

Abstract
Signaling via estrogen receptor (ER) occurs by interacting with many proteins. Nuclear interactome analysis of ERα in an embryo implantation model revealed the association of chicken tumor virus no. 10 regulator of kinase like (CrkL) with ERα, which was further validated by mammalian two-hybrid assay as well as coimmunoprecipitation and colocalization. Mutation in LPALL motif of CrkL disrupts the ERα-CrkL interaction and its transactivation potential, thereby suggesting that the interaction is mediated via its single ER binding motif, Leu-Pro-Ala-Leu-Leu (LXXLL) motif in the sarcoma homology (SH)2 domain. CrkL deletion constructs of SH2 domain target to the nucleus due to presence of nuclear localization signal. Interestingly, the SH2-SH3 (N terminal) construct shows an increased transactivation potential like CrkI. Weak interaction capability of mutated ERα-Y538F with CrkL validates that CrkL interacts with ERα via its YDLL motif at Tyr 541. In an attempt to understand the physiological relevance of this association, we investigated the impact on cell proliferation using a cancer model, because events associated in the process of pregnancy and cancer are analogous. Also, overexpression of CrkL is frequently associated with tumorigenesis. However, its significance in hormone-regulated cancers still remains obscure. Here, we demonstrate that association of ERα and CrkL directly enhances the tumorigenic potential of CrkL, thus pointing to its role in cell proliferation. In human endometrial cancers, we observed a strong association between ERα and CrkL levels. Thus, the molecular signaling set off by ERα and CrkL association may have a central role in pregnancy and cancer, two events which share parallels in growth, invasion, and immune tolerance.
Embryo implantation is an intricate process involving the activation of several signaling pathways of cell growth, cellular adhesion, and cytoskeletal restructuring. Protein-protein interactions are rife during this period, resulting in the establishment of an immunocompetent blastocyst in the uterus and thereby pregnancy. The whole process of implantation is well orchestrated by the ovarian hormones, estrogen and progesterone. An estrogen surge on a progesterone-primed uterus is crucial for successful implantation, because estrogen determines the duration of window of uterine receptivity (1). The effect of estrogen is mediated by estrogen receptor (ER)α in uterus (2). In a classical nucleus initiated steroid signaling cascade, estrogen binds one of the two specific, high affinity, ER (ERα or ERβ) located within target cells (3, 4). Upon ligand binding, the receptor undergoes a conformational change, leading to the displacement of heat-shock proteins, receptor dimerization, and subsequent interaction of ERα with specific estrogen response elements (ERE) located within the regulatory regions of target genes (5). The magnitude and the quality of the response to the estrogenic stimulus will depend on the cell-state at that point of time. The spiraling of the signal is dependent on the ability of the ER to recruit coactivator proteins (6), which directly or indirectly trigger chromatin remodeling, leading to opening of chromatin with greater accessibility to target gene promoter culminating in accelerated gene transcription. A large number of ERα-interacting proteins (coregulators), viz. p300/cAMP response element-binding protein-binding protein, members of the sarcoma (SRC) family, and proline, glutamic acid and leucine-rich protein 1, has been identified subsequently.
Chicken tumor virus no. 10 regulator of kinase like (CrkL), an Src homology (SH)2-SH3-SH3 adapter protein, is one of the major tyrosine phosphoproteins detected in cells from patients with chronic myelogenous leukemia (7). It belongs to the CRK family of adaptors and act as go-between protein in forming signaling cascades (8). CrkL is shown to be involved in numerous signaling pathways activated by T-cell activation (9), epidermal growth factor receptor activation (10), integrin cross-linking (11), and is involved in cascades leading to integrin-mediated adhesion (12). CrkL is reported to associate with signal transducer and activator of transcription (Stat)5b and function as a nuclear adaptor protein and thereby regulate gene transcription through DNA binding (13). The nuclear adaptor role of CrkL is further strengthened by our earlier report of rescue of DNA-binding ability of a mutated Stat5b associated with type 1 diabetes (14).
In a previous report, we have shown increased CrkL expression in uterus and embryo, and its estrogen triggered nuclear translocation during window of implantation (15). The presence of the LXXLL motif in CrkL enabled us to postulate it to be a coregulator of ERα (15), because most coactivators (cAMP response element-binding protein-binding protein/SRC1/receptor interacting protein 140) contain one or more LXXLL (L, leucine; X, any amino acid)-helix motifs required for interaction with the ligand-binding domain of nuclear receptors (NR) (16, 17). In the present study, we demonstrate that CrkL is an essential component in the action of ERα during the window of implantation. Consequently, we analyzed the importance of LXXLL motif in CrkL for its interaction with ERα by mutating the leucine residue of LXXLL domain with alanine by site-directed mutagenesis and subsequently assaying the interaction using far-Western blotting, coimmunopreciptation, and cotransfection techniques. We dissected the region in CrkL critical for the interaction with ERα by using CrkL deletion constructs. We proved that CrkL has a coactivator function by ERE reporter assay and analyzing the state of ERα downstream target genes. We also identify that the YDLL motif in ERα is critical for interaction with CrkL. We finally proved that the association of ERα and CrkL is evident in cancer and the molecular circuitry thereafter may involve increased proliferative and tumorigenic potential.
Results
Immunoprecipitation with ERα identifies CrkL as a component of its interactome
Because 17β-estradiol (E2) regulates high levels of uterine nuclear CrkL at d 5, 0500 h (15), we conducted immunoprecipitation with ERα antibody using the d 5 (0500 h) uterine nuclear extract. Immunoprecipitation assays with ERα (in duplicates) showed 12 bands as components of ERα nuclear interactome (Fig. 1A). ERα immunoprecipitates revealed a 66-kDa ERα positive band (band 5) (Fig. 1B) and a 37-kDa CrkL immunopositive band (band 9) (Fig. 1C and Supplemental Fig. 1A, i and ii, published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org).
Fig. 1.

ERα interacts with CrkL in vivo. A, Immunoprecipitation with ERα antibody (sc-542) using the nuclear extract of the periimplantation period. Twelve bands were obtained marked GB1–GB12. GB5 and GB9 correspond to ERα and CrkL, respectively. B, Western blot analysis with ERα immunoprecipitate using ERα antibody (sc-542) to prove the presence of ERα in the IP. C, ERα IP blot probed with CrkL (sc-319). D, Western blot analysis of ERα on ERα immunoprecipitate of different days of pregnancy using the ERα antibody and CrkL antibody. See also Supplemental Fig. 1. E, Mammalian two-hybrid assay of HEK cells transfected with pACT ERα and pBIND CrkL before and after estrogen treatment. The effect of estrogen and vectors (pACT ERα alone, pBIND CrkL alone, and pACT+ pBIND + G5Luc) on basal reporter activity is also shown. IP, Immunoprecipitate; WB, Western blot; GB, gel band.
Mascot sequence analysis of peptide fingerprints revealed that the 37-kDa CrkL immunopositive band (gel band) had a peptide fingerprint of CrkL_Mouse, Crk-like protein with a sequence coverage of 26%, whereas the 66-kDa ERα immunopositive band (gel band 5) had a peptide fingerprint of mouse ER (mER)α (Supplemental Fig. 1B). Immunoprecipitation with ERα during different days of pregnancy authenticated their interaction on late preimplantation (d 4, 1600 h) and periimplantation (d 5, 0500 h) period (Fig. 1D and Supplemental Fig. 1A, iii and iv).
Mammalian two-hybrid assay confirmed ERα-CrkL interaction
To evaluate the association between ERα and CrkL, we performed mammalian two-hybrid assays in HEK cells using the Checkmate mammalian two-hybrid system from Promega (Madison, WI). Consistent with results from the coimmunoprecipitation, pACT− ERα showed no transcriptional activity on its own or with the vector controls but strongly induced GAL4 luciferase reporter activity when cotransfected with pBIND-CrkL expression vector (Fig. 1E).
ERα and CrkL colocalize in mouse uterine sections
To establish the interaction of ERα with CrkL, colocalization studies were done in uterine sections during the different days of pregnancy. A perceptible increase in CrkL expression is paralleled with its colocalization with ERα at d 4, 1600 h, and d 5, 0500 h, as is clear from the yellow color in the overlay of CrkL and ERα (Fig. 2A). 4′,6-diamidino-2-phenylindole (DAPI) image completely superimposed with that of ERα and CrkL, thereby signifying that both the molecules get limited to the nucleus. Secondary alone controls for both Alexa Fluor 488 and Alexa Fluor 568 do not show any fluorescence.
Fig. 2.
