling better therapeutic treatments with fewer side effects for lung immaturity at birth in preterm infants.
Although glucocorticoid (GC) hormones are widely recognized as being important for normal bodily homeostasis, perhaps their first and most crucial function occurs in utero: to mature the developing respiratory system to ensure survival after birth. Lung development is highly complex, involving the coordinated actions of multiple signaling pathways, which are predominantly mediated within cells via specific nuclear transcription factors. Likewise, GC signaling acts primarily via the widely expressed glucocorticoid receptor (GR), which functions as a ligand-bound transcription factor. Once GR signaling is activated in the developing lung by a surge in endogenous GC levels during the later stages of gestation, it induces a series of morphological alterations in the pulmonary architecture, causing the interstitial mesenchymal tissue compartment, in particular, to thin considerably. This in turn brings the underlying vasculature into close proximity with the future alveolar airspaces. Upon the transition from an aqueous to a gaseous environment, which occurs rapidly at birth, the minimal blood-air barrier distance greatly enhances the ability of the lungs to oxygenate the blood and therefore allow survival of the neonate ex utero.
Perhaps the most compelling clinical evidence for the importance of GC-induced lung maturation was the discovery by Liggins and Howie in 1972 (1) showing that prenatal GC therapy reduced both morbidity and mortality in preterm infants at risk for respiratory distress syndrome. Since then, significant efforts have been directed to understand the molecular mechanisms underpinning corticosteroid function during embryonic lung development and, to a greater extent, in human neonatal lung disorders. Despite these efforts, our awareness of the important GC-regulated gene networks used in the lung remains far from complete, and to date there are no known downstream gene targets or pathways that have been definitively shown to mediate the structural and cellular transformation, which enables proper late-stage respiratory maturation and survival at birth.
Transgenic mice have proved to be an excellent resource to study these underlying processes and cellular pathways. Mouse models overexpressing or ablating GC-regulating genetic components of the hypothalamic-pituitary-adrenal (HPA) axis have been generated and used for almost a decade. Phenotypic analysis of these models has proved invaluable in our investigation of how GC signaling accelerates the maturation of the developing lung. In this article, we will primarily review current studies on both germline (total) and selective GR knockout (GR−/−) mice and the insights we have gained in the context of respiratory biology. Where appropriate, we will also discuss lung phenotypes from other mouse models of GC deficiency and how the use of exogenous GCs, particularly synthetically derived compounds such as dexamethasone (Dex), have contributed to our understanding. However, we will treat such studies with a level of caution when comparing them with findings obtained from mouse knockout models, because the molecular and physiological outcomes produced by exogenous GC treatment can vary widely depending on the species, the type of GC ligand used, the number and frequency of doses given, the strength of dose, and the method of delivery. In addition, because this review is focused specifically on GC signaling, the literature describing molecular pathways in respiratory development has been thoroughly discussed in recent reviews (2–4) and will not be analyzed in detail here.
Summary of Search Strategies Used
Relevant publications were obtained by online searches using National Center for Biotechnology Information (NCBI) PubMed and other databases that included the Nuclear Receptor Signaling Atlas (NURSA) on-line database.
Summary and Caveats of Currently Available Transgenic Mice Targeting the GR Gene
Several transgenic alleles have been generated to functionally knock out the GR gene (Nr3c1) in mice. The first described, generated by Cole et al (5), used a disruption strategy whereby a neomycin cassette was targeted to exon 2, interrupting normal GR gene transcription. When bred on an isogenic 129sv or C57BL/6 genetic background, these GR−/− mice showed a severe lung phenotype and 100% mortality at birth. Interestingly, when bred onto a mixed 129sv/C57BL/6 background, 10% of GR−/− mice survive to adulthood with no apparent abnormalities (5). The second allele, generated by Tronche et al (6), is a floxed GR transgene whereby exon 3 is flanked by loxP sites and can therefore be excised by Cre recombinase (6). Although not proven experimentally, the Tronche et al allele, once recombined, produces an out-of-frame GR mRNA species with a presumably nontranslated protein product. To our knowledge, germline GR−/− mice have not been generated from the Tronche et al allele; however, conditional deletions in lung cell layers have been produced (discussed below). The third allele, generated by Brewer et al (7), is also a floxed GR transgene with loxP sites flanking exons 1C and exon 2. When germline GR−/− mice were produced from this allele, the lung phenotype previously seen in the Cole et al GR−/− strain was again observed, and all mice died soon after birth. Perhaps because of concerns over their use of a mixed 129sv × C57BL/6 mouse background (observed in the Cole et al strain as previously discussed), the authors also assessed the degree of GR deficiency but found that GR−/− protein extracts did not bind antibodies directed to either N- or C-terminal regions. Another strain of mice termed GRdim was generated by Reichardt et al (8) to assess the specific requirement of GR monomer dimerization for GR function. GRdim mice bear a missense mutation in the second zinc finger of the GR gene, preventing dimerization of GR monomers and subsequent binding to glucocorticoid receptor elements (GREs), which is believed to be required for GR-induced transactivation. These mice live to adulthood with no apparent lung defect, suggesting that GR-DNA binding is not essential for lung development. However, because GRdim mice express wild-type mRNA levels of Pnmt, a known GR-inducible gene with a well-characterized GRE (9), it is uncertain whether GRdim mice are completely devoid of GR transactivation ability.
Other Genetic Mouse Models With GC Deficiency
Specific non-GR mouse knockouts of factors that regulate corticosterone synthesis upstream of GR function have also contributed to our understanding of GC-driven fetal lung maturation. First, a knockout of the hydroxysteroid 11β-dehydrogenase 1 (HSD11B1), which converts inactive to active corticosteroids, has been generated (10). Secondly, knockout models have been produced for CRH (11), the CRH receptor (CRHR1) (12), and the ACTH receptor (MC2R) (13), all of which regulate corticosterone levels via the HPA axis. Finally, although not a regulator of GC synthesis, the mineralocorticoid receptor (MR) can be activated by GC ligands, and, therefore, it is necessary to examine the lung phenotype of MR−/− mice (14). In this review, we only refer to studies using these knockouts, which further explore the lung phenotype.
Overview of Mammalian Lung Development
To properly understand the nature of both temporal and functional involvement of GC signaling during lung development, it is necessary to consider the stages of mammalian lung organogenesis. Morphological lung development begins with the appearance of 2 primordial buds during the embryonic phase (embryonic day [E] 9.5-E12.5 or weeks 4–7 in humans), which emerge ventrally from the foregut endoderm into the mesodermally derived mesenchyme (Figure 1A). Separation of the tracheal tube and dorsal foregut is complete by approximately E11.5, by which time airway branching has begun. The pseudoglandular stage (E10.5–E16.5 or weeks 6–16 in humans) is largely characterized by extensive dichotomous branching of the newly formed endodermal buds to form the branched respiratory tree (Figure 1B). During the branching process, the undifferentiated lung epithelium also begins to show morphological characteristics and distinct marker expression of a range of specialized cell types including secretory (Clara) cells, ciliated cells, mucosal cells, and neuroendocrine (NE) cells (4) (Figure 1C). The relative proportions and distributions of these cells vary significantly, depending on their location along the proximal-distal axis of the developing epithelium and also across different species.
Figure 1. An overview of the stages of lung development.

