

A homozygous missense mutation results in gain-of-glycosylation for ALS and leads to loss-of-function in IGF complex formation.
Abstract
The acid-labile subunit (ALS) regulates IGF bioavailability by forming heterotrimeric complexes with IGFs and IGF-binding protein-3 (IGFBP-3). A homozygous missense mutation (D440N) resulting in undetectable circulating levels of ALS with a concomitant reduction in IGF-I and IGFBP-3 has been reported to cause mild growth retardation. To understand how this particular mutation affects ALS circulating levels and IGF-transport function, we expressed recombinant ALS and its variants, D440N-ALS, T442A-ALS, and D440N/T442A-ALS, using adenovirus vectors. Compared with wild-type ALS, the secretion of D440N-ALS was 80% lower. The D440N mutation was proposed to generate an N-glycosylation site additional to the seven existing motifs in ALS. D440N-ALS appeared larger than ALS, attributable to N-linked glycans because deglycosylation with N-glycosidase F reduced both proteins to the same molecular mass. When ALS was incubated with IGF-I and IGFBP-3, 70–80% of IGF-I was detected by gel-filtration chromatography in forms corresponding to the 150-kDa ternary complex. In contrast, when D440N-ALS was tested, less than 30% of IGF-I was found in high molecular mass complexes. Two other ALS variants mutated in the same putative glycosylation site, D440N/T442A-ALS and T442A-ALS, showed similar chromatographic profiles to wild-type ALS. The D440N mutation in ALS generates a hyperglycosylated form with impaired secretion and complex formation, potentially leading to dysregulation of endocrine IGF, thus contributing to the growth retardation observed in the affected patient. This is the first study to explain how a natural mutation, D440N, in ALS impairs its function.
The IGFs, IGF-I and IGF-II, circulate predominantly as ternary complexes that also include either IGF-binding protein (IGFBP)-3 or IGFBP-5, and the acid-labile subunit (ALS), a leucine-rich glycoprotein that contributes to the regulation of IGF bioavailability (1). We have previously shown that ALS prolongs the half-life of IGF-I-IGFBP-3 complexes in the circulation (2), and that IGF-I-induced hypoglycemia is blocked by the sequestration of IGF-I into these heterotrimeric complexes.
Biochemical studies of the protein-protein interactions within the complexes have demonstrated that high-affinity binding of ALS requires the IGF-IGFBP binary complex; that is, IGF and IGFBP individually have immeasurably low affinity for ALS (3). Mutation of the homologous basic domain in the C-terminal region of either IGFBP-3 (228KGRKR) or IGFBP-5(214RGRKR) is sufficient to reduce ALS affinity by 90%, while retaining near-normal affinity for IGF (4, 5). This is consistent with our observations that charge-charge interactions are important in the binding of ALS to IGF-IGFBP complexes (6, 7). However, it is not clear how ALS physically interacts with the IGF-IGFBP complexes because the structure of ALS is yet to be solved. The major structural feature of ALS is predicted to comprise the 20 leucine-rich repeats that form a torus based on computational modeling using the solved crystal structure of porcine ribonuclease inhibitor as a template (8). Interestingly, the model showed a cluster of acidic residues, resulting in a region of negative charges, on the internal face of ALS that could interact with the basic residues identified on IGFBP-3.
There are seven N-linked glycosylation motifs in the primary sequence of ALS, some or all of which are occupied by carbohydrates because ALS can be purified as at least two glycoforms from serum. We have previously shown that enzymatic removal of N-linked carbohydrates from ALS affected its ability to form complexes where the magnitude of the effect was correlated with the level of deglycosylation such that complete deglycosylation abolished function (9). Sialic acids contribute significant negative charges on glycoproteins, and removal of these moieties from ALS also decreased its affinity for complex formation by 50–80%.
Consistent with its role in prolonging the half-life of IGF-IGFBP complexes, there is a marked reduction in the serum levels of IGF and IGFBP-3 in the absence of ALS, as evidenced by ALS-knockout mice (10) and patients with an inactivating mutation in their Igfals gene (11). Despite the marked reduction in serum levels of IGF, the impact of ALS deletion on postnatal growth is only mild. To date, at least 14 different mutations resulting in missense, duplication or frameshift have been reported in human ALS deficiency. The first homozygous Igfals missense mutation reported (c.1318G→A) resulted in a D440N substitution in the prepeptide. In this study, we recreate the D440N mutation in human cell lines and show that it introduces a new functional N-glycosylation site resulting in hyperglycosylation that causes impairment in both the secretion of ALS, and its ability to sequester IGFs.