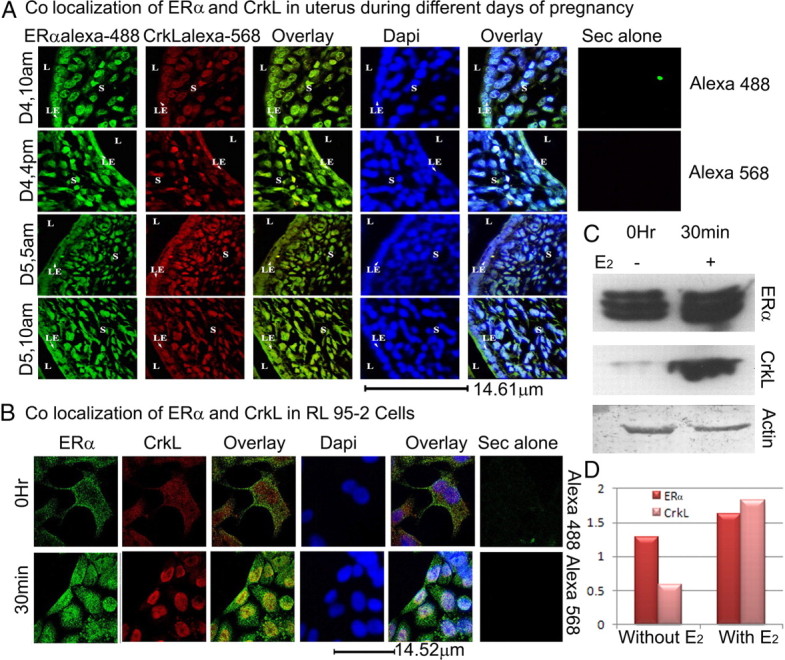
ERα and CrkL colocalize. A, Colocalization of ERα and CrkL in the uterine sections. Paraffin-embedded uterine sections of different days of pregnancy: preimplantation (d 4, 1000 h), late preimplantation (d 4, 1600 h), periimplantation (d 5, 0500 h), and postimplantation (d 5, 1000 h) periods were used. ERα probed with the primary antibody (sc-542) was labeled with antirabbit secondary Alexa Fluor 488, and CrkL probed with primary (sc-319) was labeled with Alexa Fluor 568. DAPI was used as the nuclear stain. Images were taken using the Leica TCS SP2 laser scanning confocal microscope. B, Colocalization of ERα and CrkL in RL95-2 cell line. Secondary alone controls for both Alexa Fluor 488 and Alexa Fluor 568 are shown. C, Western blot analyses of ERα and CrkL using the RL95-2 total cell extracts before and after estrogen treatment and its associated histogram (D). Actin is used as loading control.
Estrogen induces association of ERα-CrkL in human uterine epithelial cells
To test whether ERα and CrkL colocalize on estrogen treatment in human endometrium, we used a human uterine epithelial cell line, RL95-2, which expresses both estrogen and progesterone receptors (18) and is known to be estrogen responsive (19, 20). Monolayers of uterine epithelial RL95-2 cells mimic an important aspect of the in vivo situation and serve as an in vitro model for the adhesion competent receptive human uterine epithelium (21–24). At 0 h, both ERα and CrkL showed wide distribution in the cytoplasm and nucleus. But after 30 min of 1 nm E2 treatment, along with an increase in expression of ERα and CrkL, CrkL colocalized clearly with ERα in the nucleus (Fig. 2B). Secondary alone controls for both Alexa Fluor 488 and Alexa Fluor 568 do not show any fluorescence. The Western blot of ERα and CrkL using the total extracts from these two time points also revealed increased CrkL and ERα expression after the 30-min estrogen treatment (Fig. 2, C and D).
Estrogen receptor 1 expresses 5′ untranslated region (UTR) variants in the uterus
Amplification and sequencing of full-length coding sequence (CDS) of ERα from mouse uterine cDNA revealed that missing GCCGCCGCC patch near the N-terminal activation function (AF)1 region of the gene (Supplemental Fig. 2A) was responsible for the loss of three alanine residues in the protein (Supplemental Fig. 2B). This CDS was cloned into pDsRed Express C1 vector and pTYB11-N1 vector for further studies. Variation in 5′UTR region identified two 5′UTR variants in uterus (U1 and U2) (GenBank accession nos. EU791538 and EU791540).
CrkL and ERα interact via the LXXLL domain in CrkL in vitro
We have previously demonstrated the presence of a LXXLL motif (NR-box) in human and murine CrkL (15). To analyze the importance of this motif, we created mutant CrkL protein using wild-type CrkL in pTYB11 vector as template and mutated each leucine residue in LXXLL motif at position 81, 84, and 85 of CrkL with alanine creating L81A, L84A, and L85A mutant CrkL mutant proteins (as detailed in Supplemental Fig. 3, A–E). Far-Western blot analyses using purified proteins demonstrated an ERα immunopositive band with purified recombinant CrkL protein at 39 kDa, thus proving that CrkL and ERα interaction occurs in vitro and was direct (Fig. 3A). L81A CrkL mutant showed interaction with ERα similar to that of wild-type CrkL (Fig. 3A), whereas L84A and L85A mutants gave a very weak interaction signal (Fig. 3A) when the input was same (Fig. 3C). The specificity of the ERα antibody was confirmed using recombinant ERα. Stat5b interaction with CrkL was the positive control (Fig. 3B). Thus, our results indicate that L84 and L85 of CrkL are crucial for its interaction with ERα.
Fig. 3.

LXXLL motif of CrkL is crucial for its interaction with ERα in vitro. A, Far-Western blot analysis. CrkL and its mutants were used as prey, and ERα was used as the bait protein. BSA did not give any band, which confirms the specificity of the immunopositive reaction of ERα antibody. The mutants L84A and L85A did showed very weak interaction with ERα, whereas the L81A mutant CrkL showed interaction with ERα. B, As positive control, STAT5B was used as the prey and CrkL was used as bait. C, CrkL wild type and the three mutants L81A, L84A, and L85A, ERα and BSA were loaded on the SDS-PAGE to ensure that the protein input was same. See also Supplemental Fig. 3. WB, Western blot.
Assessment of role of LXXLL motif in mediating interaction of wild-type and mutant CrkL's with ERα using an in vivo model
To validate the interaction of ERα with CrkL and to substantiate the role of LXXLL motif in CrkL for estrogen-mediated interaction, cotransfection in HEK cell line with wild-type and mutant (L81A, L84A, and L85A) enhanced green fluorescent protein (EGFP)-mouse CrkL and mERα-DsRed was done (Fig. 4). Wild-type CrkL expression was limited to the cytoplasm of the cell in a spotted manner, and ERα was found in the nucleus (Fig. 4A). Upon estrogen treatment (1 nm) for 24 h, CrkL migrated to the nucleus and exhibits clear colocalization with ERα (Fig. 4B). Similarly, the cytoplasmic L81A-CrkL mutant shuttled into the nucleus and colocalized with ERα upon estrogen treatment pointing to a redundant role of the 81st leucine during ERα interaction. The L84A and L85A CrkL mutants on transfection along with ERα accorded a discrete theory. On cotransfection with ERα, both L84A and L85A CrkL mutants went into the nucleus and showed effective colocalization with ERα (Fig. 4A). Upon estrogen treatment, the mutant CrkL's displayed a spotted pattern and moved away from ERα (Fig. 4B). Differential spatiotemporal distribution of wild-type/mutated CrkL advocates that LXXLL motif in CrkL is vital for the subcellular localization of the molecule in the cell.
Fig. 4.
