

Summary
The molecular basis of sarcoidosis phenotype heterogeneity and its relationship to effective treatment of sarcoidosis have not been elucidated. Peripheral samples from sarcoidosis subjects who participated in a Phase II study of golimumab [anti‐tumour necrosis factor (TNF)‐α] and ustekinumab [anti‐interleukin (IL)−12p40] were used to measure the whole blood transcriptome and levels of serum proteins. Differential gene and protein expression analyses were used to explore the molecular differences between sarcoidosis phenotypes as defined by extent of organ involvement. The same data were also used in conjunction with an enrichment algorithm to identify gene expression changes associated with treatment with study drugs compared to placebo. Our analyses revealed marked heterogeneity among the three sarcoidosis phenotypes included in the study cohort, including striking differences in enrichment of the interferon pathway. Conversely, enrichments of multiple pathways, including T cell receptor signalling, were similar among phenotypes. We also identify differences between treatment with golimumab and ustekinumab that may explain the differences in trends for clinical efficacy observed in the trial. We find that molecular heterogeneity is associated with sarcoidosis in a manner that may be related to the extent of organ involvement. These findings may help to explain the difficulty in identifying clinically efficacious sarcoidosis treatments and suggest hypotheses for improved therapeutic strategies.
Keywords: clinical trials, human, interferon, sarcoidosis, transcriptomics
Introduction
Sarcoidosis is a multi‐organ, chronic inflammatory disease characterized by non‐caseating granuloma formation following unknown initiating events 1. Exposure of the immune system to antigen is hypothesized as the disease trigger based on genomewide association studies implicating the human leucocyte antigen loci and the identification of certain environmental risk factors 1, 2. Chronic inflammation triggered by this unknown exposure can result in progressive disease characterized by increasingly severe symptoms and tissue damage. The most frequent organ involved is the lung (approximately 90% of cases), although extrapulmonary involvement may occur anywhere in the body. Chronic sarcoidosis requiring continuous therapy occurs in up to one‐third of diagnosed patients, with some patients experiencing persistent symptoms and disability despite treatment with corticosteroids and additional immunomodulators 3. Given the complexity of disease presentation, lack of efficacy of existing anti‐inflammatory agents and poorly understood aetiology, it is generally recognized that an improved understanding of the disease‐driving signalling pathways will be a critical step in the search for more effective therapies. Identification of these key pathways and molecular drivers of disease is complicated by the extensive heterogeneity associated with sarcoidosis, including the organs involved, severity of symptoms and disease progression 1.
Assessment of molecular data from human tissues represents a powerful tool for understanding disease pathology across even heterogeneous cohorts. Previous studies of the whole blood transcriptome in sarcoidosis subjects implicated multiple immune‐related pathways, including interferon (IFN) signalling and T cell signalling 4, 5, 6. Another study analysed whole blood and tissue biopsies and implicated IFN, interleukin (IL)−12 and T helper type 17 (Th17) pathways in sarcoidosis pathology, supporting the concept of therapeutic interventions targeting tumour necrosis factor (TNF)‐α and IL‐12p40 7. Unfortunately, single‐agent interventions have not demonstrated significant efficacy in clinical trials. For example, anti‐TNF‐α treatment has had variable success in clinical trials, and treatment with anti‐IL‐12p40 failed to demonstrate clinical efficacy 8, 9, 10. The observed phenotypical disease heterogeneity may explain the lack of agreement between molecular analyses and clinical efficacy of targeted therapeutics, but the molecular implications of this heterogeneity have not been elucidated fully.
We present the molecular characterization of peripheral samples from chronic sarcoidosis subjects who participated in the Phase II clinical study of ustekinumab (anti‐IL‐12p40) and golimumab (anti‐TNF‐α) 8. Our results support previous findings from whole blood transcriptome profiling from sarcoidosis subjects 4, 5, 6, but also reveal previously unappreciated and marked molecular heterogeneity related to the specific sarcoidosis phenotypes defined by the trial protocol. Of particular interest, we observe that while subjects with skin involvement or both lung and skin involvement support the previously reported involvement of IFN signalling, this enrichment is not observed in subjects with only lung involvement. We also demonstrate that golimumab, in comparison with ustekinumab and placebo, had the greatest effect on peripheral molecular dysregulation, consistent with clinical observations.
Our findings suggest that the extent of organ involvement and disease severity may be related to molecular heterogeneity in a manner that may influence the efficacy of targeted therapeutics. Furthermore, our data suggests that stratification of sarcoidosis patients will be necessary in order to achieve meaningful clinical response rates.