Results
To understand how the D440N mutation affects ALS function, recombinant E1-deleted adenoviruses were engineered to express either ALS or D440N-ALS. When E1- complementing 911 human embryonic retinoblast cells were infected by these viruses, wild-type ALS was readily detectable in cell media whereas D440N-ALS was undetectable (Fig. 1A). Expression of green fluorescent protein (GFP), under the control of a separate cytomegalovirus (CMV) promoter in the adenoviruses, was similar in cells infected with either ALS or D440N-ALS adenoviruses (data not shown), indicating that infection was successful. Furthermore, mRNA for ALS and D440N-ALS was readily detectable by semiquantitative RT-PCR (data not shown) suggesting that the lack of D440N-ALS protein was not related to transcriptional problems and that it might be related to the specific translation of D440N-ALS protein and/or its secretion. Immunoblot analysis of the lysates of adenovirus-infected cells showed detectable levels of intracellular D440N-ALS (Fig. 1B). Given the absence of detectable secreted D440N-ALS (Fig. 1A), this suggests a potential problem in the secretion of the mutant protein. Because ALS is predominantly liver derived, we also expressed ALS and D440N-ALS in HepG2 hepatoblastoma cells (Fig. 1C). The levels of intracellular GFP were used to normalize the relative levels of ALS and D440N-ALS to correct for virus infection efficiency. The ratio of secreted to intracellular protein levels indicated a severe impairment of secretion of D440N-ALS, relative to ALS, in both cell lines (Fig. 1D).
Fig. 1.

Expression of ALS and D440N-ALS. A, 911 retinoblastoma cells were infected with either recombinant wild-type ALS or D440N-ALS adenoviruses for 48 h. Samples of cell culture media (lanes 2–5) were separated on SDS-PAGE and immunoblotted for ALS. The samples are purified 20 ng recombinant ALS protein (rALS); 0.25% and 1% of total protein secreted by cells infected with ALS (WT); or D440N-ALS (MUT) adenoviruses. B, 911 retinoblastoma cells were infected with either recombinant wild-type ALS or D440N-ALS adenoviruses for 48 h. Samples of cell lysates were separated on SDS-PAGE and immunoblotted for ALS. The samples are purified 20 ng recombinant ALS protein (rALS); 5% of lysates of cells infected with ALS (WT); or D440N-ALS (MUT) adenoviruses. C, HepG2 hepatoma cells were infected with vector (VEC), recombinant wild-type ALS (WT) or D440N-ALS (MUT) adenoviruses for 48 h. One percent of total secreted protein (cell media) and 6% of lysates (cell lysate) from the cells were separated on SDS-PAGE and immunoblotted for ALS. The panel below shows the protein levels of GFP in the cell lysates. D, Densitometric scan of the immunoblots in A, B, and C and shown as the ratio of secreted to intracellular protein levels for wild-type ALS (WT; black bars) and D440N-ALS (MUT; gray bars), expressed relative to wild-type ALS, in 911 and HepG2 cells, respectively. Data are mean ± sd for two separate experiments in each cell-line.
The D440N mutation results in the generation of a consensus motif for N-linked glycosylation (12) and notably, the secreted D440N-ALS protein appears to have increased molecular mass relative to the wild-type ALS (Fig. 1C). ALS has seven putative N-glycosylation sites and although carbohydrate occupancy at each site has not been confirmed, ALS is normally heavily glycosylated and predominantly circulates as two glycoforms of 84–86 kDa (9, 13). The apparent difference in molecular mass between ALS and D440N-ALS was no longer present when the N-linked carbohydrates were removed from the proteins by glycosidase treatment (Fig. 2A), with both proteins reduced to approximately 60 kDa, consistent with the molecular mass of 63,150 predicted for the amino acid sequence of ALS. This suggests that the additional N-glycosylation site resulting from the D440N mutation is used for glycan attachment. As further support, we found that in the presence of tunicamycin, which inhibits the glycosylation of newly synthesized proteins, cells infected with ALS or D440N-ALS adenoviruses expressed ALS proteins of similar molecular mass (Fig. 2B). Treatment of cells with tunicamycin resulted in an increased accumulation of both ALS and D440N-ALS relative to their untreated controls. Tunicamycin has previously been reported to affect the secretion of some proteins like immunoglobulins (14), but it is unclear whether the decreased secretion of D440N-ALS results solely from the lack of glycosylation per se, or from the endoplasmic reticulum stress subsequent to protein misfolding.
Fig. 2.

Deglycosylation of ALS and D440N-ALS. A, HepG2 hepatoma cells were infected with either recombinant wild-type ALS (WT) or D440N-ALS (MUT) adenoviruses for 48 h. Samples of culture media were either untreated (−) or treated (+) with N-glycosidase F (PNGase F) for 16 h before separation on SDS-PAGE and immunoblotted for ALS. B, HepG2 hepatoma cells were either untreated (lanes 2, 6, and 10), or treated with 1 μm (lanes 3, 7, and 11), 2.5 μm (lanes 4, 8, and 12) and 5 μm (lanes 5, 9, and 13) of tunicamycin for 2 h before infection with vector (VEC; lanes 2–5), recombinant wild-type ALS (WT; lanes 6–9) or D440N-ALS (MUT; lanes 10–13) adenoviruses for 18 h. The viruses were removed, and tunicamycin treatment was continued for another 8 h before cell lysates were processed for immunoblotting. As a control, 10 ng of purified recombinant wild-type ALS protein (rALS) were loaded in lane 1.