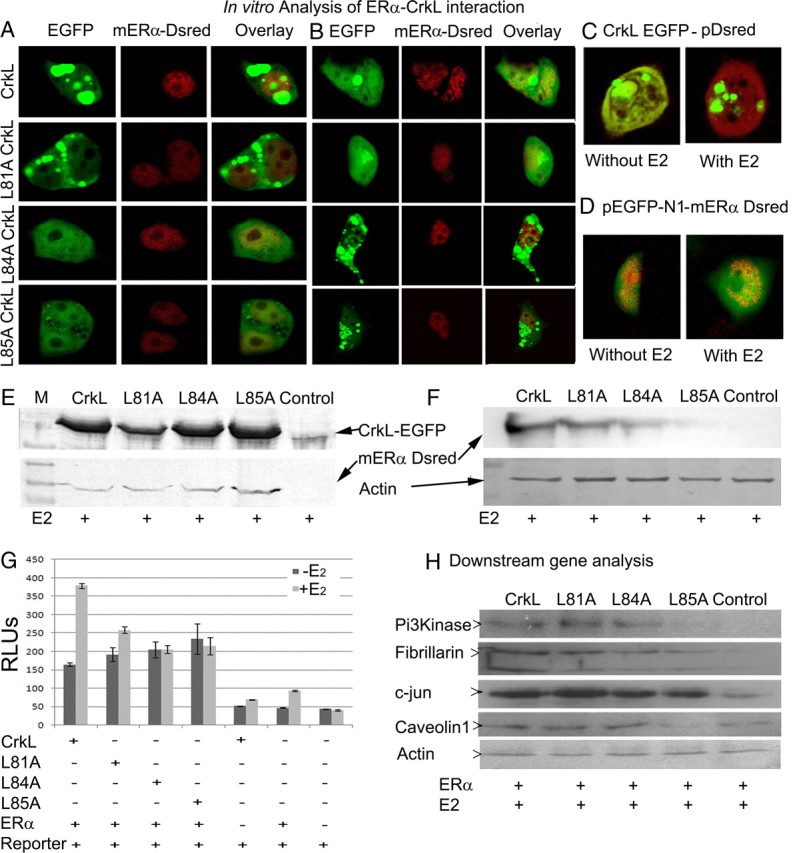
LXXLL motif of CrkL is key for transactivation potential of ERα in vivo. A, Cotransfection of mERα-DsRed with CrkL-EGFP, L81A, L84A, and L85A mutant CrkL-EGFP in HEK cell line. Cells were imaged using Leica SP2 confocal microscope. The panel shows localization of wild-type CrkL and ERα before estrogen treatment. CrkL is found in the cytoplasm, whereas ERα gets confined to the nucleus. The L81A mutant CrkL shows similar pattern of distribution, whereas the other mutants colocalize with ERα in the nucleus. B, After 24 h of estrogen (1 nm) treatment, wild-type CrkL colocalizes with ERα in the nucleus. L81A mutant shows similar pattern, which proves that the interaction of ERα with CrkL is not mediated by the 81st leucine. L84A and L85A show a spotty pattern after estrogen treatment and do not colocalize with ERα. The data affirm the far-Western blotting results. Control cotransfection with empty pDsRed-C1 vector (C) and CrkL-EGFP empty EGFP with mERα-DsRed (D) was done. E, Western blot analysis with GFP antibody to show the presence of fusion protein in the CrkL and mutant-transfected cell lines and with ERα antibody to show the presence of fusion protein in the transfected cell lines. F, IP with GFP antibody to show the pull down of ERα in the wild-type and mutant-transfected cell lines using the total cell extract. G, Reporter assay to show the efficiency of transactivation of ERα by wild-type and mutant CrkL (n = 6). H, Expression of ERα downstream genes in wild-type and mutant CrkL-transfected cells. See also Supplemental Fig. 2. RLU, Relative luminescence unit; M, protein marker.
Immunoprecipitation assay demonstrated that ERα interacts with CrkL and L81A CrkL-EGFP after estrogen (1 nm) treatment, whereas L84A showed weak interaction, and L85A CrkL-EGFP was unable to interact with ERα (Fig. 4F), thereby revalidating our far-Western blotting data. Together, the transfection data chime in with the far-Western blotting result, and all these portray CrkL as a new coregulator for ERα in the uterus during implantation.
CrkL modulates the transcriptional activity of ERα via its LXXLL motif
To evaluate whether CrkL is a coactivator or repressor of ER function, we tested the coactivator potential of CrkL by luciferase assay and downstream genes analysis. The assay showed a 2.3-fold increase in the ERE activity on treatment with 1 nm estrogen, in cells transfected with wild-type CrkL (P < 0.000001) in comparison with cells without estrogen treatment. Compared with the wild type, the mutant L81A CrkL showed less activity as is evident from a 1.35-fold increase (P < 0.003). The reporter activity did not change significantly in L84A and L85A mutants after E2 administration (Fig. 4G). Appropriate controls, viz. transfection with CrkL, ERα, and empty reporter vectors, show basal luciferase activity.
To identify the ability of CrkL as a coactivator of ERα and the impact of mutations in LXXLL domain, we performed Western blot of ERα-regulated genes in the uterus (25), viz. fibrillarin, phosphatidylinositol 3 kinase (PI3K), caveolin 1, and c-Jun. Transfection with CrkL (wild type) and L81A mutant showed increased expression of all the genes analyzed when compared with the control (ERα-transfected E2-treated cells). All groups were cotransfected with ERα to analyze the estrogen response. The L84A and L85A CrkL mutants-transfected cells show a decrease in the expression of these genes in comparison with Crkl-transfected cells (Fig. 4H). It is important to note that PI3K and caveolin 1 were absent in L85A-transfected cells when compared with wild type, and the levels were lower than that of control.
The SH2 and N-terminal SH3 [SH3(N)] domain of CrkL interacts with ERα and is important for its transcriptional activation potential
To scrutinize which region of CrkL is important for interaction with ERα deletion, constructs of CrkL were created (Fig. 5A) and cotransfected with full-length ERα in HEK cells (Fig. 5B). Estrogen treatment led to nuclear translocation of SH2-CrkL and SH2-SH3(N) and their colocalization with ERα (Fig. 5C), suggesting its potential interactions with ERα via its SH2 domain encompassing the LXXLL motif. The SH3(N) domain alone did not change after estrogen treatment, although it colocalizes with ERα in the nucleus (Fig. 5, B and C). The C-terminal SH3 [SH3(C)] domain transfection revealed its inability to be retained in the nucleus and thus confirms the finding of Smith et al. (26) that it contains a nuclear export signal.
Fig. 5.
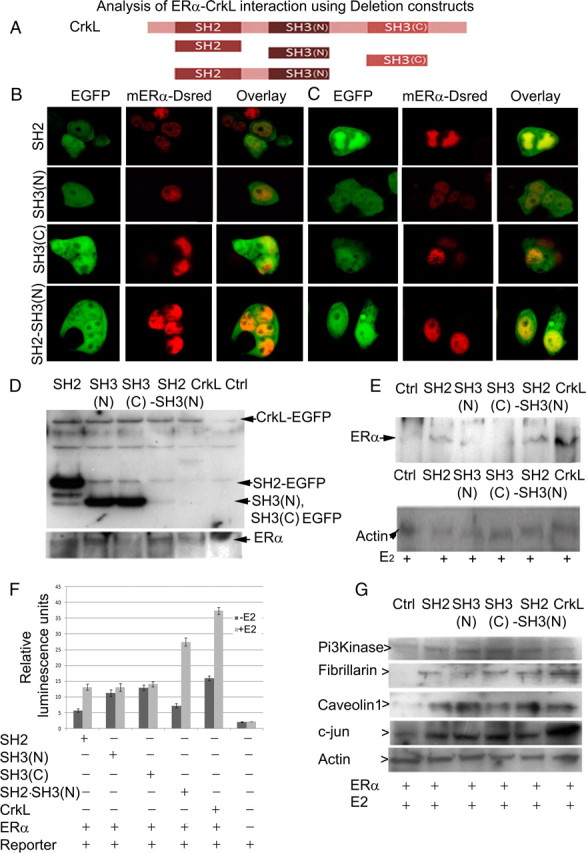
SH2 and SH3 domain of CrkL play an integral part in acquisition of transactivation potential of ERα. A, Schematic representation of CrkL deletion constructs used in experiments. B, Cotransfection of CrkL deletion constructs with mERα-DsRed to analyze which part of CrkL interacts with ERα. C, After 24 h of estrogen (1 nm) treatment, SH2 domain alone and in combination with SH3 translocates to the nucleus and colocalizes with ERα. SH3(N) domain shows no change before and after E2 treatment. SH3(C) domain showed its inability to be retained in nucleus when E2 was given. D, Western blot analysis with GFP antibody to show the presence of fusion protein in the CrkL and the CrkL deletion constructs' transfected cell lines and Western blot analysis with ERα antibody to show the presence of fusion protein in the transfected cell lines. E, IP with the GFP antibody to show the pull down of ERα in the wild type and deletion construct-transfected cell lines using total cell extract. F, Reporter assay showing ERE activity by different deletion constructs of CrkL. G, Western blot analysis of ERα downstream genes in ERα and CrkL deletion constructs' [Crk, SH2-CrkL, and SH2-SH3(N)-CrkL] cotransfected cell lines. IP, Immunoprecipitate; Ctrl, control.
Immunoprecipitation assay pulled down ERα as interacting partner in CrkL, SH2, and SH2-SH3(N) EGFP-transfected cell lines after (1 nm) estrogen treatment, restrengthening the importance of SH2 domain (Fig. 5, D and E). Surprisingly, SH3(N)-CrkL EGFP also pulled down ERα, and this is in agreement with our colocalizaton data, which showed a partial nuclear presence. SH3(C)-CrkL EGFP did not show any interaction with ER. Untransfected estrogen-treated HEK cells served as negative control.