A, The embryonic phase (E9.5–E12.5) begins with the appearance of 2 primordial buds, which evaginate ventrally from the foregut endoderm. Separation of the trachea and esophagus is complete by ∼E11.5, by which time airway branching has begun. B, The pseudoglandular stage is characterized by extensive dichotomous branching of the newly formed buds to form the respiratory tree. C, During the pseudoglandular phase, the epithelium begins to differentiate into the various cell lineages of the conducting airway including mucosal, neuroendocrine, ciliated, and secretory cells. Differentiation of the epithelium progresses in a proximal to distal manner, with proximal progenitors losing expression of the distal multipotent cell marker Sox9, but gaining expression of the proximal marker Sox2 as well as specific markers of conducting airway lineages. D, During the canalicular /saccular phase (E16.5–PN5), the distal lung epithelium expands to form “sacculi” while the mesenchyme thins. Differentiation of distal epithelial progenitors leads to formation of the BADJ (the boundary between the conducting airway and the alveolar epithelium), as well as type I and type II AECs. E, In the alveolar phase (PN5–PN30), elastin fiber deposition by myofibroblasts drives the formation of secondary septae, which subdivide sacculi into alveoli (i). Secondary septae initially include dual capillary layers (ii), which finally fuse via microvascularization to form a single capillary layer (iii). A, anterior; D, dorsal; L, left; P, posterior; R, right; V, ventral. Epithelial markers used the following: Sox2, SRY-box containing gene 2; Scgb1a1, secretoglobin, family 1A, member 1 (uteroglobin); FoxJ1, forkhead box J1; Spdef, SAM pointed domain containing ets transcription factor; CGRP, calcitonin/calcitonin-related polypeptide, α; Sox9, SRY-box containing gene 9; Aqp5, aquaporin 5; Sftpb, surfactant associated protein B; Sftpc, surfactant-associated protein C; αSMA, actin, α 2, smooth muscle, aorta; CD31, platelet/endothelial cell adhesion molecule 1.
During the canalicular (E16.5-E17.5 or weeks 16–24 in humans) and saccular (E17.5–postnatal [P]5 or week 24 until birth in humans) stages, extensive remodeling of the distal lung gives rise to the gas exchange regions and the first appearance of an expanded airway lumen, whereas the mesenchymal tissue content begins to decrease (Figure 1D). The distal epithelial progenitors begin to differentiate into both thin type I and cuboidal type II alveolar epithelial cells (AECs), which are responsible for gas exchange and surfactant production, respectively. A discrete region dividing the distal and proximal epithelium is marked by the bronchoalveolar duct junction (BADJ), which harbor bronchoalveolar stem cells (15). These cells express markers of both bronchiolar and alveolar epithelium and may serve as a source of reparative stem cells after lung injury but can also act as the cell of origin for certain types of lung cancers (4).
The structural changes associated with final maturation of the developing lung occur during the alveolar stage (P5–P30 or ∼32 weeks until ∼2 years in humans). “Saccules” formed during the saccular phase are further subdivided by secondary septae (or secondary crests) to form alveoli, whereas the mesenchymal content continues to decrease, allowing capillaries to come into closer alignment with the epithelium (Figure 1E). Importantly, during all these stages, the mesenchymal compartment has played an important role in directing the epithelial cell fate as well as branching morphogenesis via mesenchymal-epithelial cell signaling. Although less well studied compared with the lung epithelium, the mesenchyme is also composed of many cell lineages that differentiate into the mesothelium, vascular endothelium, airway smooth muscle, and vascular smooth muscle as well as alveolar fibroblasts, pericytes, and lipofibroblasts.
The Respiratory Phenotype of Mouse Models With Global GC or GR Deficiency
Collectively, the different mouse models of body-wide GC or GR deficiency show either very similar or identical morphological lung phenotypes in late gestation. As shown first by the Cole et al (5) isogenic strain, GR−/− mice die soon after birth and appear cyanotic. By histological analysis, the lungs of fetal GR−/− mice appear normal until ∼E15.5 but from E16.5 show progressive hypercellularity within the saccular walls with little or no airway luminal dilation (Figure 2, A and B). The latter observation is often attributed to airway collapse or atelectasis in the respiratory literature. Newborn mouse lungs from the Brewer et al (7) GR−/− strain show an almost identical lung phenotype and also die of respiratory failure at birth. Other knockout models, such as CRH−/−, CRHR1−/−, and MC2R−/− mice, which have disrupted HPA axis function (and hence defective corticosterone synthesis), also display these traits but only when born from dams that are themselves homozygous knockouts for their respective gene (11–13). In dams with normal HPA axis function, the transfer of maternal corticosterone across the placenta compensates for a lack in the fetus, inducing sufficient lung maturation to allow survival at birth (16). HSD11B1 knockout mice have normal survival at birth and normal fetal lung morphology, although minor differences in surfactant synthesis are observed (10, 17). Finally, MR−/− mice survive for up to 8 to 10 days after birth without any described lung defect and instead die from dehydration due to renal sodium and water loss (14). This suggests that GC signaling via MR has no crucial role in fetal lung development.
Figure 2. Endogenous and exogenous GC effects on murine lung maturation.

A, Lung morphology in E16.5 and E18.5 GR−/− fetal mice. B, Endogenous GC function during late lung development. From E16.5 to E18.5, GC-mediated remodeling of the distal lung results in thinner saccular walls and expansion of the airway lumen. In mouse models of GC deficiency, saccular walls are thicker with less luminal expansion. C, Left lung lobe morphology in E16.5 wild-type mice treated with saline (left) or Dex (right). Insets show magnified regions of the distal lung parenchyma. D, Lung morphology in PN7 mice treated with saline (left) or Dex (right).
In general, the findings obtained via exogenous GC treatment in the lung concur with those derived from GR−/− and GC-deficient mouse models. GC treatment stimulates the expansion of both proximal and distal airways and thins the intervening mesenchymal tissue (Figure 2C). This has been demonstrated in GC-treated fetal lung explant systems and by maternal GC treatment in rodents (18–22), rabbit (23), sheep (24), and rhesus monkey (25). These characteristics are consistent with an arrest of normal proximal lung morphogenesis along with precocious acceleration of the structural maturation of the distal lung. On the other hand, exogenous GCs have been shown to impair the formation of secondary septae during alveolarization, producing an emphysematous phenotype (Figure 2D) (26, 27). As alveolarization occurs postnatally in the rodent lung, it is not possible to investigate this phase of development in GC- and GR-deficient mice, which die at birth.
Before the onset of the GR−/− respiratory phenotype at ∼E16.5, little is known about endogenous GC function for lung development. The lungs of CRH−/− mice (born to CRH−/− mothers) as early as E14.5 show no gross morphological differences from controls, although earlier events such as lung bud formation or branching have not been thoroughly investigated for subtle phenotypes. Nevertheless, the possible existence of such minor defects is unlikely because the surge in fetal GC levels does not occur until late gestation. For example, elevated corticosterone levels in fetal mice are not seen until approximately E14.5 (28), toward the end of the pseudoglandular phase. In addition, lung epithelial cell–specific staining of the respiratory tree in developing Brewer et al (7) GR−/− mice has not revealed any major defects in the lung branching program (29, 30).
After ∼E15.5, the overall lung phenotype from GC deficiency appears to be primarily due to hypercellularity. However, the lungs of GR−/− mice do not appear grossly hypertrophic relative to those of controls at any gestational age examined (our unpublished observations). This observation suggests that from ∼E16.5 until birth, increased saccular wall growth (and not atelectasis) occurs at the expense of luminal volume expansion (Figure 2B). Stereological measurements performed in the lungs of Cole et al (5) isogenic E18.5 GR−/− mice in 2 separate studies agree with this, showing identical degrees of increases and decreases in the percentages of tissue and air, respectively (31, 32). Similar findings, which argue against a strictly atelectatic phenotype were also noted in the lungs of E18.5 CRH−/− mice (born to CRH−/− mothers) (33). Furthermore, although it is well known that GCs induce cell death, particularly in immune cell lineages (34), it is almost certain that the hypercellular lung phenotype seen in mouse models of GC deficiency is the direct result of an increased rate of cell proliferation, rather than a lack of GC-induced cell apoptosis. Studies in the lung of Cole et al isogenic GR−/− mice and CRH−/− mice show no differences from controls in numbers of apoptotic cell nuclei, yet show a large increase in the percentage of dividing cells (33, 35). Based on this finding, much of the ongoing investigation into developmental GC signaling in the lung has focused on its role as an inhibitor of cell proliferation.