To investigate the effect of the mutation on ALS function, an in vitro ternary complex formation assay was employed. Due to the poor secretion of D440N-ALS, it was not feasible to purify the secreted protein and instead, the cell culture media were concentrated and then dialyzed extensively to remove excess salts. To ensure that the composition of the cell culture medium containing wild-type ALS was similar to that of D440N-ALS, it was initially diluted with medium derived from cells infected with a control adenovirus, before concentration and dialysis in the same way. When [125I]IGF-I and IGFBP-3 were incubated with culture media containing wild-type ALS and then separated by size exclusion chromatography, the majority (70–80%) of [125I]IGF-I was found in the 150-kDa fraction, indicating that it was complexed to IGFBP-3 and ALS (Fig. 3). In contrast, [125I]IGF-I was mainly found in the 50-kDa fraction in the presence of medium containing D440N-ALS, with less than 30% corresponding to the high molecular weight complex form. The chromatographic profile was identical to that seen in the absence of any ALS, indicating that D440N-ALS was unable to bind to the 50-kDa [125I]IGF-I-IGFBP-3 binary complexes.
Fig. 3.

Ternary complex formation by ALS and D440N-ALS. Culture media from HepG2 cells infected with adenoviruses, containing 3 pmol of either recombinant wild-type ALS (triangles) or D440N-ALS (circles) estimated by immunoblot, were preincubated with 1 pmol of IGFBP-3 and 100,000 cpm of [125I]IGF-I before fractionation on a Superose-12 column. The amount of radioactivity was determined in each fraction. As a control, a similar reaction was performed with an equal volume of culture media from cells infected with empty vector virus (squares).
We hypothesized that the inability of the mutant protein to form ternary complexes is due to the additional carbohydrate potentially obscuring the IGFBP-3 binding site on ALS. However, the D440N mutation also replaces an acidic residue that contributes toward the electronegatively charged surface on ALS that we have proposed as the IGFBP-3 binding site (8). To test whether the mutation of the primary sequence, or the posttranslational modification resulting from that mutation, was the cause of impaired secretion and complex formation, we generated two additional ALS mutant proteins: D440N/T442A-ALS, which results in the loss of the acidic aspartate at residue 440 but without the generation of an N-glycosylation motif, and T442A-ALS to be used as a control. It was not possible to produce a mutant to test the effect of glycosylation in the absence of D440 substitution because the asparagine residue is obligatory in the consensus N-glycosylation motif, N-X-S/T. Both D440N/T442A-ALS and T442A-ALS migrated on SDS-PAGE as similarly sized proteins to wild-type ALS (Fig. 4), indicating that neither protein has increased glycosylation. Both D440N-ALS and D440N/T442A-ALS showed reduced secreted levels compared with ALS and T442A-ALS (Fig. 4A), thus indicating that the loss of an acidic aspartate residue contributes to poor secretion even in the absence of additional glycosylation. The poor yield of secreted protein was not due to increased proteolysis because it was not improved in the presence of broad-spectrum protease inhibitors (data not shown). Proportionally more protein was found intracellularly for all three mutant ALS compared with wild-type ALS (Fig. 4B). The ratio of intracellular to secreted protein levels indicated significant accumulation of intracellular D440N-ALS and D440N/T442A-ALS, relative to ALS (P ≤ 0.0001; Fig. 4D). Interestingly, the ratio for D440N-ALS was approximately 2-fold more than that for D440N/T442A-ALS (P = 0.0002), suggesting that the additional glycosylation as a result of the D440N mutation is also a contributing factor in the poor secretion and increased intracellular pool of D440N-ALS.
Fig. 4.
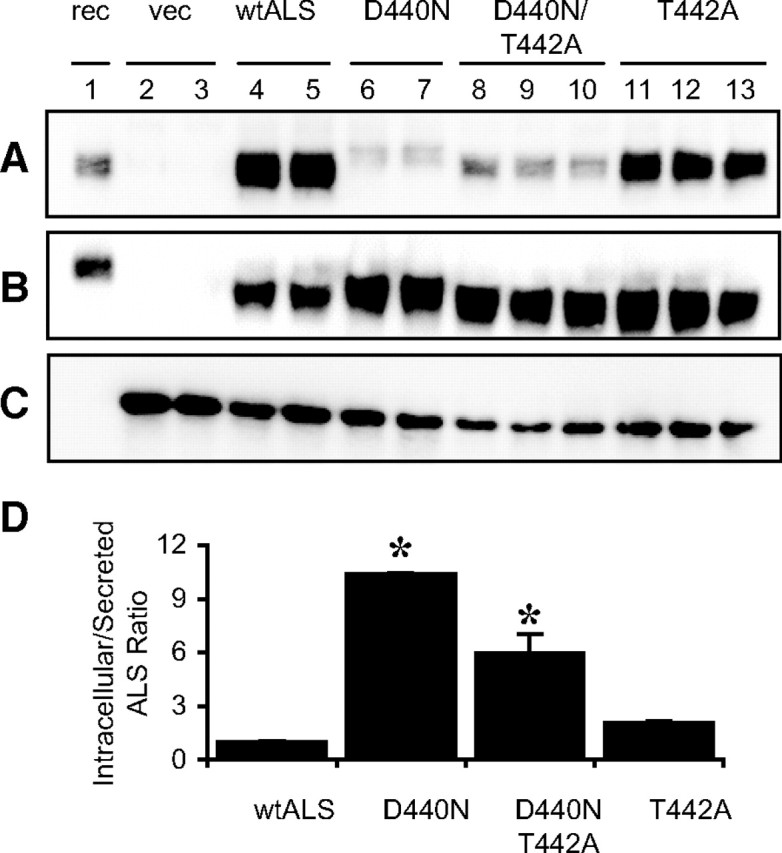
Expression of ALS analogs. HepG2 cells were infected with vector (vec; lanes 2 and 3), recombinant wild-type ALS (wtALS; lanes 4 and 5), D440N-ALS (lanes 6 and 7), D440N/T442A-ALS (lanes 8–10), or T442A-ALS (lanes 11–13) adenoviruses for 24 h. Samples of culture media (panel A) or cell lysates (panel B) were separated on SDS-PAGE and immunoblotted for ALS. Samples of cell lysates were also immunoblotted for GFP as a marker of infection and protein expression efficiency (panel C). As a control, 10 ng of purified recombinant wild-type ALS protein (rec) was loaded in lane 1. D, Densitometric scan of the immunoblots above. ALS levels are normalized to GFP levels and expressed as the ratio of intracellular/secreted levels. The data shown are mean ± sd expressed relative to recombinant wild-type ALS from at least two separate experiments. *, P ≤ 0.0001 compared with wild-type ALS.