Methods
Study design
Whole blood and serum samples were collected from participants in a published clinical trial of golimumab and ustekinumab in sarcoidosis (NCT00955279; see 8). Briefly, subjects were enrolled who had had histologically confirmed sarcoidosis for at least 2 years previously based on criteria appropriate for either lung involvement or skin involvement. Criteria for entry into the lung group included evidence of lung parenchymal disease on chest radiograph, a percentage of predicted forced vital capacity of 45–80%, a Medical Research Council 9 dyspnoea score of > 2 and a 6‐min walking distance of 100–550 m. Criteria for entry into the skin group included unresolved chronic skin lesions for ≥ 3 months despite therapy, a single lesion of ≥ 2 cm or multiple (≥ 3) lesions with at least one lesion of ≥ 1 cm in longest dimension, and a skin physician's Global Assessment Score ≥ 2. Subjects were randomized to receive treatment with golimumab, ustekinumab or placebo 8.
Nearly 95% of study participants consented for the collection of whole blood at baseline for the present analysis. This cohort was approximately 35% black or African American and 60% white, similar to the racial makeup of the full trial cohort 8. Control whole blood samples were matched to the sarcoidosis cohort based on age, sex and race (Bioreclamation, Westbury, NY, USA).
Serum proteins
Baseline levels of serum proteins from this cohort were presented in a previous publication 8. Analytes included TNF‐α and IL‐12p40, and sarcoidosis‐associated biomarkers 1, 11 angiotensin converting enzyme (ACE) and regulated upppon activation, normal T cell expressed and secreted protein (RANTES).
Transcriptomics
Total RNA was extracted with PAXgene Blood RNA MDx Kit plus customized reagent BM3 (#762431; Qiagen Inc., Valencia, CA, USA). Microarray was performed as described previously 12.
Outlier samples were excluded following identification using principle component analysis, image artefact analysis and chip‐based quality assessments. Expression values were normalized using robust multi‐array average normalization 13. Established quality control procedures identified race as a possible covariate, thus general linear models used to perform t‐tests included race as a covariate. P‐values were corrected for multiple hypotheses using the Benjamini–Hochberg procedure. Unless noted otherwise, significant differences were those with a fold‐change > 1·5 and adjusted P‐value < 0·05.
Pathway enrichment was performed using Ingenuity Pathway Analysis (IPA; Ingenuity Systems, Redwood City, CA, USA). Pathways with Fisher's exact P‐value < 0·05 were considered enriched significantly. Where noted, enrichment with custom signatures was performed with gene set variation analysis (GSVA; see 14).
Results
Whole blood transcriptomics
In order to explore molecular heterogeneity across the sarcoidosis cohort, we used the definitions in the study protocol to describe three patient phenotypes (see Methods and 8); namely, the lung phenotype (subjects meeting lung involvement criteria only), the skin phenotype (subjects meeting skin involvement criteria only) and the combined phenotype (subjects meeting both lung and skin involvement criteria). Next, we compared whole blood microarray gene expression data collected prior to administration of study drug (baseline) to demographically matched healthy controls (Table 1). This comparison was performed for all sarcoidosis subjects and then separately for each phenotype. Separate analysis of phenotypes increased the numbers of probesets that differentiated sarcoidosis subjects significantly from healthy controls for two of the three phenotypes, despite reduced numbers of subjects per group (Table 1). We next compared the gene expression signatures (Table 1) for each phenotype, which revealed a ‘core’ signature of 497 probesets common to all three phenotype signatures and that also shared directionality relative to normal controls (Fig. 1a). Furthermore, despite the small number of samples, 658 probesets distinguished the combined phenotype from the other two phenotypes, suggesting that these subjects are molecularly distinct from the skin phenotype and lung phenotype.
Table 1.
Microarray comparison of whole blood from sarcoidosis subjects at baseline and healthy controls
| Phenotype | Subjects | Controls | Probesets |
|---|---|---|---|
| All | 164 | 45 | 990 |
| Lung | 108 | 45 | 1197 |
| Skin | 39 | 45 | 684 |
| Combined | 17 | 45 | 1651 |
Microarray data generated from whole blood from sarcoidosis subjects prior to administration of study drug were compared to those generated from health controls. This comparison was repeated for each of four groups of subjects: all sarcoidosis subjects (All), subjects with the lung phenotype (Lung), subjects with the skin phenotype (Skin) and subjects with the combined phenotype (Combined). Subjects with the combined phenotype were not included in the lung or skin phenotype groups as they were in the original clinical study [8]. Probesets significantly different between each sarcoidosis phenotype and control were defined as those meeting a fold‐change cut‐off of 1·5, a corrected P‐value cut‐off of 0·05 and an LS mean cut‐off of 5. The numbers of subjects in each group are also shown.
Figure 1.