Models of Conditional GR Deletion in the Mouse Lung and Phenotypic Variability
Although many molecular pathways that regulate lung morphogenesis do so via paracrine interactions and are driven by morphogens and receptors localized to either the epithelium or mesenchyme, GC hormones diffuse freely into all cells to activate the GR, which is expressed almost ubiquitously (36, 37). However, it is generally accepted that within the developing lung, separate functions are performed by compartmentally specific GR in response to the uniformly distributed GC ligands. In addition, it is likely that epithelial or mesenchymal GR activity can induce paracrine interactions between compartments as suggested by early work from Post et al (38) and Smith (39). These studies proposed the existence of a GC-induced “fibroblast pneumocyte factor” (FPF) secreted from the mesenchyme to account for stimulated surfactant biosynthesis in the lung epithelium. Although to date the FPF has not been identified, it has prompted further investigation of selective GC function within lung compartments. The severe lung phenotype observed in GR−/− mice also then raises the issue of whether GC-induced lung maturation is mediated by GR activity within single or multiple tissue compartments, and, if the latter, what relative contribution or biological function is provided by each compartment?
Although somewhat controversial, recent studies predominantly indicate that of the functions of all lung compartments, GR activity derived from the mesenchyme is the most crucial for producing the GC-induced changes necessary for proper lung development and survival at birth (Figure 3, A and B). Mesenchymal GR deletion using either of the 2 available GR floxed strain results in a lung phenotype very similar to germline GR−/− mice, including highly thickened saccular walls, markedly reduced airway luminal space, and highly increased or complete neonatal lethality (Figure 3C) (32, 40, 41). On the other hand, with one notable exception (as discussed below), studies investigating the phenotype of GR deletion in lung epithelium find either little or no effect on pulmonary architecture and do not report a loss of survival at birth (Figure 3C) (30, 32, 40). Similarly, no overt lung phenotype or loss of survival is observed in mice lacking GR in the endothelium (32, 40).
Figure 3. Variation in respiratory phenotype between conditional GR knockout mice.

A, Representation of GR deletion in the developing lung epithelium and mesenchyme and globally in all compartments. Black layers depict GR deletion. B, E18.5 lung morphology in the respective GR knockout mice. Note that representative lung images shown are from the mouse strains described in Bird et al (40). C, Summary of the neonatal survival and morphological lung phenotypes observed in (1) Manwani et al (42), (2) Bird et al (40), (3) Habermehl et al (32), and (4) Li et al (41).
Interestingly, one study casts doubt on the assertion that lung epithelial GR activity is dispensable for lung structural maturation and neonatal survival. Work conducted by Manwani et al (42) using the Brewer et al (7) strain and a doxycycline (Dox)-inducible human surfactant protein C (hSFTPC)-Cre system showed that loss of lung epithelial GR in mice resulted in approximately 50% reduced viability at birth and produced moderately thickened saccular walls. In support of this finding, hSFTPC-driven overexpression of lung epithelial GR on a Cole et al (5) GR−/− homozygote background reduced lung cellularity to levels comparable with those of wild-type mice, although this did not prevent neonatal lethality (43). Several other studies however, contradict these findings. For example, whereas Habermehl et al (30) did observe thicker saccular walls in epithelial GR knockout lungs generated from a separate hSFTPC-Cre strain, no neonatal lethality was observed. In addition, work by Martinez Alanis et al (30) using epithelial knockouts derived from a sonic hedgehog (Shh)-Cre strain did not report any lung phenotype, although survival in this case was not assessed. However, the findings by Manwani et al (42) are particularly surprising given the results from Bird et al (40), who generated identical knockout mice also using the Brewer et al (7) strain and the same Dox-inducible system of deletion, yet did not report any increased lethality at birth or a respiratory phenotype. Furthermore, both studies were careful to assess the degree of lung epithelial recombination, and each shows almost complete GR deletion. Therefore, the observed discrepancy is probably due to a combination of differing genetic mouse backgrounds and also Dox treatment intervals used between the 2 studies. Dox and also reverse tetracycline transactivator (rtTA) expression, which together drive the recombination system, have known deleterious effects on lung development that can lead to lethality, the severity of the effect being heavily dependent on the genetic background of the mice used (44). C57BL/6 mice, as used in the Bird et al study, are more resistant, whereas the background used by Manwani et al is not named and may therefore be of a more susceptible variety. Combined with the lengthier Dox treatment interval used by Manwani et al, it appears likely that the reduced viability is a consequence of these factors, rather than a requirement of lung epithelial GR expression. Together, these results suggest that although lung epithelial GR activity may partially reduce lung tissue cellularity during development, it is not essential for neonatal survival.
Separate investigations of the respiratory phenotype in mesenchymal GR knockout mice are much more in agreement; however, the degree of severity appears to differ slightly, depending on which floxed GR line and type of mesenchymal-specific Cre are used. Work by Habermehl et al (32), using the Tronche et al (6) strain crossed with a collagen, type I, α2 (Col1a2)-Cre mouse, shows that mesenchymal GR deletion results in complete lethality at birth and a hypercellular lung morphology almost devoid of airspace. Similarly, Li et al (41), also using the Tronche et al strain but crossed with a Dermo1 (also known as twist homolog 2 [Twist2])-Cre mouse, found a comparable degree of lung hypercellularity although only a very small number of mesenchymal knockout mice (∼3%) survive beyond birth. In contrast, deletion via the Dermo1-Cre in the Brewer et al (7) strain as shown by Bird et al (40) results in a slightly milder phenotype with a greater number of surviving knockouts (∼25%) and a less condensed, although still significantly hypercellular, lung morphology. Rather than attribute these small percentages of surviving knockouts to possible compensatory GR activity from other lung compartments, it is more likely that the limited survival seen in models using the Dermo1-Cre strain is due to an incomplete deletion of GR in the lung mesenchyme. Double-labeling experiments clearly show GR-positive cells within the lung mesenchyme in Dermo1-Cre derived knockout mice (30, 40). Alternatively, it is possible that these GR-positive mesenchymal cells have arisen due to compensatory transdifferentiation from other lung compartments, although if true this would also be expected to occur in mesenchymal knockouts derived from the Col1a2-Cre strain and thus allow some measure of survival, yet this was not observed (32). Whereas the origin of the remaining GR-positive mesenchymal cells in Dermo1-Cre–derived lungs remains, thus far, unanswered, their presence and resulting milder phenotype compared with that of GR−/− lungs actually provide a useful internal control and argue that a threshold of GR mesenchymal activity exists to provide sufficient lung maturation for neonatal survival.
An important objective is to determine which subset of mesenchymal cells harbors the GR activity critical for proper lung maturation and survival. As previously mentioned, the lung is composed of many mesenchymal lineages including airway and vascular smooth muscle, mesothelial cells, endothelial cells, and several types of fibroblasts. However, compared with the role of the relatively well-studied lung epithelium, the precise roles of lung mesenchymal cell types and their corresponding gene expression patterns have been poorly described. As a result, selective GR deletion in vivo within the entire range of lung mesenchymal lineages is not currently possible because the respective Cre recombinase strains have yet to be generated. Some mesenchymal lineages, however, can be reliably discounted. For example, endothelial cells almost certainly do not contribute important GR activity as shown by a lack of a lung phenotype in Tie2-Cre derived knockout mice (32, 40). On the other hand, vascular smooth muscle may be an important source of mesenchymal GR activity, given that considerable HSD11B1 expression has been noted in these cells during late lung development in the mouse (our unpublished observations).