Using the same protocol as in Fig. 3, we compared the ability of D440N/T442A-ALS and T442A-ALS to that of D440N-ALS and wild-type ALS to form ternary complexes with IGFBP-3 and [125I]IGF-I. In contrast to D440N-ALS, the other two mutants showed similar chromatographic profiles to wild-type ALS, where approximately 70–80% of [125I]IGF-I was found in the 150-kDa fraction (Fig. 5). Because the additional N-glycosylation motif is not present in D440N/T442A-ALS, we conclude that the additional Asn-linked glycosylation on D440N-ALS, and not the substitution of Asp with Asn, is the main factor influencing its ability to form ternary complexes with IGFBP-3 and IGF-I. We propose that, as a result of its impaired ternary complex formation, any D440N-ALS that reaches the circulation might be cleared rapidly. This could not be tested directly because the poor secretion of the mutant ALS in vitro prevented its purification. However, Supplemental Fig. 1 published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org demonstrates that iv administration of uncomplexed wild-type human ALS in GH-deficient spontaneous dwarf rats is cleared more rapidly than when injected as complexed forms with IGF-I and IGFBP-3, implying that ternary complex formation affects ALS stability in the circulation.
Fig. 5.
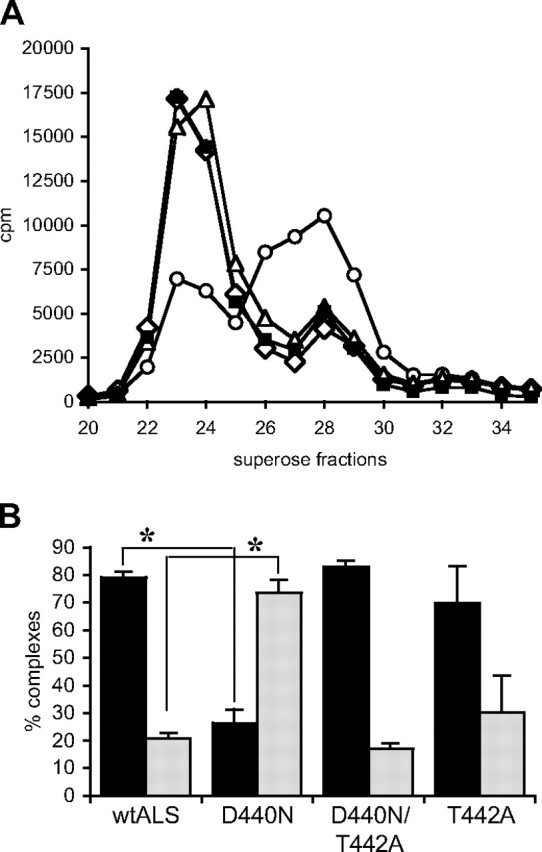
Ternary complex formation by ALS analogs. A, Culture media from HepG2 cells infected with adenoviruses, containing 3 pmol of recombinant wild-type ALS (solid squares), D440N-ALS (open circles), D440N/T442A-ALS (open diamonds), and T442A-ALS (open triangles) estimated by immunoblot, were preincubated with 1 pmol of IGFBP-3 and 100,000 cpm of [125I]IGF-I before fractionation on a Superose-12 column. The amount of radioactivity was determined in each fraction. B, The relative proportion of [125I]IGF-I in the peaks representing ternary (fractions 22–27; black bars) and binary (fractions 27–30; gray bars) complexes, from two separate experiments, is shown as mean ± sd. *, P = 0.002. wtALS, Wild-type ALS.