Reporter assay with CrkL deletion constructs (Fig. 5F) validate that the SH2 region is important for estrogen-mediated interaction, as seen by a significant 2.30-fold increase (P < 0.000001) in luciferase activity after estrogen treatment. Estrogen-mediated transcriptional activity is further significantly potentiated by 3.81-fold (P < 0.0000001) in the presence of SH3 domain with SH2 [SH2-SH3(N)-CrkL], reaching levels almost equal to the activation with full-length CrkL. In contrast, the full-length CrkL showed a 2.34-fold (P < 0.0000001) increase in the activation of ERE similar to that with SH2 deletion construct (Fig. 5F). SH3(N) or SH3(C)-CrkL was unable to activate ERE after E2 induction, thus enabling us to speculate that they are not important in this activation by themselves. Downstream target genes of ERα revealed variable profile in cells transfected with deletion constructs (Fig. 5G). SH3(N)-transfected cells showed low fibrillarin expression, whereas SH3(C)-transfected cells had decreased caveolin 1 expression when compared with full-length CrkL and SH2-SH3(N)-transfected cells upon E2 treatment.
Tyrosine phosphorylation and CrkL-ERα interaction
Because the SH2 domain of CrkL is known to have a high affinity for tyrosine phosphorylated motif (pYXXP/L), we analyzed the presence of a YXXP/L motif in ERα. Both mERα and human ERα contain a conserved YXXP/L (YDLL) motif at Tyr 541 (mouse) and Tyr 538 (human) (Fig. 6A). Sodium orthovanadate (inhibitor of tyrosine phosphatase activity)-treated cells show improved detection of endogenous ERα-CrkL association (Fig. 6B). To further confirm the interaction, far-Western blot analysis with wild-type and mutated ERα, viz. ERY538F and CrkL, was done. Figure 6C shows the mutated sequence of ERα. CrkL was used as the bait protein, whereas ERα was the prey protein and was loaded onto gels. CrkL was also loaded on to the gel to show the antibody specificity The mutated ERα showed no band when compared with wild type (Fig. 6D), proving that ERα binds to CrkL via its AF2 domain through the YDLL motif (Fig. 6E).
Fig. 6.

Model showing CrkL-ERα interaction via CrkL's SH2 motif and ERα's SH2 binding motif (YXXP/L). A, Alignment showing the SH2 binding motif (YXXP/L) motif in ERα of mouse and human. B, Endogenous ERα coprecipitates with endogenous CrkL in orthovanadate-treated HEK cells. Cells were treated with 50 μm sodium orthovanadate and/or 10 nm E2 for 6 h as required. Cells were lysed, and lysates (∼1 mg of total protein per sample) were subjected to immunoprecipitation as detailed in Materials and Methods. Bound proteins were detected by immunoblotting with anti-ERα and CrkL antibody. C, Sequence of ERα showing the mutated tyrosine residue (ERY538F). D, Far-Western blot analysis using the wild-type and mutated ERα as prey. CrkL was used as the bait protein and BSA as negative control. Mutated ERα did not give a signal in the far-Western blot analysis. Lower panel shows ERα (wild type), ERα mutant (ERY538F), CrkL, and BSA loaded on the SDS-PAGE to ensure that the protein input was same. E, Cartoon representing motifs involved in ERα-CrkL interaction.
ERα and CrkL association potentiates transforming activity in MCF-7 cells
The role of CrkL as a potential oncogene to transform cells when over expressed made us to further investigate its ability when it is combined with ERα. Reports suggest that CrkL overexpressed cells can transform fibroblasts (27). MCF-7 cells were transfected with CrkL, ERα, and CrkL and ERα in combination and then plated individually onto soft agar. Cells transfected with vector alone served as control. Colonies were formed in CrkL alone and ERα alone plates. Increased colony formation efficiency was evident when ERα-CrkL was cotransfected when compared with the other two (Fig. 7, A and B). Control plate did not show any colonies. This again reiterates that presence of CrkL with ER enhances the proliferative signaling pathway.
Fig. 7.
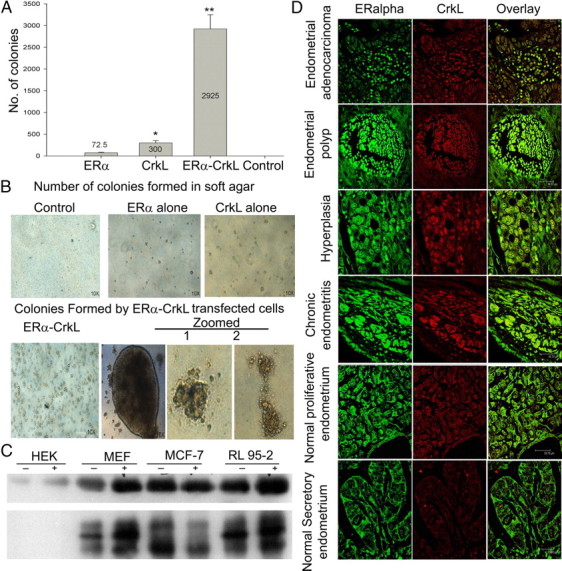
Association of ERα and CrkL in cancer and impact on tumorigenesis. A, MCF-7 cells transfected with ERα alone, CrkL alone, and ERα and CrkL in combination and vector alone were plated into soft agar, and colonies were counted after 3 wk. Error bars represent the average of two experiments. *, P < 0.02; **, P < 0.006. B, Photographs of colonies are shown after 2 wk. C, Western blot analysis of immunoprecipitation with ERα in different cell lines, viz. HEK, MEF, MCF-7, and RL-95-2, with and without estrogen treatment and probed with ERα and CrkL. D, Colocalization of ERα and CrkL in the endometrial cancer tissue array: representative sections of different stages. ERα probed with the primary antibody (sc-542) was labeled with antirabbit secondary Alexa Fluor 488, and CrkL probed with primary (sc-319) was labeled with Alexa Fluor 568. Images were taken using the Leica TCS SP2 laser scanning confocal microscope.
ERα and CrkL associate in endometrial cancer
We analyzed the association of ERα and CrkL with coimmunoprecipitation with ERα using total extracts from cell lines. CrkL was pulled down by ERα in cell lines, viz. MEF and RL95-2, thus showing a strong association in human endometrial cancer cell line (RL95-2) (Fig. 7C). There was no band in the case of CrkL in HEK total cell extract. These results pointed to the possible association of ERα and CrkL in endometrial cancer. Colocalization of ERα and CrkL in endometrial cancer tissue array (US Biomax, Inc., Rockville, MD) was also performed to understand the physiological relevance of the molecule. CrkL expression and colocalization with ERα was seen both in the cytosol and nucleus in endometrial polyp, hyperplasia, and chronic endometritis stages (Fig. 7D). Normal proliferative endometrium stage also showed similar pattern of ERα expression like hyperplasia but lacks the distinctive nuclear localization of ERα, suggesting that CrkL plays a prominent role in the transformation and proliferation of cancer tissue.
Discussion
In this study, we have established a role for CrkL in augmenting ER-mediated nucleus-initiated steroid signaling through its ability to act as a coactivator. We could undoubtedly demonstrate the physical interaction of CrkL with ERα by coimmunoprecipitation (with confirmation by peptide mass fingerprinting), mammalian two-hybrid assay and colocalization during the late pre- and periimplantation period in uterine sections, and far-Western blotting and cotransfection studies. These results are in agreement with earlier report from our lab of an increased CrkL expression and its nuclear translocation under estrogen stimulation (15). CrkL is involved in signaling pathways that regulate cellular adhesion through integrins (12). Cellular adhesion via integrins is crucial during embryo implantation and thus CrkL could play an important role during this process. All these data collate together to support the hypothesis that CrkL is a new interacting partner for ERα in the nucleus during embryo implantation.
The NR-coactivator interaction requires the NR-box motifs (LXXLL) within the coactivators, which give the ability to bind into the coactivator binding groove within the ligand-binding domain of the receptor (16, 28). SRC proteins bind directly to liganded NR using the NR-box (16, 29). A wide array of ER coregulators, which modulate ER at target promoters, harbor an LXXLL motif, viz. coactivator independent of AF-2 function (30), estrogen receptor-associated protein with a predicted molecular mass of 75 kDa (31), and metastasis-associated protein 1 (32, 33). We have previously demonstrated the presence of a LXXLL motif (NR-box) in human and murine CrkL (15). On further analyzing the primary amino acid sequence of various Crk for a similar motif, we found that all Crk except Drosophila Crk had a LXXLL motif (Supplemental Fig. 4A). Interestingly, CrkL (mouse, rat, and human) could be further classified as class I peptide containing a positively charged residue at the −1 position based on sequences flanking the core LXXLL motif, because it has a histidine (positive charge) at −1. Thus, CrkL can be classified as class I peptide containing a positively charged residue at the −1 position, placing it alongside glutamate receptor-interacting protein 1 and SRC1-type (p160s) cofactors, which have the charged residue lysine at position −1 (34, 35).