GR Signaling and Epithelial Cell Differentiation During Lung Development
GC-mediated regulation of lung epithelial cell differentiation has been investigated in several studies via the use of GC-treated in vivo, in vitro, and lung explant models in a variety of species (21, 45–47), as well as in a more limited number of studies using GC-deficient mouse knockouts. Often this analysis has been performed via direct assessment of epithelial cell morphology, the presence/absence of cell-defining organelles (such as lamellar bodies within type II AECs), or levels of intracellular glycogen, which is prominent in undifferentiated but not differentiated distal epithelium. In more recent studies, the degree of epithelial cell differentiation is determined indirectly by expression analysis of epithelial cell markers, either by qualitative visual evaluation of immuno-labeled cells or by quantitative analysis of mRNA or protein levels in whole lung homogenates. In the latter case, some caution is advised in interpretation of the data. First, the complete range of genes whose transcription is regulated by GC signaling in the developing lung has not been clearly defined, and, therefore, any differential expression of putative epithelial markers cannot be reliably attributed solely to cell differentiation. Secondly, many studies have used markers that have since been shown to be inappropriate for epithelial cell–specific quantitation. For example, genes such as podoplanin (Pdpn, also known as T1α) and surfactant protein C (Sftpc) are often used as mature type I and II AEC markers, respectively, although both are also expressed in bipotent progenitor AECs (48, 49). This example highlights the need for correct and preferably multiple markers to support a conclusion of altered cell differentiation in the lung.
In the distal lung epithelium, most studies agree that GC signaling stimulates the differentiation of immature multipotent progenitors into committed lineages. In mouse models of GC deficiency, this has been investigated primarily during late lung developmental phases (late pseudoglandular to saccular) when the levels of endogenous GCs dramatically increase. At this time, differentiation of the more proximal conducting airway epithelium is largely complete, although most distal epithelial progenitors are bipotent and have the potential to become either type I or type II AECs (50). As opposed to factors such as N-myc (Mycn), which inhibits AEC differentiation and may therefore play an important role in preventing precocious distal epithelial differentiation in the lung (51), GC signaling promotes this process. In vivo this is suggested first by reduced mRNA levels of type I and II AEC markers in saccular-stage CRH−/− and GR−/− mouse lungs (31, 33). Second, very recent work by Martinez Alanis et al (30) also demonstrates that GC signaling dictates the positioning of the BADJ along the proximal-distal axis of the mouse lung epithelium. In saccular-stage GR−/− lungs, the BADJ was localized more distally, whereas in Dex-treated lungs, the BADJ was localized more proximally and resulted in the precocious emergence of cells immuno-positive for AEC markers. GR−/− lungs also possess relatively larger branch tips composed of cells almost completely immuno-positive for SOX9, a marker of undifferentiated distal lung epithelium (30). Together these findings suggest that during late lung development GCs can both switch the fate of distal progenitors destined to become conducting airway epithelium to instead become AECs and also act on progenitors located even more distally to commit to a defined AEC lineage (Figure 4B).
Figure 4. GR-mediated mechanisms in the distal mouse lung during late gestation (late pseudoglandular to saccular phases).
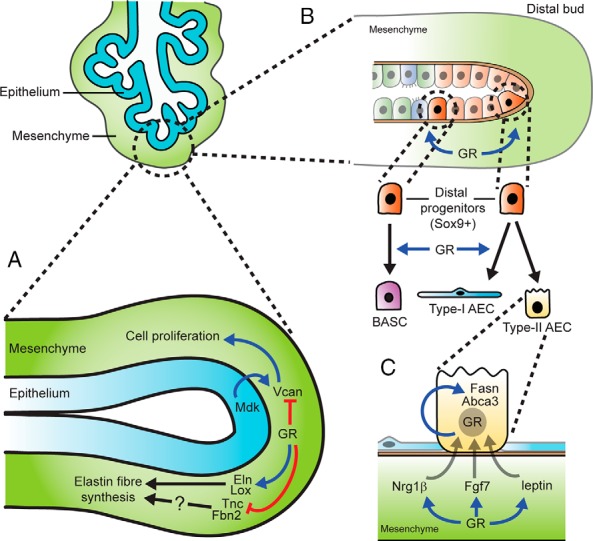
A, Mesenchymal GR restrains cell proliferation in the lung via inhibition of Vcan expression and promotes elastin fiber synthesis via induction of Eln and Lox expression. GR-mediated inhibition of Tnc and Fbn2 expression may also participate in correct elastin fiber synthesis. B, Model of mesenchymal GR-regulated epithelial cell differentiation in the distal lung. The distal lung epithelium contains a pool of Sox9+ progenitors, which are stimulated by mesenchymal GR signaling to become either bronchoalveolar stem cells (BASCs), forming the BADJ, or more distally, type I AECs. Type II AEC differentiation is not affected by GR signaling. C, Model of GR-mediated type II AEC maturation by induction of surfactant-associated factors. Mesenchymal GR induces expression of genes including Nrg1β, Fgf7, and leptin, which act on type II AECs via paracrine signaling. Epithelial GR induces expression of genes including Fasn and Abca3 via autocrine signaling.
Other studies further suggest that GC signaling may selectively promote type I AEC differentiation. Direct quantification of AEC types and intermediates in saccular-stage GR−/− lungs by electron microscopy showed markedly reduced type I AEC proportions, whereas type II AEC numbers remained unaffected (31). Similarly, type II AEC differentiation is unaffected in fetal rat lung explants treated with the GR inhibitor RU486 (52). Whether GC-induced AEC differentiation is regulated directly by epithelial GR activity, via cross talk from mesenchymal GR activity, or possibly by a combination of both is as yet unclear. Work by Manwani et al (42) and Li et al (41) showed reduced mRNA levels of the well-validated type I AEC marker aquaporin 5 (Aqp5) in both epithelial and mesenchymal GR knockout lungs, respectively (41, 42), suggesting that both compartments are involved. Both studies also showed reduced mRNA levels of surfactant proteins to indicate that type II AEC differentiation is similarly induced by epithelial and mesenchymal GR activity. However, because GCs are known to regulate many of the surfactant proteins at the level of gene transcription (53–55), it is uncertain whether these findings in conditional GR knockouts are truly due to altered type II AEC differentiation. Instead, aside from type I AEC differentiation, it is more likely that GR function from either compartment is limited to maturation of previously committed type II AECs by inducing the mechanisms associated with surfactant synthesis and processing (Figure 4C). This observation is also supported by a wealth of studies showing GC-mediated stimulation of surfactant protein gene transcription (reviewed in Ref. 56) and also expression of factors that regulate surfactant-associated lipid synthesis such as mesenchymal-derived FGF7 (also known as KGF) (54), neuregulin-1β (NRG1β) (57), and leptin (58) as well as epithelial cell–derived fatty acid synthase (Fasn) (59) and ABCA3 (60).
In contrast, little is known about possible GC involvement in regulating differentiation of the proximal lung epithelium. Because most proximal epithelial lineages differentiate primarily during the pseudoglandular phase (61), before the endogenous GC surge and onset of the GR−/− respiratory phenotype, it appears unlikely that GCs play an important role. Although some isolated studies show that exogenous GC treatment can influence NE, ciliated, and secretory cell proportions during lung development (46, 62, 63), immuno-labeling of these lineages by cell-specific markers in saccular-stage GR−/− lungs has not revealed any major differences (our unpublished observations). Interestingly, contradictory findings were observed in saccular-stage CRH−/− lungs, which showed complete loss of cells immuno-positive for the secretory cell marker CC10 (Scgb1a1) and abnormal localization of the NE cell marker PGP9.5 (33). These findings raise the possibility that CRH expression or GC ligand synthesis, independent of GR function, may be important for stimulation of pathways that regulate lung epithelial differentiation.