Discussion
The IGF system controls an evolutionarily conserved signaling pathway fundamental to normal embryonic and fetal development, cellular and somatic growth, and metabolic regulation. IGFs form heterotrimeric complexes in the circulation by binding to either of two IGFBPs, IGFBP-3 and IGFBP-5, and a liver-derived glycoprotein, ALS (3, 7). The function of ALS is to stabilize these heterotrimeric complexes and prolong their half-life in circulation. Consistent with this, mice lacking both igfals alleles have greatly reduced IGFBP-3 and IGF-I levels, presumably due to increased clearance (10) but surprisingly, these ALS knockout mice only showed mild postnatal growth retardation. Similarly, mutations in the human IGFALS gene that result in total loss of circulating ALS also cause severe deficiencies in serum IGF-I and IGFBP-3 levels, but patients harboring these mutations exhibit only mild reduction in growth and delayed puberty, as well as insulin resistance that was not observed in the ALS knockout mice (12, 15–20). To date, 14 different mutations of the human IGFALS gene have been identified in 17 patients (11), all of whom presented clinically with short stature. These mutations resulted in profound deficiency of circulating ALS and, consequently, reduced levels of IGF-I and IGFBP-3 as well. When the sera of approximately half of these patients were incubated with radiolabeled IGF-I, the radiolabeled IGF-I was either sequestered in 50-kDa complexes, presumably by IGFBPs, or remained unbound, thus indicating that there was no functional serum ALS present.
Approximately two thirds of the ALS protein structure is made up of 20 repeating domains of 24 amino acids, all of which contain the consensus motif for the leucine-rich repeat (LRR) protein superfamily. Cysteine-rich amino- and carboxyl-terminal domains flank the LRR central domain. Of the 14 reported mutations in IGFALS, four resulted in frameshifts and premature stop codons that would produce truncated proteins that are plausibly nonfunctional. Six other mutations involved conserved Leu or Asn residues of the LRR, and a missense mutation each in the flanking amino- and carboxyl-terminal regions involved Cys residues that would conceivably impact on the protein structure. Of the remaining two missense mutations involving nonconserved residues in the LRR domain, D440N and P73L, we have now shown that N-linked glycans occupy the N-glycosylation site introduced by the D440N mutation.
An intriguing aspect of this study is that the additional carbohydrate had a profound inhibitory effect on ALS complex formation with IGFBP-3 and IGF-I. Because we observed aberrant glycosylation in mutant ALS expressed in two different human cell lines, including a liver-derived line, we assume that our results reflect the form and function of the mutant ALS in the patient harboring the same IGFALS mutation. However, this cannot be confirmed with absolute certainty because no ALS was detectable in the patient. Our previous studies into the role of carbohydrates in ALS function showed clearly that the loss of carbohydrate, whether by enzymatic removal or by mutation of each of the seven naturally occurring N-glycosylation sites, also caused loss of ALS complex formation function (9). It is difficult to reconcile how both addition and loss of carbohydrates can result in the impairment of complex formation. According to the toroidal structure of the central LRR domain of ALS that was modeled based on homology to the porcine ribonuclease inhibitor, six of the seven N-glycosylation sites are located near the junction of LRR1 and LRR20 and the N- and C-terminal domains (8). It was suggested that such clustering of N-linked carbohydrates may result in a concentration of negatively charged sialic acid moieties, the loss of which was shown to affect ternary complex formation (9). The isolated seventh site is located in LRR13, distal to the LRR1 and LRR20 junction, and is predicted to face directly into the lumen of the torus. Asp440 is predicted to be located in LRR16 on the surface of the ALS molecule, and it is possible that the N-linked glycans in the D440N mutant obscure the binding site on ALS for the IGF- IGFBP binary complexes. When the N-linked glycans are absent from Asn440, as in the D440N/T442A-ALS mutant, ternary complex formation activity is restored to a level similar to that of wild-type ALS. The lack of effect of the D440N/T442A mutation on ALS binding activity suggests that Asp440 is not an essential component of the binding site.
Mutation of Asp440 to Asn, with or without the addition of N-linked glycans, resulted in impaired secretion and intracellular accumulation of the protein. Increased glycosylation may result in impaired secretion due to altered folding of the protein but generally, such misfolded protein will be targeted for degradation (21). Our data suggest that the lack of circulating ALS in the patient harboring the D440N mutation (12) results, in part, from poor secretion of the mutant protein by hepatocytes. Further, any secreted D440N-ALS protein would potentially be cleared from the circulation rapidly because we have shown that it fails to form ternary complexes. In support of this concept, Supplemental Fig. 1. suggests that uncomplexed ALS has a shorter circulating half-life than complexed ALS when injected into spontaneous dwarf rats that are totally GH deficient. It is interesting to note that when human ALS was injected alone into partially GH-deficient rats, its clearance kinetics were no different than when coinjected with IGF-I and IGFBP-3 (22). The partially GH-deficient rats had higher endogenous levels of IGF-I, IGFBP-3, and ALS (∼30–40% of normal levels) than the totally GH-deficient rats (<10% of normal), that would influence the proportion of human ALS in complexes and, hence, its circulating half-life. In the present study, measurement of in vivo clearance rates of D440N-ALS was not feasible due to the low production levels of the mutant protein. Nevertheless, we may conclude that in the patient with the D440N mutation, clearance of any mutant protein from the circulation occurred at least as rapidly as its secretion into the circulation, because no ALS was detectable.