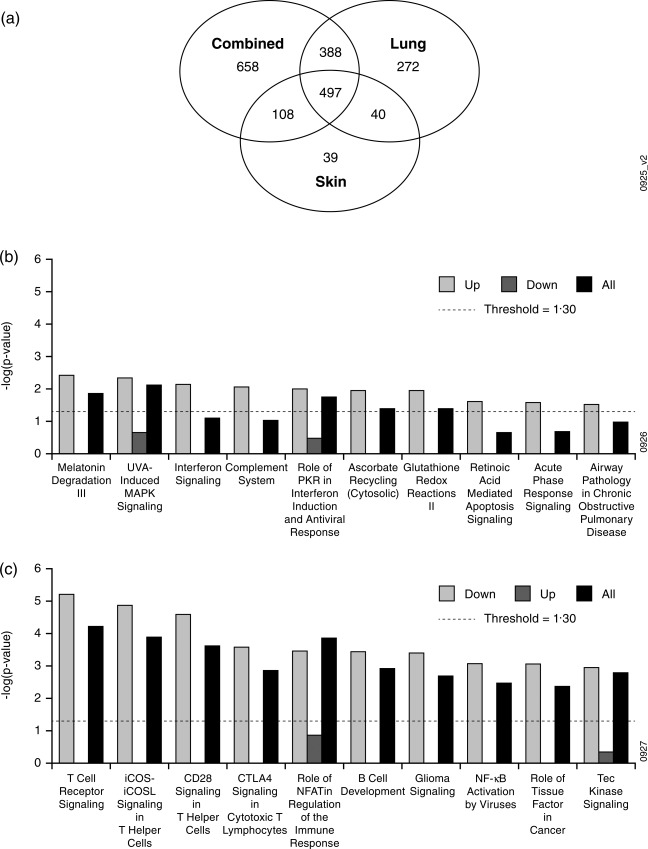
Identification and analysis of the core sarcoidosis signature. (a) Probesets derived from whole blood microarray comparison of sarcoidosis subjects and healthy controls (Table 1) were compared for each phenotype. (b,c) Probesets included in the core signature (497 probesets shared by all three phenotypes) were analysed with Ingenuity Pathway Analysis. The core signature was analysed in its entirety (All) and also separately as up‐ and down‐regulated genes (Up, which contained 110 probesets, and Down, which contained 387 probesets, respectively) relative to healthy controls. Pathway enrichments were sorted for the top 10 most significantly enriched pathways for the (b) up‐regulated genes and (c) down‐regulated genes. The y‐axes indicate the significance of enrichment for a given signature from Ingenuity's knowledge base. Enrichment scores less than the value indicated by the dashed line were not considered significant.
In order to explore the biology that might be common to all sarcoidosis subjects, we computed pathway enrichment scores for the entire core sarcoidosis signature and also separately for the up‐ and down‐regulated genes (Fig. 1b,c). Enrichment scores for the top 10 enriched pathways in the up‐regulated core sarcoidosis genes were relatively weak, but implicated IFN signalling (Fig. 1b) consistent with previous studies 4, 7. Conversely, the down‐regulated genes were characterized by strong enrichment, particularly for pathways related to signalling processes in T cells (Fig. 1c); however, enrichment of these pathways was driven by highly overlapping lists of T cell‐related genes (including AKT3, ATM, CALM1, CAMK4, CD28, CTLA4, FYN, ITK, RASGRP1, PLEKHA1, TRAT1 and RCAN3), suggesting that this enrichment pattern may be explained by a general reduction in T cell numbers rather than a specific change in peripheral T cell signalling processes. This observation is consistent with at least one previous transcriptomic study in sarcoidosis 4 and the common observation of lymphopenia in sarcoidosis subjects 15.
Next, we compared the pathway enrichment scores among the three sarcoidosis phenotypes in our study. Consistent with the core sarcoidosis signature, each phenotype signature was enriched for T cell receptor (TCR) signalling (Fig. 2), with the majority of genes down‐regulated compared to healthy controls (Table 2a). Interestingly, we noticed that enrichment for IFN signalling was not shared across phenotypes, as enrichment was highest for the combined phenotype, markedly lower but still enriched significantly for the skin phenotype and not enriched significantly for the lung phenotype (Fig. 2). This pattern is not explained by differences in phenotype group sizes, given that the enrichment scores are related inversely to the number of subjects (Table 1). While the observation of enrichment for IFN signalling in peripheral blood from sarcoidosis subjects using various knowledge bases is consistent with previous studies 4, 7, heterogeneity in this enrichment in a manner related to phenotype has not been reported.
Figure 2.