GR Signaling and Regulation of Cell Proliferation During Lung Development
As previously mentioned, the hypercellular respiratory phenotype seen in mouse models of total GC deficiency is probably due to an increased rate of cell proliferation rather than a lack of cell apoptosis. Because this phenotype is recapitulated primarily in mesenchymal but not epithelial GR knockouts it is expected that mesenchymal GR activity is mostly responsible for restraining lung cell proliferation during development, allowing tissue to thin. To verify this, a cell proliferation index has been determined in saccular-stage lungs from epithelial and mesenchymal GR knockout mice (32, 40). In agreement with the hypercellular lung morphology observed, work by Habermehl et al (32) showed that cell proliferation is significantly increased only in mesenchymal GR knockout lungs. Surprisingly, however, work by Bird et al (40) shows that although increased cell proliferation is indeed observed in mesenchymal GR knockout lungs, a similar increase is also found in epithelial GR knockout lungs. As discussed previously, it is likely that the discrepancies observed between these studies is largely due to the different GR floxed mice and genetic deletion systems used. Taken together, these findings confirm that inhibition of cell proliferation in the developing lung is driven primarily by mesenchymal GR activity although it is unclear whether epithelial GR is also partially responsible.
GR-mediated signaling pathways that regulate cell proliferation in the lung also probably involve paracrine interactions between compartments. The studies by Bird et al (40) and Habermehl et al (32) show that mesenchymal GR activity inhibits cell proliferation not only in the mesenchyme but also in the epithelium. Furthermore, Bird et al (40) observed that lungs from epithelial GR knockout mice also exhibit increased cell proliferation in both compartments. Although epithelial GR involvement remains unclear, these findings strongly suggest that a GC-mediated mechanism of cross talk exists in the developing lung, which acts to restrain cell proliferation in multiple compartments. To date, the gene expression networks regulated by GR in the developing lung have been poorly defined. However, recent microarray data generated from saccular-stage GR−/− and conditional GR knockout lungs have identified a number of putative GR-mediated targets that are known to regulate cell proliferation (32, 35, 64). One striking target, identified in 2 separate microarray analyses is the cyclin-dependent kinase inhibitor p21CIP1 (Cdkn1a), which showed reduced expression in the lungs of both GR−/− mice (35) and mesenchymal GR knockout mice (32). Cyclin-dependent kinase inhibitors are well-known inducers of cell cycle arrest and GC-mediated stimulation of p21CIP1 gene and protein expression in lung cells has been described (65, 66). However, p21CIP1 mRNA and protein levels in saccular-stage lungs were unaffected in a separate strain of mesenchymal GR knockout mice, although interestingly mRNA levels were reduced in endothelial GR knockout lungs (40). Although some inconsistencies between studies may again be due to strain differences, other evidence suggests that p21CIP1 is not an important GR target for preventing lung hyperplasia. First, p21CIP1 mouse knockouts show normal survival (67) and lung morphology (68). Second, in the fetal lung, induction of p21CIP1 expression by GCs has been shown primarily in epithelial cells, whereas to our knowledge similar findings have not been described in mesenchymal cells, which are the likely source of critical GR activity for respiratory maturation.
Other potential microarray-identified GR targets that may influence cell proliferation in the developing lung include the growth factor midkine (Mdk) and its receptor versican (Vcan, also known as CSPG2), an extracellular matrix (ECM) proteoglycan. Both factors have known stimulatory roles in cell proliferation, cell migration, and angiogenesis (reviewed in Refs. 69 and 70). Increased mRNA levels of Mdk and Vcan were found in saccular-stage lungs from GR−/− (35, 64) and mesenchymal GR knockout mice (40), suggesting that GCs normally repress expression of these factors. However, although widespread midkine protein distribution was noted in saccular-stage GR−/− lungs compared with distal epithelial cell–specific localization in control lungs (64), no obvious changes were found in mesenchymal GR knockout lungs (40). Together with the finding that transgenic overexpression of midkine in lung epithelium does not lead to any major defect in lung morphology or survival (71), these findings suggest that Mdk is not an essential GR target for respiratory maturation. In contrast, versican protein, normally restricted to bronchiolar and vascular smooth muscle cells in saccular stage lungs, was detected throughout the distal mesenchyme in mesenchymal GR knockout lungs (40). This observation suggests that GCs normally prevent versican expression within distal lung mesenchymal cells, such as fibroblasts surrounding developing saccules, to prohibit binding and growth signals from midkine and/or other mitogens (Figure 4A).
GR Signaling and Regulation of ECM Components During Lung Development
In addition to ECM factors such as proteoglycans (ie, versican), which possess known signaling roles in respiratory development, GCs are known to regulate expression of proteins that are essential for the synthesis of structural lung ECM components, such as elastin and collagen fibers (Figure 4A). In the lung, these fibers surround the conducting airways, blood vessels, and alveoli and function in different capacities to provide either rigid mechanical support (collagen) or a potential for stretch and recoil (elastin) which is required during breathing movements (reviewed in Ref. 72). In particular, GC-mediated regulation of genes that contribute to elastogenesis in the lung is a well-studied topic as either a loss or excess of pulmonary elastin fibers can lead to emphysema (73) or fibrosis (74), respectively. Several in vivo and in vitro studies show that in the fetal lung, GCs stimulate gene expression of elastin (Eln), which encodes the tropoelastin monomeric subunit of elastin fibers (75–77). Reduced Eln gene expression is also observed in saccular-stage lungs from GR−/− as well as 3 separate strains of mesenchymal GR knockout mice (32, 40, 41), suggesting that GC-mediated stimulation of the Eln gene expression is due to mesenchymal GR activity. However, it is unlikely that reduced mesenchymal elastin gene expression contributes significantly to the GR−/− respiratory phenotype because ELN−/− mice do not show any lung defect until the alveolar phase, several days after birth (78).
Similar evidence also indicates that in the fetal lung GCs, acting via the mesenchymal GR, stimulate gene expression of lysyl oxidase (Lox), which catalyzes the cross-linking of tropoelastin monomers to form mature elastin fibers (32, 79). Not surprisingly, reduced elastin fiber content, as assessed by histological staining, is observed in lungs from mesenchymal GR knockouts (32, 40, 41), although studies differ on whether this loss is found throughout the lung or is restricted to the distal mesenchyme. LOX is also required for proper cross-linking of collagen fibers in the developing lung, as shown by poorly formed collagen fibers in lungs from saccular-stage LOX−/− knockout mice (80). Therefore, LOX may be an important GR target in the lung mesenchyme to promote collagen fiber processing and maturation. Interestingly, higher levels of collagen I/III fiber immunostaining were observed in saccular-stage mesenchymal GR knockout lungs than in control lungs (32). However, this may be due to more collagen-expressing cells in the mesenchymal compartment of mesenchymal GR knockout lungs rather than to any indication of changes in collagen fiber maturation, which cannot be detected by immunostaining.
Several other ECM factors have been identified as GR targets in the lung; however, their role during the critical window of development corresponding to the GR−/− respiratory phenotype has not been thoroughly investigated. The glycoproteins fibrillin 2 and tenascin C, encoded by the Fbn2 and Tnc genes, respectively, have increased mRNA levels in saccular-stage lungs from mesenchymal GR knockout mice (Fbn2 [32)] and Tnc [unpublished observations]). FBN2 is believed to contribute to elastic fiber synthesis during early lung development (81). On the other hand, TNC, previously shown to be down-regulated by Dex in rat lung explants (82), is implicated in elastic fiber synthesis during the alveolar phase (83). Both FBN2 and TNC may also participate in branching morphogenesis. Based on these roles, it is possible that GCs down-regulate Fbn2 and Tnc to maintain an appropriate balance of elastogenic factors (Figure 4A).