Congenital disorders of glycosylation (CDG) are frequently associated with the loss of N- or O-glycosylation due to mutations in the genes coding for the enzymes involved in the glycosylation process, but mutations that lead to the loss of glycosylation in specific proteins have also been reported (23). The poor nutritional status of CDG patients could potentially impact on the GH-IGF axis, and studies have shown that although serum levels of the GH-dependent components of the IGF ternary complex are decreased, GH levels are either normal or elevated (24, 25), consistent with a state of GH resistance. In one such study, impaired glycosylation of ALS in children with CDG was associated with decreased circulating levels of IGF-I, IGF-II, IGFBP-3, and ALS (25). Oral mannose therapy of one child increased ALS glycosylation and improved growth velocity, raising the possibility that ALS glycosylation as well as nutritional status may influence GH sensitivity. Mutations involving gain-of-glycosylation for specific proteins are rare, and the pathogenic role of the new glycan is not well studied (26). Vogt et al. (27) described a missense mutation that introduced a new N-glycosylation motif in the interferon-γR2 chain that resulted in loss of function. It was shown biochemically that the loss of function was attributable to the new glycan chain because function was restored by the use of chemical glycosylation inhibitors.
Similarly, in the first study to explore biochemically how a natural ALS mutation affects its biological function, we have now shown that the D440N mutation in human ALS leads to hyperglycosylation that causes loss of function of the protein at two levels: decreased cell secretion resulting from the Asp-Asn substitution and impaired ternary complex formation resulting from the added glycan. We conclude that the moderate growth retardation associated with this particular mutation (12) is likely to be due to the lack of ternary complex formation and consequent dysregulation of endocrine IGFs.
Materials and Methods
Materials
Recombinant IGF-I was a gift from Tercica, Inc. (Brisbane, CA), and recombinant IGFBP-3 was expressed and purified in house as described previously (28). Anti-ALS antiserum is a goat polyclonal antibody raised against C-terminal residues 551–578 of human ALS (Diagnostic Systems Laboratories, Inc., Webster, TX). It reacts with both wild-type ALS and D440N-ALS. The anti-GFP antibody is a mouse monoclonal raised against recombinant GFP (Roche Applied Science, Mannheim, Germany). Horseradish peroxidase-conjugated antigoat and antimouse secondary antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA) and GE Healthcare Life Sciences Australia (Rydalmere, New South Wales, Australia), respectively.
Expression of recombinant wild-type and mutant ALS
The adenoviral-mediated expression system, AdEasy-1 (29), was used for the heterologous expression of ALS protein. Human ALS cDNA (30) was cloned into pAdTrack-CMV and used in homologous recombination with pAdEasy-1 to generate recombinant ALS adenoviruses. The mutations, D440N, D440N/T442A, and T442A, were introduced by site-directed mutagenesis using oligonucleotides (5′-GAGCTGCTGGAGCTCAACCTGGACTCCAC-CAG, 5′-GAGCTGCTGGAGCTCAACCTGGCCTCCAACCAG, and 5′-GAGCTGCTGGAGCTCG-ACCTGGCCTCCACCAG, respectively) and pAdTrack-CMV-ALS as the mutagenesis vector, essentially as described previously (4). Recombinant ALS or vector adenoviruses were used to infect either 911 human embryonic retinoblast or HepG2 hepatoblastoma cells for 24–48 h. Culture medium was harvested and stored at −20 C. Recombinant wild-type ALS was purified from culture media using our published protocol (13).
Deglycosylation of ALS and immunoblot analysis
Culture media containing either ALS or D440N-ALS were treated with 20 U of recombinant N-glycosidase F (Roche Diagnostics GmbH, Mannheim, Germany) in 5 mm sodium phosphate buffer containing 0.1% (vol/vol) n-octyl glucoside for 16 h at 37 C before analysis by immunoblot.
HepG2 cells were treated with increasing concentrations (0–5 μm) of tunicamycin (Sigma-Aldrich, St. Louis, MO) for 2 h. Culture medium was then changed before infection with recombinant ALS adenoviruses, in the presence of tunicamycin, for another 18 h. Culture medium was changed to remove the viruses, and tunicamycin treatment was continued for an additional 8 h, after which cell lysates were harvested in 20 mm Tris-HCl, pH 7.4, 10 mm EDTA, 1% vol/vol Nonidet P-40, 137 mm NaCl, 100 U/ml aprotinin, 1 mm phenylmethylsulfonyl fluoride, and 2 mm Na3VO4 for immunoblot analysis.
Samples of culture media or cell lysates were separated by SDS-PAGE under reducing conditions and transferred onto nitrocellulose. The blot was blocked for 2 h with Tris-buffered saline with 0.1% (vol/vol) Tween 20 containing 5% skim milk, incubated with anti-ALS antiserum at 1:1000 for 16 h at 4 C, washed three times with Tris-buffered saline-Tween 20 before being probed with peroxidase-conjugated antigoat secondary antibody at 1:2000 and subjected to enhanced chemiluminescence detection, using Amersham ECL Plus (GE Healthcare Life Sciences Australia), by LAS-3000 imaging system (Berthold Australia Pty Ltd, Bundoona, Victoria, Australia). The blots were stripped and reprobed with anti-GFP antiserum at 1:1000 followed by peroxidase-conjugated antimouse secondary antibody at 1:2,000. The relative intensities of each protein band of interest were quantified by densitometry using Fujifilm Multi Gauge software (Berthold Australia Pty Ltd).