Far-Western blot and cotransfection analyses suggest that the LXXLL motif in CrkL is very important for the interaction of CrkL with ERα especially, the last two leucine residues in the LXXLL. Even a single mutation in the 2nd and 3rd leucine impairs CrkL-ERα interaction. LXXLL motif illustrates itself as the center for protein-protein interaction, the specificity of interaction being determined by the surrounding amino acids (28).
Our in vivo transfection results with LXXLL mutation revealed that LXXLL motif in CrkL contributes to the nuclear localization of the molecule. LXXLL mutations in CrkL influence their targeting in the cellular compartments. L84A and L85A CrkL go into the nucleus without any activation and upon estrogen treatment disassociates from ERα. This is in line with earlier observations that mutation in LXXLL motif might affect the localization of the molecule inside the cell. The interaction between dosage-sensitive sex reversal-adrenal hypoplasia congenita critical region on the X chromosome, gene 1 (Dax-1) and the adrenal 4 binding protein/steroidogenic factor 1 via the LXXLL motif of Dax-1 had been critical for targeting Dax-1 into the nucleus (36). The LXXLL motif in aryl hydrocarbon receptor is important for its subcellular localization and transcriptional activity (37). Our reporter assay and downstream gene product expression analysis clearly specify the importance of the 2nd and 3rd leucine of LXXLL motif of CrkL, whose mutation causes decrease in ERα-mediated transcriptional activation consistent with earlier reports of importance of LXXLL in transcriptional activation (37, 38).
Of special significance is the observation that transfection with CrkL-SH2 and SH2-SH3(N)-EGFP showed significant nuclear migration under estrogen treatment. This clear migration of SH2 domain alone or with SH3(N) can be explained by the presence of a monopartite NLS (Supplemental Fig. 4B) found by PSORT II (an online in silico analytic tool to predict the subcellular localization sites of proteins) analysis. The ability for estrogen response is obvious, because the LXXLL domain also resides in the SH2 domain of CrkL (Supplemental Fig. 4C). Our findings consolidate the hypothesis of the importance of the SH2 domain of CrkL in the interaction with mERα-DsRed, thereby potentiating transcriptional activity of ERα.
Reporter assays with SH2-SH3(N)-CrkL caused an extrapolation of the interaction and transactivation potential as evidenced by increased promoter activity in comparison with SH2 and SH3 alone. Because SH3(C) is linked through long loop region, it could probably interfere with the interaction of “LPALL,” thereby adding to the hypothesis that the main function of SH3(C) domain is to stabilize CrkL in a conformation that negatively regulates its biological function (39, 40). CrkI, which has high transforming activity, is a spliced variant of Crk comprising of SH2-SH3(N) only and lacks the SH3(C).
SH2 domains of proteins bind with the pY-X-X-L motif containing tyrosine-phosphorylated proteins (10, 41). Interestingly, the known site of mERα phosphorylation that conforms to a Crk-SH2 binding consensus (YXXP/L) appears to be conserved in human and others (Fig. 6A) and is present in the AF2 domain of ERα. Our results of loss of interaction with CrkL on mutation of Y538F signify its importance. It is interesting to note that Y537S mutation occurs in endometrial cancer (42) and Y537N (43) mutation is found in ER-negative metastatic breast cancer patient. It would be exciting to see how CrkL association is modified due to these mutations in human cancers.
The transition of Crk, initially identified as oncogene v-crk in 1980s to a family of adaptors available for their interaction with other proteins, thereby occupying a central position in networking signals has been quick. Earlier work suggested CrkL to be capable of modulating the DNA-binding ability of Stat5b (14). This report, leading to ordainment of CrkL with a coactivator function, opens up wide implications in biology. The differential nuclear ER signaling in various tissues under physiological and pathological conditions could be a consequence of the coregulators expressed in them. The relevance of CrkL as a coactivator would have far-reaching impact in disease biology as well, because dysregulation of Crk proteins is associated with human disease, including cancer, diabetes, and susceptibility to pathogen infection. Our results have provided the first evidence for regulation of endometrial cancer by virtue of ERα-CrkL association leading to increased tumorigenesis in vitro (Fig. 7, A and B) and increased risk of a highly proliferative state of endometrial cancers in (viz. endometrial polyp and chronic endometritis) (Fig. 7D). It is important to remember that uterine cancer is the fourth most common cancer among women. Thus, a better understanding of ERα-CrkL association may lead to more rational treatment strategies for human diseases associated with aberrant estrogen signaling. Needless to say that augmentation of ERα-triggered pathways due to ERα and CrkL association may have a defining role in pregnancy and cancer, two events that involve cell migration, adhesion, and invasion.
Materials and Methods
Reagents
Polyclonal antibodies against ERα (MC-20, sc-542), CrkL (C-20, sc-319), fibrillarin (sc-25397), PI3K (sc-1637), caveolin 1 (sc-894), c-Jun (sc-44), GFP(FL) (sc-8334), and goat antirabbit horseradish peroxidase (sc-2004) were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Goat antirabbit [F(ab)2] secondary antibody conjugated with Alexa Fluor 488 or 568 were from Invitrogen (Carlsbad, CA).
Animals
All experiments were performed with SWISS strain mice housed and bred in our animal facility. All animals were given food and water ad libitum and were housed in strict regimens of temperature (27 ± 1 C) and light (14 h light, 10 h dark). All animal protocols were approved by Institutional Animal Ethical Committee.
Pregnancy model
Regularly cycling, sexually mature, 3- to 4-month-old virgin female mice were housed with males of proven fertility, and mating was confirmed by the presence of the vaginal plug, the day after. The day of the vaginal plug was designated as d 1 of pregnancy. Females were killed by cervical dislocation at early preimplantation (d 4, 1000 h), late preimplantation (d 4, 1600 h), periimplantation (d 5, 0500 h), and late periimplantation (d 5, 1000 h) stages. The uteri were excised, washed with PBS (10 mm), and flushed for collecting embryos to determine the developmental stage of the embryos to stage the time of pregnancy.
Preparation of nuclear extract and pull-down assays
Nuclear extracts from the uteri of different days of pregnancy were prepared using NXTRACT nuclei extraction kit (Sigma, St. Louis, MO) according to manufacturer's instructions. The nuclear extract was precleared with protein A agarose and then incubated with primary antibody (1–5 μg for 200 μg of protein) on the Tarson Rotospin for 60 min. It was further incubated with protein A agarose at room temperature for 60 min and then centrifuged in Eppendorf 5820R centrifuge (Eppendorf, Hamburg, Germany) at 14,000 rpm at 4 C for 30 min. The pellet was denatured in Laemmli buffer, and proteins were separated electrophoretically by SDS-PAGE by the method of Laemmli (44). The gels were stained using coomassie, and the protein bands cut from the gel and processed as detailed in next section.
Preparation of samples for matrix-assisted laser desorption/ionization (MALDI)
The protein bands cut from the gel were digested using the Trypsin Profile in-gel digestion kit from Sigma as per the manufacturer's instruction. Briefly, the bands were first destained using the destaining solution twice by incubating the bands with 200 μl of the solution at 37 C for 30 min. The gel piece was then dried, and 20 μl of trypsin was added along with 50 μl of trypsin reaction buffer and incubated at 37 C for overnight. After the incubation, the liquid containing the tryptic peptides (solution I) was transferred to another tube; 50 μl of peptide extraction solution were added to the gel piece and incubated for 30 min at 37 C. This solution was mixed with solution I and was subjected to MALDI analysis using a MALDI-time of flight (Kratos, Chestnut Ridge, NY). The peptide fingerprints thus obtained were analyzed in Mascot and PepMAPPER sequence analysis softwares.
SDS-PAGE, Western blotting, and immunoreaction
Immunoprecipitated samples were denatured, separated by SDS-PAGE and transferred to sequiblot polyvinylidene fluoride (PVDF) (0.2 microns; Bio-Rad, Hercules, CA). Blots were developed with suitable primary and secondary antibodies using enhanced chemiluminescence plus Western blotting system of GE Healthcare (Princeton, NJ). Images were captured by Bio-Rad FluorS Multi Imager.
Colocalization studies in uterine sections
Paraffin-embedded uterine sections of different stages were processed as described earlier (45). The sections were double stained with ERα and CrkL using suitable primary antibodies. ERα was labeled with Alexa Fluor 488 and CrkL with Alexa Fluor 568. They were further processed for nuclear staining with DAPI (50 μg/ml) and mounted in glycerol. Results were observed and images acquired at room temperature using Leica TCS SP2 Confocal Laser Scanning Microscope (Leica Microsystems, Wetzlar, Germany). Each histological analysis was repeated three times.