Concluding Remarks and Future Studies
Currently, GC therapy still remains a standard treatment of mothers at risk of preterm delivery and has improved the survival for preterm infants in neonatal care worldwide. However, the known clinical value of GCs in accelerating fetal lung maturity is not matched by an appropriate level of research to explore the underlying molecular mechanisms. Much of the information available has been obtained via the use of GC-deficient mouse models, and recent conditional GR knockouts have identified the specific roles of GC signaling within lung tissue compartments. Considering the critical role of the mesenchyme for directing GC-mediated lung maturation, researchers are now able to focus on mesenchymal cell subsets to identify not only the essential GR-regulated gene targets responsible but also which mesenchyme-expressed cofactors are potentially involved. It is likely that this research will uncover new factors that modulate surfactant activity in the lung epithelium via cell-cell cross talk, such as the unidentified FPF.
The importance of mesenchymal GR then prompts several other questions. What are the mesenchymal cell subset(s) that mediate critical GR activity? Furthermore, are the GR-activated signaling pathways within these cell subsets specific to the process of sacculation or are they a reiteration of early branching programs? Answering these questions will require a greater understanding of the complex heterogeneity and progenitor differentiation relationships within the lung mesenchymal cell population. Hopefully, this will lead to the discovery of novel mesenchymal markers as well as to the generation of additional Cre-recombinase mouse strains, allowing gene deletion of GR and other factors in specific lung mesenchymal lineages. It would also be useful if such Cre-recombinase systems could be created to allow for temporal control, such as the newly developed lung mesenchymal Tbx4-rtTA line (84). This would enable mesenchymal deletion of GR after birth, which is currently precluded by the neonatal lethality in the available mesenchymal GR knockout lines. This would be particularly useful in studying mesenchymal GR involvement in postnatal alveolarization or in lung disorders such as chronic obstructive pulmonary disease, pulmonary fibrosis, and asthma.
Finally, a better understanding of how different GC ligands affect GR activity and function in the lung would be extremely beneficial. It is now clear that different GC ligands elicit a variety of different GR-regulated responses, probably due to different effects on GR dimerization, GRE DNA binding, cofactor recruitment, and also activation of other nuclear receptors such as the MR (85). Responses produced from different GC ligands in specific tissues are not well characterized. Further characterization of their specific effects in the fetal and mature adult lung would allow the development of a new generation of selective GC ligands enabling better therapeutic treatments for fetal and adult lung disorders, with fewer undesirable side effects.
Acknowledgments
This work was supported by program grant 606789 from the Australian National Health and Medical Research Council.
Disclosure Summary: The authors have nothing to disclose.
Funding Statement
This work was supported by program grant 606789 from the Australian National Health and Medical Research Council.
Footnotes
- AEC
- alveolar epithelial cell
- BADJ
- bronchoalveolar duct junction
- Dex
- dexamethasone
- Dox
- doxycycline
- E
- embryonic day
- ECM
- extracellular matrix
- FPF
- fibroblast pneumocyte factor
- GC
- glucocorticoid
- GR
- glucocorticoid receptor
- GRE
- glucocorticoid receptor element
- HPA
- hypothalamic-pituitary-adrenal
- HSD11B1
- hydroxysteroid 11β-dehydrogenase 1
- MR
- mineralocorticoid receptor
- NE
- neuroendocrine
- P
- postnatal day.
References
- 1. Liggins GC, Howie RN. A controlled trial of antepartum glucocorticoid treatment for prevention of the respiratory distress syndrome in premature infants. Pediatrics. 1972;50:515–525. [PubMed] [Google Scholar]
- 2. Morrisey EE, Hogan BL. Preparing for the first breath: genetic and cellular mechanisms in lung development. Dev Cell. 2010;18:8–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ornitz DM, Yin Y. Signaling networks regulating development of the lower respiratory tract. Cold Spring Harb Perspect Biol. 2012;4(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Herriges M, Morrisey EE. Lung development: orchestrating the generation and regeneration of a complex organ. Development. 2014;141:502–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cole TJ, Blendy JA, Monaghan AP, et al. Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes Dev. 1995;9:1608–1621. [DOI] [PubMed] [Google Scholar]
- 6. Tronche F, Kellendonk C, Kretz O, et al. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23:99–103. [DOI] [PubMed] [Google Scholar]
- 7. Brewer JA, Kanagawa O, Sleckman BP, Muglia LJ. Thymocyte apoptosis induced by T cell activation is mediated by glucocorticoids in vivo. J Immunol. 2002;169:1837–1843. [DOI] [PubMed] [Google Scholar]
- 8. Reichardt HM, Kaestner KH, Tuckermann J, et al. DNA binding of the glucocorticoid receptor is not essential for survival. Cell. 1998;93:531–541. [DOI] [PubMed] [Google Scholar]
- 9. Ross ME, Evinger MJ, Hyman SE, et al. Identification of a functional glucocorticoid response element in the phenylethanolamine N-methyltransferase promoter using fusion genes introduced into chromaffin cells in primary culture. J Neurosci. 1990;10:520–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kotelevtsev Y, Holmes MC, Burchell A, et al. 11β-Hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proc Natl Acad Sci USA. 1997;94:14924–14929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Muglia L, Jacobson L, Dikkes P, Majzoub JA. Corticotropin-releasing hormone deficiency reveals major fetal but not adult glucocorticoid need. Nature. 1995;373:427–432. [DOI] [PubMed] [Google Scholar]
- 12. Smith GW, Aubry JM, Dellu F, et al. Corticotropin releasing factor receptor 1-deficient mice display decreased anxiety, impaired stress response, and aberrant neuroendocrine development. Neuron. 1998;20:1093–1102. [DOI] [PubMed] [Google Scholar]
- 13. Chida D, Sato T, Sato Y, et al. Characterization of mice deficient in melanocortin 2 receptor on a B6/Balbc mix background. Mol Cell Endocrinol. 2009;300:32–36. [DOI] [PubMed] [Google Scholar]
- 14. Berger S, Bleich M, Schmid W, et al. Mineralocorticoid receptor knockout mice: pathophysiology of Na+ metabolism. Proc Natl Acad Sci USA. 1998;95:9424–9429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim CF, Jackson EL, Woolfenden AE, et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121:823–835. [DOI] [PubMed] [Google Scholar]
- 16. Venihaki M, Carrigan A, Dikkes P, Majzoub JA. Circadian rise in maternal glucocorticoid prevents pulmonary dysplasia in fetal mice with adrenal insufficiency. Proc Natl Acad Sci USA. 2000;97:7336–7341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hundertmark S, Dill A, Ebert A, et al. Foetal lung maturation in 11β-hydroxysteroid dehydrogenase type 1 knockout mice. Horm Metab Res. 2002;34:545–549. [DOI] [PubMed] [Google Scholar]