Gel-filtration chromatography
Culture media containing either wild-type or mutant forms of ALS (3 pmol)) were preincubated with 1 pmol of IGFBP-3 and 100,000 cpm [125I]IGF-I for 2 h. Reaction mixtures were fractionated at 0.5 ml/min in 50 mm sodium phosphate, 0.1 m NaCl (pH 6.5) buffer on a Superose-12 column using an AKTA Purifier (GE Healthcare Life Sciences Australia). Radioactivity in each fraction was measured in a Wizard γ-counter (PerkinElmer Australia, Glen Waverley, Victoria, Australia).
Statistical analysis
Statistical analysis was carried out using Stat-View 4.02 (Abacus Concepts, Inc., Berkeley, CA). Differences between groups were evaluated by Fisher's Protected Least Significant Difference test after ANOVA, and a significant difference was defined as P < 0.05.
Acknowledgments
Address all correspondence and requests for reprints to: Sue M. Firth, Kolling Institute of Medical Research, Royal North Shore Hospital, St Leonards, New South Wales 2065, Australia. E-mail: sue.firth@sydney.edu.au.
This work was supported by Australian Research Council Grant DP071559 (to S.M.F. and R.C.B.), and a Cancer Institute New South Wales Fellowship (to S.M.F.).
Disclosure Summary: The authors have nothing to disclose.
Both S.M.F. and X.Y. contributed equally to this work.
- ALS
- Acid-labile subunit
- CDG
- congenital disorders of glycosylation
- CMV
- cytomegalovirus
- GFP
- green fluorescent protein
- IGFBP
- IGF-binding protein
- LRR
- leucine-rich repeat.
References
- 1. Firth SM , Baxter RC. 2002. Cellular actions of the insulin-like growth factor binding proteins. Endocr Rev 23:824–854 [DOI] [PubMed] [Google Scholar]
- 2. Firth SM , McDougall F , McLachlan AJ , Baxter RC. 2002. Impaired blockade of insulin-like growth factor I (IGF-I)-induced hypoglycemia by IGF binding protein-3 analog with reduced ternary complex-forming ability. Endocrinology 143:1669–1676 [DOI] [PubMed] [Google Scholar]
- 3. Baxter RC , Martin JL. 1989. Structure of the Mr 140,000 growth hormone-dependent insulin-like growth factor binding protein complex: determination by reconstitution and affinity-labeling. Proc Natl Acad Sci USA 86:6898–6902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Firth SM , Ganeshprasad U , Baxter RC. 1998. Structural determinants of ligand and cell surface binding of insulin-like growth factor binding protein-3. J Biol Chem 273:2631–2638 [DOI] [PubMed] [Google Scholar]
- 5. Firth SM , Clemmons DR , Baxter RC. 2001. Mutagenesis of basic amino acids in the carboxyl-terminal region of insulin-like growth factor binding protein-5 affects acid-labile subunit binding. Endocrinology 142:2147–2150 [DOI] [PubMed] [Google Scholar]
- 6. Holman SR , Baxter RC. 1996. Insulin-like growth factor binding protein-3: factors affecting binary and ternary complex formation. Growth Regul 6:42–47 [PubMed] [Google Scholar]
- 7. Twigg SM , Baxter RC. 1998. Insulin-like growth factor (IGF) binding protein-5 forms an alternative ternary complex with IGFs and the acid-labile subunit. J Biol Chem 273:6074–6079 [DOI] [PubMed] [Google Scholar]
- 8. Janosi JB , Ramsland PA , Mott MR , Firth SM , Baxter RC , Delhanty PJ. 1999. The acid-labile subunit of the serum insulin-like growth factor-binding protein complexes. Structural determination by molecular modeling and electron microscopy. J Biol Chem 274:23328–23332 [DOI] [PubMed] [Google Scholar]
- 9. Janosi JB , Firth SM , Bond JJ , Baxter RC , Delhanty PJ. 1999. N-Linked glycosylation and sialylation of the acid-labile subunit. Role in complex formation with insulin-like growth factor (IGF)-binding protein-3 and the IGFs. J Biol Chem 274:5292–5298 [DOI] [PubMed] [Google Scholar]
- 10. Ueki I , Ooi GT , Tremblay ML , Hurst KR , Bach LA , Boisclair YR. 2000. Inactivation of the acid labile subunit gene in mice results in mild retardation of postnatal growth despite profound disruptions in the circulating insulin-like growth factor system. Proc Natl Acad Sci USA 97:6868–6873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Domené HM , Hwa V , Argente J , Wit JM , Camacho-Hübner C , Jasper HG , Pozo J , van Duyvenvoorde HA , Yakar S , Fofanova-Gambetti OV , Rosenfeld RG. 2009. Human acid-labile subunit deficiency: clinical, endocrine and metabolic consequences. Horm Res 72:129–141 [DOI] [PubMed] [Google Scholar]
- 12. Hwa V , Haeusler G , Pratt KL , Little BM , Frisch H , Koller D , Rosenfeld RG. 2006. Total absence of functional acid labile subunit, resulting in severe insulin-like growth factor deficiency and moderate growth failure. J Clin Endocrinol Metab 91:1826–1831 [DOI] [PubMed] [Google Scholar]