Construction of vectors
Total RNA was prepared from mouse uteri using Trizol reagent from Sigma. Purity of total RNA was established by determining the 260/280 ratio and concentration adjusted (100 ng to 5 μg). cDNA from total RNA was prepared using Ready To Go T-primed first strand synthesis kit, which uses the Moloney murine leukemia virus reverse transcriptase and an oligo(dt)18 primer. All oligonucleotides were purchased from Sigma and Ocimum Biosolutions (Hyderabad, India).
Mouse uterine CrkL was PCR amplified and sequence obtained showed complete homology to the available GenBank sequence (accession no. NM_007764). The product was cloned into pEGFP-N1 vector (CLONTECH, Mountain View, CA) (enzymes, XhoI and SalI) using forward primer, 5′-CTCGAGATGTCCTCCGCCAGGTTTGATTCTTCAG-3′ and reverse primer, 5′-GTCGACCACTCGTTGTCATCGGGGTTCTGAG-3′.
For recombinant CrkL protein expression in pTYB11-N1 (New England Biolabs, Ipswich, MA), the cloning was done using forward primer, 5′-GGTGGTACTAGTATGTCCTCCGCCAGGTTTGATTCTTCAG-3′ and reverse primer, 5′-GGTGGTCTCGAGTCACTCGTTGTCATCGGGGTTCTGAG-3′.
CrkL deletion constructs in pEGFP-N1 were generated to identify specifically the interaction domain in CrkL. CrkL deletions include the SH2 domain (amino acids 12–94) created using primers SH2 forward, 5′-GGTGGTGTCGACATGGTCTGCCTGGTACATGGGGCC-3′ and SH2 reverse, 5′-GGTGGTGGATCCCGGTCCAGGTAGTGGATCTTGT-3′ and the two different SH3 domains SH3(N) (amino acids 126–182) [using primers SH3(N) forward, 5′-GGTGGTGTCGACATGGAATATGTACGGACCCTT-3′ and SH3(N) reverse, 5′-GGTGGTGGATCCCGCACAAGCTTTTCAACGTA-3′] and SH3(C) (amino acids 238–295) [using primers SH3(C) forward, 5′-GGTGGTGTCGACATGGTCTTTGCAAAAGCAATC-3′ and SH3(C) reverse, 5′-GGTGGTGGATCCCGGTCAAAGATTTTAACATG-3′].
Full-length ERα was amplified from mouse uterine cDNA using mERα_forward, 5′-AGCCTTGTGATGCCAGGAGAG-3′ and mERα_reverse, 5′ GCTCTCAGATCGTGTTGGGG 3′ and cloned into pDsRed Express-C1 vector (CLONTECH) using mERα_forward, 5′-GGTGGTGAATTCTATGACCATGACCCTCCACAC-3′ and mERα_ reverse, 5′-GGTGGTGGATCCTCAGACCGTGGCAGGGAAACCCTCT-3′. For recombinant mERα protein production, the full-length mERα was cloned into an expression vector pTYB11 (New England Biolabs) using mERα_forward, 5′-GGTGGTACTAGTATGACCATGACCCTCCACAC-3′ and mERα reverse, 5′-GGTGGTGAATTCTCAGACCGTGGCAGGGAAACCCTCT-3′.
Site-directed mutagenesis
The LXXLL motif was disrupted by alanine substitution using the GeneTailor site-directed mutagenesis kit (Invitrogen) as per manufacturer's instructions and primers designed using the PrimerX software. Each leucine residue in LXXLL motif at position 81, 84, and 85 of CrkL was mutated with alanine creating L81A, L84A, and L85A CrkL mutants. CrkL created earlier (14) in pTYB11 vector was used as template. CrkL L81A, CrkL L84A, and CrkL L85A mutants were created in pTyb11 for mutant CrkL protein production using the below mentioned primers.
L81A forward, 5′-GACCAGGAGTTTGACCATGCACCGGCCTTGTTA-3′
L81A reverse, 5′-ATGGTCAAACTCCTGGTCCCCGATCTTAAA-3′;
L84A forward, 5′-AGTTTGACCATTTGCCGGCCGCATTAGAGTTCT-3′
L84A reverse, 5′-GGCCGGCAAATGGTCAAACTCCTGGTCCCC-3′;
L85A forward, 5′-GACCATTTGCCGGCCTTGGCTGAGTTCTACAAG-3′
L85A reverse, 5′-CAAGGCCGGCAAATGGTCAAACTCCTGGTC-3′.
The mutated sequence in the vector was confirmed by sequencing (Fig. 3A). The different mutants were then amplified and subcloned into pEGFP-N1 vector generating CrkL L81A EGFP, CrkL L84A EGFP, and CrkL L85A EGFP.
Far-Western blot analysis
We used pTYB11 vector for recombinant protein production, in which the N terminus of the target protein is fused to an intein tag. The self-cleavage activity of the intein allows the release of the target protein from the chitin-bound intein tag, resulting in expression and purification of a native target protein without any vector-derived residues. Thus, such a recombinant protein would be more useful for interaction and functional studies. mERα protein was created by cloning in the CDS amplified above into pTYB11. The wild-type recombinant CrkL whose expression has been validated earlier (14), the three mutant CrkL recombinant proteins (L81A, L84A, and L85A CrkL) (Fig. 3, B and C), and mERα protein (Fig. 3, D and E) were purified using the IMPACT CN purification system and identity of mERα confirmed by Western blotting (Fig. 3E). Interaction of CrkL with ERα was analyzed in vitro by far-Western blotting technique (46, 47). Equal amounts (1 μg) of CrkL (wild type and mutant) were loaded onto SDS-PAGE gels and were subsequently blotted onto PVDF membrane. The membrane was subjected to a denaturation-renaturation using gradients of guanidine hydrochloride (6–0 m) and then blocked with 5% milk. The membrane was incubated with recombinant ERα for 4 h, and after washing with PBST, it was subjected to conventional immunoblotting protocol, including blocking, incubation in primary antibody against ERα, and suitable secondary antibody. STAT5B interaction with CrkL was used as the positive control (14). Far-Western blot analysis was also done using wild-type and mutated ERα (ERα wild type and ERαY538F) as prey and CrkL as bait protein. Antibody specificity was confirmed by loading recombinant CrkL in the far-Western blotting. BSA served as negative control.
Cell line
HEK cell line was maintained in the laboratory according to the American Type Culture Collection (Manassas, VA) and transient cotransfections with the CrkL-EGFP and mER-DsRed; L81A CrkL-EGFP, L84A CrkL-EGFP, and L85A CrkL-EGFP each with mERα-DsRed was done in 35-mm glass bottom culture dishes (In Vitro Scientific, Sunnyvale, CA) using Lipofectamine 2000 reagent (Invitrogen). Analysis of cotransfection was done using Leica Confocal microscopy after 2 d of incubation at 37 C in CO2 incubator. These transfected cell lines were treated with 1 nm E2 for 24 h and again imaged using the confocal microscope. Transient transfections were also done for CrkL deletion constructs and mERα-DsRed. HEK cells transfected with empty EGFP and mERα-DsRed and Crkl-EGFP, and empty DsRed served as the negative control. The live cell imaging was done at 37 C.
Pull-down assay with the transfected cells
Total cell extracts were prepared by using a high salt detergent buffer. Briefly, the cells were washed with PBS and resuspended in two-cell volume of Totex buffer containing 20 mm HEPES (pH 7.9), 350 mm Nacl, 20% glycerol, 1% Nonidet P-40, 1 mm MgCl2, 0.5 mm EDTA, 0.1 mm EGTA, 0.5 mm dithiothreitol, 0.1% phenylmethylsulfonylfluoride, and protease inhibitor cocktail (Roche, Indianapolis, IN). The cell lysate was incubated on ice for 30 min and centrifuged for 5 min at 14,000 rpm at 4 C. The protein content of the lysate was estimated, and equal amount of protein was loaded on to a 10% SDS-PAGE and blotted onto PVDF membrane. The membrane was checked for the fusion protein using the GFP antibody, and immunoprecipitation with GFP antibody was done to confirm the interaction.