- 18. Kauffman SL. Acceleration of canalicular development in lungs of fetal mice exposed transplacentally to dexamethasone. Lab Invest. 1977;36:395–401. [PubMed] [Google Scholar]
- 19. Massoud EA, Sekhon HS, Rotschild A, Thurlbeck WM. The in vitro effect of triamcinolone acetonide on branching morphogenesis in the fetal rat lung. Pediatr Pulmonol. 1992;14:28–36. [DOI] [PubMed] [Google Scholar]
- 20. Melnick M, Choy HA, Jaskoll T. Glucocorticoids, tumor necrosis factor-α, and epidermal growth factor regulation of pulmonary morphogenesis: a multivariate in vitro analysis of their related actions. Dev Dyn. 1996;205:365–378. [DOI] [PubMed] [Google Scholar]
- 21. Oshika E, Liu S, Ung LP, et al. Glucocorticoid-induced effects on pattern formation and epithelial cell differentiation in early embryonic rat lungs. Pediatr Res. 1998;43:305–314. [DOI] [PubMed] [Google Scholar]
- 22. Rotschild A, Solimano A, Sekhon HS, Massoud EA, Thurlbeck WM. Effect of triamcinolone acetonide on the development of the pulmonary airways in the fetal rat. Pediatr Pulmonol. 1997;23:76–86. [DOI] [PubMed] [Google Scholar]
- 23. Snyder JM, Rodgers HF, O'Brien JA, Mahli N, Magliato SA, Durham PL. Glucocorticoid effects on rabbit fetal lung maturation in vivo: an ultrastructural morphometric study. Anat Rec. 1992;232:133–140. [DOI] [PubMed] [Google Scholar]
- 24. Boland R, Joyce BJ, Wallace MJ, et al. Cortisol enhances structural maturation of the hypoplastic fetal lung in sheep. J Physiol. 2004;554(Pt 2):505–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bunton TE, Plopper CG. Triamcinolone-induced structural alterations in the development of the lung of the fetal rhesus macaque. Am J Obstet Gynecol. 1984;148:203–215. [DOI] [PubMed] [Google Scholar]
- 26. Massaro D, Teich N, Maxwell S, Massaro GD, Whitney P. Postnatal development of alveoli. Regulation and evidence for a critical period in rats. J Clin Invest. 1985;76:1297–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Massaro GD, Massaro D. Formation of alveoli in rats: postnatal effect of prenatal dexamethasone. Am J Physiol. 1992;263(1 Pt 1):L37–L41. [DOI] [PubMed] [Google Scholar]
- 28. Salomon DS, Gift VD, Pratt RM. Corticosterone levels during midgestation in the maternal plasma and fetus of cleft palate-sensitive and -resistant mice. Endocrinology. 1979;104:154–156. [DOI] [PubMed] [Google Scholar]
- 29. Groenman F, Unger S, Post M. The molecular basis for abnormal human lung development. Biol Neonate. 2005;87:164–177. [DOI] [PubMed] [Google Scholar]
- 30. Martinez Alanis D, Chang DR, Akiyama H, Krasnow MA, Chen J. Two nested developmental waves demarcate a compartment boundary in the mouse lung. Nat Commun. 2014;5:3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cole TJ, Solomon NM, Van Driel R, et al. Altered epithelial cell proportions in the fetal lung of glucocorticoid receptor null mice. Am J Respir Cell Mol Biol. 2004;30:613–619. [DOI] [PubMed] [Google Scholar]
- 32. Habermehl D, Parkitna JR, Kaden S, et al. Glucocorticoid activity during lung maturation is essential in mesenchymal and less in alveolar epithelial cells. Mol Endocrinol. 2011;25:1280–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Muglia LJ, Bae DS, Brown TT, et al. Proliferation and differentiation defects during lung development in corticotropin-releasing hormone-deficient mice. Am J Respir Cell Mol Biol. 1999;20:181–188. [DOI] [PubMed] [Google Scholar]
- 34. Schlossmacher G, Stevens A, White A. Glucocorticoid receptor-mediated apoptosis: mechanisms of resistance in cancer cells. J Endocrinol. 2011;211:17–25. [DOI] [PubMed] [Google Scholar]
- 35. Bird AD, Tan KH, Olsson PF, et al. Identification of glucocorticoid-regulated genes that control cell proliferation during murine respiratory development. J Physiol. 2007;585(Pt 1):187–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Speirs HJ, Seckl JR, Brown RW. Ontogeny of glucocorticoid receptor and 11β-hydroxysteroid dehydrogenase type-1 gene expression identifies potential critical periods of glucocorticoid susceptibility during development. J Endocrinol. 2004;181:105–116. [DOI] [PubMed] [Google Scholar]
- 37. Kitraki E, Kittas C, Stylianopoulou F. Glucocorticoid receptor gene expression during rat embryogenesis. An in situ hybridization study. Differentiation. 1997;62:21–31. [DOI] [PubMed] [Google Scholar]
- 38. Post M, Floros J, Smith BT. Inhibition of lung maturation by monoclonal antibodies against fibroblast-pneumonocyte factor. Nature. 1984;308:284–286. [DOI] [PubMed] [Google Scholar]
- 39. Smith BT. Lung maturation in the fetal rat: acceleration by injection of fibroblast-pneumonocyte factor. Science. 1979;204:1094–1095. [DOI] [PubMed] [Google Scholar]
- 40. Bird AD, Choo YL, Hooper SB, McDougall AR, Cole TJ. Mesenchymal glucocorticoid receptor regulates the development of multiple cell layers of the mouse lung. Am J Respir Cell Mol Biol. 2014;50:419–428. [DOI] [PubMed] [Google Scholar]
- 41. Li A, Hardy R, Stoner S, Tuckermann J, Seibel M, Zhou H. Deletion of mesenchymal glucocorticoid receptor attenuates embryonic lung development and abdominal wall closure. PLoS One. 2013;8:e63578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Manwani N, Gagnon SP, Post M, et al. Reduced viability of mice with lung epithelial-specific knockout of glucocorticoid receptor. Am J Respir Cell Mol Biol. 2010;43:599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gagnon S, Atmodjo W, Humes D, McKerlie C, Kaplan F, Sweezey NB. Transgenic glucocorticoid receptor expression driven by the SP-C promoter reduces neonatal lung cellularity and midkine expression in GRhypo mice. Biol Neonate. 2006;90:46–57. [DOI] [PubMed] [Google Scholar]
- 44. Morimoto M, Kopan R. rtTA toxicity limits the usefulness of the SP-C-rtTA transgenic mouse. Dev Biol. 2009;325:171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lohninger AK, Böck P, Salzer H, Sevelda P, Lohninger AF. Antenatal betamethasone-dose-effects on fetal rat lung morphology and surfactant. J Perinat Med. 1994;22:319–328. [DOI] [PubMed] [Google Scholar]
- 46. Massaro GD, Massaro D. Development of bronchiolar epithelium in rats. Am J Physiol. 1986;250(5 Pt 2):R783–R788. [DOI] [PubMed] [Google Scholar]
- 47. Gonzales LW, Ballard PL, Ertsey R, Williams MC. Glucocorticoids and thyroid hormones stimulate biochemical and morphological differentiation of human fetal lung in organ culture. J Clin Endocrinol Metab. 1986;62:678–691. [DOI] [PubMed] [Google Scholar]
- 48. Desai TJ, Brownfield DG, Krasnow MA. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature. 2014;507:190–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Treutlein B, Brownfield DG, Wu AR, et al. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature. 2014;509:371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yang J, Chen J. Developmental programs of lung epithelial progenitors: a balanced progenitor model. Wiley Interdiscip Rev Dev Biol. 2014;3:331–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Okubo T, Knoepfler PS, Eisenman RN, Hogan BL. Nmyc plays an essential role during lung development as a dosage-sensitive regulator of progenitor cell proliferation and differentiation. Development. 2005;132:1363–1374. [DOI] [PubMed] [Google Scholar]