- 13. Baxter RC , Martin JL , Beniac VA. 1989. High molecular weight insulin-like growth factor binding protein complex. Purification and properties of the acid-labile subunit from human serum. J Biol Chem 264:11843–11848 [PubMed] [Google Scholar]
- 14. Elbein AD. 1981. The tunicamycins—useful tools for studies on glycoproteins. Trends Biochem Sci 6:219–221 [Google Scholar]
- 15. Domené HM , Bengolea SV , Martinez AS , Ropelato MG , Pennisi P , Scaglia P , Heinrich JJ , Jasper HG. 2004. Deficiency of the circulating insulin-like growth factor system associated with inactivation of the acid-labile subunit gene. N Engl J Med 350:570–577 [DOI] [PubMed] [Google Scholar]
- 16. Domené HM , Scaglia PA , Lteif A , Mahmud FH , Kirmani S , Frystyk J , Bedecarrás P , Gutiérrez M , Jasper HG. 2007. Phenotypic effects of null and haploinsufficiency of acid-labile subunit in a family with two novel IGFALS gene mutations. J Clin Endocrinol Metab 92:4444–4450 [DOI] [PubMed] [Google Scholar]
- 17. Heath KE , Argente J , Barrios V , Pozo J , Diaz-González F , Martos-Moreno GA , Caimari M , Gracia R , Campos-Barros A. 2008. Primary acid-labile subunit deficiency due to recessive IGFALS mutations results in postnatal growth deficit associated with low circulating insulin growth factor (IGF)-I, IGF binding protein-3 levels, and hyperinsulinemia. J Clin Endocrinol Metab 93:1616–1624 [DOI] [PubMed] [Google Scholar]
- 18. van Duyvenvoorde HA , Kempers MJ , Twickler TB , van Doorn J , Gerver WJ , Noordam C , Losekoot M , Karperien M , Wit JM , Hermus AR. 2008. Homozygous and heterozygous expression of a novel mutation of the acid-labile subunit. Eur J Endocrinol 159:113–120 [DOI] [PubMed] [Google Scholar]
- 19. Fofanova-Gambetti OV , Hwa V , Kirsch S , Pihoker C , Chiu HK , Högler W , Cohen LE , Jacobsen C , Derr MA , Rosenfeld RG. 2009. Three novel IGFALS gene mutations resulting in total ALS and severe circulating IGF-I/IGFBP-3 deficiency in children of different ethnic origins. Horm Res 71:100–110 [DOI] [PubMed] [Google Scholar]
- 20. David A , Rose SJ , Miraki-Moud F , Metherell LA , Savage MO , Clark AJ , Camacho-Hübner C. 2010. Acid-labile subunit deficiency and growth failure: description of two novel cases. Horm Res Paediatr 73:328–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Parodi AJ. 2000. Protein glucosylation and its role in protein folding. Annu Rev Biochem 69:69–93 [DOI] [PubMed] [Google Scholar]
- 22. Lewitt MS , Saunders H , Phuyal JL , Baxter RC. 1994. Complex formation by human insulin-like growth factor-binding protein-3 and human acid-labile subunit in growth hormone-deficient rats. Endocrinology 134:2404–2409 [DOI] [PubMed] [Google Scholar]
- 23. Schachter H , Freeze HH. 2009. Glycosylation diseases: quo vadis? Biochim Biophys Acta 1792:925–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. de Zegher F , Jaeken J. 1995. Endocrinology of the carbohydrate-deficient glycoprotein syndrome type 1 from birth through adolescence. Pediatr Res 37:395–401 [DOI] [PubMed] [Google Scholar]
- 25. Miller BS , Khosravi MJ , Patterson MC , Conover CA. 2009. IGF system in children with congenital disorders of glycosylation. Clin Endocrinol (Oxf) 70:892–897 [DOI] [PubMed] [Google Scholar]
- 26. Vogt G , Vogt B , Chuzhanova N , Julenius K , Cooper DN , Casanova JL. 2007. Gain-of-glycosylation mutations. Curr Opin Genet Dev 17:245–251 [DOI] [PubMed] [Google Scholar]
- 27. Vogt G , Bustamante J , Chapgier A , Feinberg J , Boisson Dupuis S , Picard C , Mahlaoui N , Gineau L , Alcaïs A , Lamaze C , Puck JM , de Saint Basile G , Khayat CD , Mikhael R , Casanova JL. 2008. Complementation of a pathogenic IFNGR2 misfolding mutation with modifiers of N-glycosylation. J Exp Med 205:1729–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Firth SM , Ganeshprasad U , Poronnik P , Cook DI , Baxter RC. 1999. Adenoviral-mediated expression of human insulin-like growth factor-binding protein-3. Protein Expr Purif 16:202–211 [DOI] [PubMed] [Google Scholar]
- 29. He TC , Zhou S , da Costa LT , Yu J , Kinzler KW , Vogelstein B. 1998. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA 95:2509–2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Leong SR , Baxter RC , Camerato T , Dai J , Wood WI. 1992. Structural and functional expression of the acid-labile subunit of the insulin-like growth factor binding protein complex. Mol Endocrinol 6:870–876 [DOI] [PubMed] [Google Scholar]