Reporter assay
To substantiate the importance of CrkL as a potential coactivator dual luciferase assay was performed. We used Cignal ERE Reporter Assay kit from SABiosciences (Valencia, CA). The ERE reporter is a mixture of an inducible ER-responsive luciferase construct and a constitutively expressing renilla construct (40:1). The cells (1 million per well) were seeded in a 12-well tissue culture plate the day before transfection. The cells were then transfected with 100 ng of reporter plasmid, coactivator CrkL plasmid, and renilla. Each sample was done in triplicates. The CrkL mutants were transfected in the same manner. After transfection, the cells were incubated with 1 nm estrogen. The cells were lysed using the passive lysis buffer (dual luciferase assay kit; Promega) 24 h after transfection. Luciferase assays were performed with 20 μl of the cell extract and 100 μl of luciferase assay buffer. The enzyme activity was measured for 2 sec using a TD-20/20 luminometer.
Mammalian two-hybrid assay
ERα- CrkL interaction was confirmed using Checkmate Mammalian two-hybrid assays from Promega as per manufacturer's instructions. Briefly, CrkL was inserted in to BamHI-SalI site of the pBIND vector, which contains the yeast GAL4 DNA-binding domain. ERα was put in the pACT vector containing the herpes simplex virus VP16 activation domain. In addition, the pBIND vector expresses the Renilla reniformis luciferase, which was used for normalizing differences in transfection efficiency. The pG5luc vector contains five GAL4 binding sites upstream of a minimal TATA-box, which in turn is upstream of the firefly luciferase gene (luc+). The pGAL4 (pBIND-CrkL) and pVP16 (pACT-ERα) fusion constructs were transfected along with the pG5luc vector into HEK cells using lipofectamine 2000 (from Invitrogen). The cells were treated with 1 nm estrogen for 24 h, 2 d after transfection, were lysed and the amount of Renilla luciferase and firefly luciferase were quantitated using the Dual-Luciferase Reporter Assay System.
Soft agarose assay
MCF-7 cells transfected with ERα alone, CrkL alone, ERα-CrkL in combination, and the control vector alone were used for the experiment. Soft agar experiments were done according to the protocol developed in Flemington Lab. The cells were plated on to a thin layer of 0.7% agarose, respectively, at 1 × 106 cells per dish in duplicate in 10-cm2 dishes with the growth medium. The medium was replaced every 3 d. After 3 wk, the colonies were counted with a ×10 objective on a Nikon TS100 microscope (Nikon, Melville, NY).
Statistical analysis
For luciferase assays done in transfected cells, P values were obtained by applying a two-tailed, type 2 t test using Microsoft excel.
Confocal image acquisition
Results were observed and images acquired using Leica TCS SP2 Confocal Laser Scanning Microscope DMIRE2 using the objective lens hcxPLAPOlbd.BL63.0x1.40OI, ×63 magnification and numerical aperture 1.4 NA and Leica camera (DC350F) using acquisition software of Leica.
Acknowledgments
We thank technical assistance provided by Ms. Jiji V and Ms. Bindu for confocal imaging and Dr. Manoj P. Kumar of sequencing facility, Rajiv Gandhi Centre for Biotechnology, Thiruvananthapuram for infrastructural support. HEK cell lines were a gift from Dr. Ruby John Anto.
This work was supported by the Council of Scientific and Research Council, India Grant via sanction No. 37(1241)/05/EMR-II (December 16, 2005, to March 31, 2010) and Rajiv Gandhi Centre for Biotechnology core funds from the Department of Biotechnology (M.L.). A.P.R. was a junior research fellow and L.N. was a senior research fellow (Extended) in the grant. A.P.R. received a Junior Research fellowship from the University Grants Commission, New Delhi, India, via sanction no. 10-2(5)/2006(i)-E.U.II.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AF
- Activation function
- CDS
- coding sequence
- CrkL
- chicken tumor virus no. 10 regulator of kinase like
- DAPI
- 4′,6-diamidino-2-phenylindole
- Dax-1
- dosage-sensitive sex reversal-adrenal hypoplasia congenita critical region on the X chromosome, gene 1
- E2
- 17β-estradiol
- EGFP
- enhanced green fluorescent protein
- ER
- estrogen receptor
- ERE
- estrogen response element
- MALDI
- matrix-assisted laser desorption/ionization
- mER
- mouse ER
- NR
- nuclear receptor
- PI3K
- phosphatidylinositol 3 kinase
- PVDF
- polyvinylidene fluoride
- SH
- Src homology
- SH3(N)
- N-terminal SH3
- SRC
- sarcoma
- Stat
- signal transducer and activator of transcription
- UTR
- untranslated region.
References
- 1. Ma WG , Song H , Das SK , Paria BC , Dey SK. 2003. Estrogen is a critical determinant that specifies the duration of the window of uterine receptivity for implantation. Proc Natl Acad Sci USA 100:2963–2968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Couse JF , Lindzey J , Grandien K , Gustafsson JA , Korach KS. 1997. Tissue distribution and quantitative analysis of estrogen receptor-α (ERα) and estrogen receptor-β (ERβ) messenger ribonucleic acid in the wild-type and ERα-knockout mouse. Endocrinology 138:4613–4621 [DOI] [PubMed] [Google Scholar]
- 3. Couse JF , Korach KS. 1999. Estrogen receptor null mice: what have we learned and where will they lead us? Endocr Rev 20:358–417 [DOI] [PubMed] [Google Scholar]
- 4. Korach KS , Couse JF , Curtis SW , Washburn TF , Lindzey J , Kimbro KS , Eddy EM , Migliaccio S , Snedeker SM , Lubahn DB , Schomberg DW , Smith EP. 1996. Estrogen receptor gene disruption: molecular characterization and experimental and clinical phenotypes. Recent Prog Horm Res 51:159–186; discussion 186–188 [PubMed] [Google Scholar]
- 5. Grandien K , Berkenstam A , Gustafsson JA. 1997. The estrogen receptor gene: promoter organization and expression. Int J Biochem Cell Biol 29:1343–1369 [DOI] [PubMed] [Google Scholar]
- 6. Klinge CM. 2000. Estrogen receptor interaction with co-activators and co-repressors. Steroids 65:227–251 [DOI] [PubMed] [Google Scholar]
- 7. ten Hoeve J , Morris C , Heisterkamp N , Groffen J. 1993. Isolation and chromosomal localization of CRKL, a human crk-like gene. Oncogene 8:2469–2474 [PubMed] [Google Scholar]
- 8. Birge RB , Kalodimos C , Inagaki F , Tanaka S. 2009. Crk and CrkL adaptor proteins: networks for physiological and pathological signaling. Cell Commun Signal 7:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sawasdikosol S , Ravichandran KS , Lee KK , Chang JH , Burakoff SJ. 1995. Crk interacts with tyrosine-phosphorylated p116 upon T cell activation. J Biol Chem 270:2893–2896 [DOI] [PubMed] [Google Scholar]
- 10. Birge RB , Fajardo JE , Mayer BJ , Hanafusa H. 1992. Tyrosine-phosphorylated epidermal growth factor receptor and cellular p130 provide high affinity binding substrates to analyze Crk-phosphotyrosine-dependent interactions in vitro. J Biol Chem 267:10588–10595 [PubMed] [Google Scholar]
- 11. Sattler M , Salgia R , Shrikhande G , Verma S , Uemura N , Law SF , Golemis EA , Griffin JD. 1997. Differential signaling after β1 integrin ligation is mediated through binding of CRKL to p120(CBL) and p110(HEF1). J Biol Chem 272:14320–14326 [DOI] [PubMed] [Google Scholar]
- 12. Uemura N , Griffin JD. 1999. The adapter protein Crkl links Cbl to C3G after integrin ligation and enhances cell migration. J Biol Chem 274:37525–37532 [DOI] [PubMed] [Google Scholar]
- 13. Fish EN , Uddin S , Korkmaz M , Majchrzak B , Druker BJ , Platanias LC. 1999. Activation of a CrkL-stat5 signaling complex by type I interferons. J Biol Chem 274:571–573 [DOI] [PubMed] [Google Scholar]
- 14. Laloraya M , Davoodi-Semiromi A , Kumar GP , McDuffie M , She JX. 2006. Impaired Crkl expression contributes to the defective DNA binding of Stat5b in nonobese diabetic mice. Diabetes 55:734–741 [DOI] [PubMed] [Google Scholar]