- 52. Guettari N, Dufour ME, Marin L. Effects of the antiglucocorticoid RU 486 on the initiation of ultrastructural type-II cell differentiation in fetal rat lung. Biol Neonate. 1990;58:173–180. [DOI] [PubMed] [Google Scholar]
- 53. Huang HW, Bi W, Jenkins GN, Alcorn JL. Glucocorticoid regulation of human pulmonary surfactant protein-B mRNA stability involves the 3′-untranslated region. Am J Respir Cell Mol Biol. 2008;38:473–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mouhieddine-Gueddiche OB, Pinteur C, Chailley-Heu B, et al. Dexamethasone potentiates keratinocyte growth factor-stimulated SP-A and SP-B gene expression in alveolar epithelial cells. Pediatr Res. 2003;53:231–239. [DOI] [PubMed] [Google Scholar]
- 55. Ogasawara Y, Kuroki Y, Tsuzuki A, Ueda S, Misaki H, Akino T. Pre- and postnatal stimulation of pulmonary surfactant protein D by in vivo dexamethasone treatment of rats. Life Sci. 1992;50:1761–1767. [DOI] [PubMed] [Google Scholar]
- 56. Mendelson CR. Role of transcription factors in fetal lung development and surfactant protein gene expression. Annu Rev Physiol. 2000;62:875–915. [DOI] [PubMed] [Google Scholar]
- 57. King G, Maker GL, Berryman D, Trengove RD, Cake MH. Role of neuregulin-1β in dexamethasone-enhanced surfactant synthesis in fetal type II cells. FEBS Lett. 2014;588:975–980. [DOI] [PubMed] [Google Scholar]
- 58. Torday JS, Sun H, Wang L, Torres E, Sunday ME, Rubin LP. Leptin mediates the parathyroid hormone-related protein paracrine stimulation of fetal lung maturation. Am J Physiol Lung Cell Mol Physiol. 2002;282:L405–L410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Beneke S, Rooney SA. Glucocorticoids regulate expression of the fatty acid synthase gene in fetal rat type II cells. Biochim Biophys Acta. 2001;1534:56–63. [DOI] [PubMed] [Google Scholar]
- 60. Yoshida I, Ban N, Inagaki N. Expression of ABCA3, a causative gene for fatal surfactant deficiency, is up-regulated by glucocorticoids in lung alveolar type II cells. Biochem Biophys Res Commun. 2004;323:547–555. [DOI] [PubMed] [Google Scholar]
- 61. Rawlins EL. The building blocks of mammalian lung development. Dev Dyn. 2011;240:463–476. [DOI] [PubMed] [Google Scholar]
- 62. Dayer AM, Kapanci Y, Rademakers A, Rusy LM, De Mey J, Will JA. Increased numbers of neuroepithelial bodies (NEB) in lungs of fetal rhesus monkeys following maternal dexamethasone treatment. Cell Tissue Res. 1985;239:703–705. [DOI] [PubMed] [Google Scholar]
- 63. Nord M, Andersson O, Brönnegård M, Lund J. Rat lung polycholorinated biphenyl-binding protein: effect of glucocorticoids on the expression of the Clara cell-specific protein during fetal development. Arch Biochem Biophys. 1992;296:302–307. [DOI] [PubMed] [Google Scholar]
- 64. Kaplan F, Comber J, Sladek R, et al. The growth factor midkine is modulated by both glucocorticoid and retinoid in fetal lung development. Am J Respir Cell Mol Biol. 2003;28:33–41. [DOI] [PubMed] [Google Scholar]
- 65. Corroyer S, Schittny JC, Djonov V, Burri PH, Clement A. Impairment of rat postnatal lung alveolar development by glucocorticoids: involvement of the p21CIP1 and p27KIP1 cyclin-dependent kinase inhibitors. Pediatr Res. 2002;51:169–176. [DOI] [PubMed] [Google Scholar]
- 66. Greenberg AK, Hu J, Basu S, et al. Glucocorticoids inhibit lung cancer cell growth through both the extracellular signal-related kinase pathway and cell cycle regulators. Am J Respir Cell Mol Biol. 2002;27:320–328. [DOI] [PubMed] [Google Scholar]
- 67. Deng C, Zhang P, Harper JW, Elledge SJ, Leder P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995;82:675–684. [DOI] [PubMed] [Google Scholar]
- 68. Zhang P, Wong C, Liu D, Finegold M, Harper JW, Elledge SJ. p21CIP1 and p57KIP2 control muscle differentiation at the myogenin step. Genes Dev. 1999;13:213–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wight TN. Versican: a versatile extracellular matrix proteoglycan in cell biology. Curr Opin Cell Biol. 2002;14:617–623. [DOI] [PubMed] [Google Scholar]
- 70. Muramatsu T. Midkine, a heparin-binding cytokine with multiple roles in development, repair and diseases. Proc Jpn Acad Ser B Phys Biol Sci. 2010;86:410–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Reynolds PR, Mucenski ML, Le Cras TD, Nichols WC, Whitsett JA. Midkine is regulated by hypoxia and causes pulmonary vascular remodeling. J Biol Chem. 2004;279:37124–37132. [DOI] [PubMed] [Google Scholar]
- 72. Ahlfeld SK, Conway SJ. Aberrant signaling pathways of the lung mesenchyme and their contributions to the pathogenesis of bronchopulmonary dysplasia. Birth Defects Res A Clin Mol Teratol. 2012;94:3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Shifren A, Mecham RP. The stumbling block in lung repair of emphysema: elastic fiber assembly. Proc Am Thorac Soc. 2006;3:428–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hoff CR, Perkins DR, Davidson JM. Elastin gene expression is upregulated during pulmonary fibrosis. Connect Tissue Res. 1999;40:145–153. [DOI] [PubMed] [Google Scholar]
- 75. Anceschi MM, Palmerini CA, Codini M, Luzi P, Cosmi EV. Collagen and elastin in rabbit fetal lung: ontogeny and effects of steroids. J Dev Physiol. 1992;18:233–236. [PubMed] [Google Scholar]
- 76. Noguchi A, Firsching K, Kursar JD, Reddy R. Developmental changes of tropoelastin synthesis by rat pulmonary fibroblasts and effects of dexamethasone. Pediatr Res. 1990;28:379–382. [DOI] [PubMed] [Google Scholar]
- 77. Pierce RA, Mariencheck WI, Sandefur S, Crouch EC, Parks WC. Glucocorticoids upregulate tropoelastin expression during late stages of fetal lung development. Am J Physiol. 1995;268(3 Pt 1):L491–L500. [DOI] [PubMed] [Google Scholar]
- 78. Wendel DP, Taylor DG, Albertine KH, Keating MT, Li DY. Impaired distal airway development in mice lacking elastin. Am J Respir Cell Mol Biol. 2000;23:320–326. [DOI] [PubMed] [Google Scholar]
- 79. Chinoy MR, Zgleszewski SE, Cilley RE, Krummel TM. Dexamethasone enhances ras-recision gene expression in cultured murine fetal lungs: role in development. Am J Physiol Lung Cell Mol Physiol. 2000;279:L312–L318. [DOI] [PubMed] [Google Scholar]
- 80. Mäki JM, Sormunen R, Lippo S, Kaarteenaho-Wiik R, Soininen R, Myllyharju J. Lysyl oxidase is essential for normal development and function of the respiratory system and for the integrity of elastic and collagen fibers in various tissues. Am J Pathol. 2005;167:927–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Yang Q, Ota K, Tian Y, et al. Cloning of rat fibrillin-2 cDNA and its role in branching morphogenesis of embryonic lung. Dev Biol. 1999;212:229–242. [DOI] [PubMed] [Google Scholar]
- 82. Zhao Y. Tenascin is expressed in the mesenchyme of the embryonic lung and down-regulated by dexamethasone in early organogenesis. Biochem Biophys Res Commun. 1999;263:597–602. [DOI] [PubMed] [Google Scholar]
- 83. Roth-Kleiner M, Hirsch E, Schittny JC. Fetal lungs of tenascin-C-deficient mice grow well, but branch poorly in organ culture. Am J Respir Cell Mol Biol. 2004;30:360–366. [DOI] [PubMed] [Google Scholar]
- 84. Zhang W, Menke DB, Jiang M, et al. Spatial-temporal targeting of lung-specific mesenchyme by a Tbx4 enhancer. BMC Biol. 2013;11:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Cole TJ, Mollard R. Selective glucocorticoid receptor ligands. Med Chem. 2007;3:494–506. [DOI] [PubMed] [Google Scholar]