- 15. Nautiyal J , Kumar PG , Laloraya M. 2004. 17β-estradiol induces nuclear translocation of CrkL at the window of embryo implantation. Biochem Biophys Res Commun 318:103–112 [DOI] [PubMed] [Google Scholar]
- 16. Heery DM , Kalkhoven E , Hoare S , Parker MG. 1997. A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature 387:733–736 [DOI] [PubMed] [Google Scholar]
- 17. Heery DM , Hoare S , Hussain S , Parker MG , Sheppard H. 2001. Core LXXLL motif sequences in CREB-binding protein, SRC1, and RIP140 define affinity and selectivity for steroid and retinoid receptors. J Biol Chem 276:6695–6702 [DOI] [PubMed] [Google Scholar]
- 18. Way DL , Grosso DS , Davis JR , Surwit EA , Christian CD. 1983. Characterization of a new human endometrial carcinoma (RL95-2) established in tissue culture. In Vitro 19:147–158 [DOI] [PubMed] [Google Scholar]
- 19. Wadehra M , Mainigi M , Morales SA , Rao RG , Gordon LK , Williams CJ , Braun J. 2008. Steroid hormone regulation of EMP2 expression and localization in the endometrium. Reprod Biol Endocrinol 6:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang JZ , O'Flatharta C , Harvey BJ , Thomas W. 2008. Membrane ERα-dependent activation of PKCα in endometrial cancer cells by estradiol. Steroids 73:1110–1122 [DOI] [PubMed] [Google Scholar]
- 21. Heneweer C , Kruse LH , Kindhäuser F , Schmidt M , Jakobs KH , Denker HW , Thie M. 2002. Adhesiveness of human uterine epithelial RL95-2 cells to trophoblast: rho protein regulation. Mol Hum Reprod 8:1014–1022 [DOI] [PubMed] [Google Scholar]
- 22. Hohn HP , Linke M , Denker HW. 2000. Adhesion of trophoblast to uterine epithelium as related to the state of trophoblast differentiation: in vitro studies using cell lines. Mol Reprod Dev 57:135–145 [DOI] [PubMed] [Google Scholar]
- 23. John NJ , Linke M , Denker HW. 1993. Quantitation of human choriocarcinoma spheroid attachment to uterine epithelial cell monolayers. In Vitro Cell Dev Biol Anim 29A:461–468 [PubMed] [Google Scholar]
- 24. Thie M , Denker HW. 2002. In vitro studies on endometrial adhesiveness for trophoblast: cellular dynamics in uterine epithelial cells. Cells Tissues Organs 172:237–252 [DOI] [PubMed] [Google Scholar]
- 25. Hong SH , Nah HY , Lee JY , Gye MC , Kim CH , Kim MK. 2004. Analysis of estrogen-regulated genes in mouse uterus using cDNA microarray and laser capture microdissection. J Endocrinol 181:157–167 [DOI] [PubMed] [Google Scholar]
- 26. Smith JJ , Richardson DA , Kopf J , Yoshida M , Hollingsworth RE , Kornbluth S. 2002. Apoptotic regulation by the Crk adapter protein mediated by interactions with Wee1 and Crm1/exportin. Mol Cell Biol 22:1412–1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Senechal K , Halpern J , Sawyers CL. 1996. The CRKL adaptor protein transforms fibroblasts and functions in transformation by the BCR-ABL oncogene. J Biol Chem 271:23255–23261 [DOI] [PubMed] [Google Scholar]
- 28. McInerney EM , Rose DW , Flynn SE , Westin S , Mullen TM , Krones A , Inostroza J , Torchia J , Nolte RT , Assa-Munt N , Milburn MV , Glass CK , Rosenfeld MG. 1998. Determinants of coactivator LXXLL motif specificity in nuclear receptor transcriptional activation. Genes Dev 12:3357–3368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Torchia J , Rose DW , Inostroza J , Kamei Y , Westin S , Glass CK , Rosenfeld MG. 1997. The transcriptional co-activator p/CIP binds CBP and mediates nuclear-receptor function. Nature 387:677–684 [DOI] [PubMed] [Google Scholar]
- 30. Sauvé F , McBroom LD , Gallant J , Moraitis AN , Labrie F , Giguère V. 2001. CIA, a novel estrogen receptor coactivator with a bifunctional nuclear receptor interacting determinant. Mol Cell Biol 21:343–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen M , Ni J , Zhang Y , Muyan M , Yeh S. 2008. ERAP75 functions as a coactivator to enhance estrogen receptor α transactivation in prostate stromal cells. Prostate 68:1273–1282 [DOI] [PubMed] [Google Scholar]
- 32. Kumar R , Wang RA , Mazumdar A , Talukder AH , Mandal M , Yang Z , Bagheri-Yarmand R , Sahin A , Hortobagyi G , Adam L , Barnes CJ , Vadlamudi RK. 2002. A naturally occurring MTA1 variant sequesters oestrogen receptor-α in the cytoplasm. Nature 418:654–657 [DOI] [PubMed] [Google Scholar]
- 33. Singh RR , Kaluarachchi K , Chen M , Rayala SK , Balasenthil S , Ma J , Kumar R. 2006. Solution structure and antiestrogenic activity of the unique C-terminal, NR-box motif-containing region of MTA1s. J Biol Chem 281:25612–25621 [DOI] [PubMed] [Google Scholar]
- 34. Chang C , Norris JD , Grøn H , Paige LA , Hamilton PT , Kenan DJ , Fowlkes D , McDonnell DP. 1999. Dissection of the LXXLL nuclear receptor-coactivator interaction motif using combinatorial peptide libraries: discovery of peptide antagonists of estrogen receptors α and β. Mol Cell Biol 19:8226–8239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gaillard S , Dwyer MA , McDonnell DP. 2007. Definition of the molecular basis for estrogen receptor-related receptor-α-cofactor interactions. Mol Endocrinol 21:62–76 [DOI] [PubMed] [Google Scholar]
- 36. Kawajiri K , Ikuta T , Suzuki T , Kusaka M , Muramatsu M , Fujieda K , Tachibana M , Morohashi K. 2003. Role of the LXXLL-motif and activation function 2 domain in subcellular localization of Dax-1 (dosage-sensitive sex reversal-adrenal hypoplasia congenita critical region on the X chromosome, gene 1). Mol Endocrinol 17:994–1004 [DOI] [PubMed] [Google Scholar]
- 37. Ikuta T , Watanabe J , Kawajiri K. 2002. Characterization of the LxxLL motif in the aryl hydrocarbon receptor: effects on subcellular localization and transcriptional activity. J Biochem 131:79–85 [DOI] [PubMed] [Google Scholar]
- 38. Litterst CM , Pfitzner E. 2002. An LXXLL motif in the transactivation domain of STAT6 mediates recruitment of NCoA-1/SRC-1. J Biol Chem 277:36052–36060 [DOI] [PubMed] [Google Scholar]
- 39. Muralidharan V , Dutta K , Cho J , Vila-Perello M , Raleigh DP , Cowburn D , Muir TW. 2006. Solution structure and folding characteristics of the C-terminal SH3 domain of c-Crk-II. Biochemistry 45:8874–8884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ogawa S , Toyoshima H , Kozutsumi H , Hagiwara K , Sakai R , Tanaka T , Hirano N , Mano H , Yazaki Y , Hirai H. 1994. The C-terminal SH3 domain of the mouse c-Crk protein negatively regulates tyrosine-phosphorylation of Crk associated p130 in rat 3Y1 cells. Oncogene 9:1669–1678 [PubMed] [Google Scholar]
- 41. Waksman G , Kumaran S , Lubman O. 2004. SH2 domains: role, structure and implications for molecular medicine. Expert Rev Mol Med 6:1–18 [DOI] [PubMed] [Google Scholar]
- 42. Kohler MF , Berkholz A , Risinger JI , Elbendary A , Boyd J , Berchuck A. 1995. Mutational analysis of the estrogen-receptor gene in endometrial carcinoma. Obstet Gynecol 86:33–37 [DOI] [PubMed] [Google Scholar]
- 43. Zhang QX , Borg A , Wolf DM , Oesterreich S , Fuqua SA. 1997. An estrogen receptor mutant with strong hormone-independent activity from a metastatic breast cancer. Cancer Res 57:1244–1249 [PubMed] [Google Scholar]
- 44. Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 45. Saxena D , Purohit SB , Kumer GP , Laloraya M. 2000. Increased appearance of inducible nitric oxide synthase in the uterus and embryo at implantation. Nitric Oxide 4:384–391 [DOI] [PubMed] [Google Scholar]
- 46. Guichet A , Copeland JW , Erdélyi M , Hlousek D , Závorszky P , Ho J , Brown S , Percival-Smith A , Krause HM , Ephrussi A. 1997. The nuclear receptor homologue Ftz-F1 and the homeodomain protein Ftz are mutually dependent cofactors. Nature 385:548–552 [DOI] [PubMed] [Google Scholar]
- 47. Schwartz CJ , Sampson HM , Hlousek D , Percival-Smith A , Copeland JW , Simmonds AJ , Krause HM. 2001. FTZ-factor1 and fushi tarazu interact via conserved nuclear receptor and coactivator motifs. EMBO J 20:510–519 [DOI] [PMC free article] [PubMed] [Google Scholar]