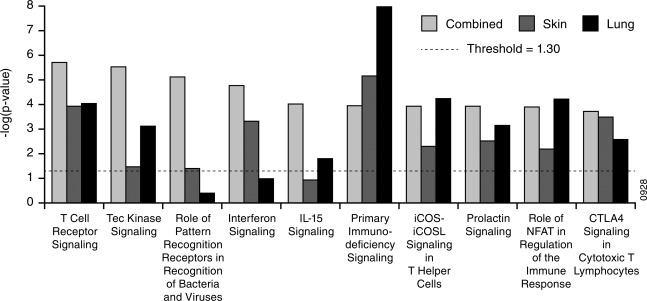
Comparison of sarcoidosis phenotype signatures. Pathway enrichment was performed using Ingenuity Pathway Analysis for the phenotype signatures listed in Table 1; y‐axes indicate the significance of enrichment for a given signature from Ingenuity's knowledge base. Enrichment scores less than the value indicated by the dashed line were not considered significant. The number of subjects per group can be found in Table 1.
Table 2.
(a) T cell receptor signalling
| Gene symbol | Combined (n = 17) | Skin (n = 39) | Lung (n = 108) |
|---|---|---|---|
| ATM | –(1·6) | –(1·5) | –(1·5) |
| BMX | +1·5 | ||
| CAMK4 | –(2·7) | –(2·0) | –(2·3) |
| CARD11 | –(1·5) | ||
| CD28 | –(2·3) | –(1·6) | –(2·3) |
| CD3E | –(1·8) | –(1·7) | |
| CD8A | –(1·5) | –(1·5) | |
| CD8B | –(1·5) | –(1·6) | |
| CTLA‐4 | –(2·1) | –(1·9) | –(2·3) |
| FYN | –(1·7) | –(1·6) | –(1·6) |
| ITK | –(2·0) | –(1·7) | –(1·8) |
| LCK | –(1·5) | ||
| PIK3C2B | –(1·5) | –(1·6) | |
| PLCG1 | –(1·6) | –(1·6) | |
| PRKCQ | –(1·7) | –(1·6) | |
| RASGRP1 | –(1·7) | –(1·5) | –(1·8) |
| RRAS2 | –(1·5) | –(1·6) |
| (b) Interferon signalling | |||
|---|---|---|---|
| Gene symbol | Combined (n = 17) | Skin (n = 39) | Lung (n = 108) |
| BCL2 | –(1·8) | –(1·7) | |
| IFI35 | +1·7 | +1·6 | |
| IFIT3 | +2·6 | +2·0 | +1·8 |
| IRF1 | +1·8 | +1·6 | |
| JAK2 | +1·7 | ||
| OAS1 | +1·5 | +1·5 | |
| SOCS1 | +1·6 | ||
| STAT1 | +2·4 | +1·9 | +1·6 |
| TAP1 | +1·5 | ||
Genes that overlapped between the indicated sarcoidosis signature from Table 1 and interferon signalling signatures from Ingenuity's knowledge base are shown. The fold‐change for comparison of sarcoidosis to control are shown for each gene and phenotype. Data showing bold + and –(x.x) indicate up‐ and down‐regulation compared to control, respectively, and blank cells indicate a gene that did not meet statistical significance for that comparison. The number of sarcoidosis subjects per group from Table 1 is also shown.
Examining the genes driving enrichment of the IFN pathway revealed that the majority were up‐regulated compared to healthy controls (Table 2b), suggesting an increase in IFN signalling in the combined and skin phenotypes but no increase in IFN signalling in the lung phenotype compared to healthy controls. Such differences could be explained by differences in corticosteroid use among phenotypes, which has been shown to affect gene expression in asthma 16. In particular, FKBP51 expression was shown to change with corticosteroids 16, 17, 18. We note that FKBP51 was indeed expressed differentially in the lung phenotype compared to healthy controls, suggesting that increased corticosteroids in the lung phenotype could be one possible contributing factor to the observed gene expression differences (Table 1). However, we do not believe that this hypothesis explains the pronounced pathway enrichment differences we observed, because the lung and the combined phenotypes did not differ statistically in corticosteroid use (Supporting information, Fig. S1) but did represent the strongest molecular differences (Fig. 2).
Serum proteins
We extended our analysis of sarcoidosis phenotypes by re‐analysing published protein levels in baseline serum samples from the same sarcoidosis cohort 8. IL‐12p40 and RANTES were elevated for the combined phenotype and for the skin phenotype compared to the lung phenotype (Fig. 3a,b, respectively). This former finding is consistent with significant enrichment of IFN signalling for the combined phenotype and skin phenotype and no significant enrichment of IFN signalling for the lung phenotype and the directionality of the probesets relative to healthy controls driving those enrichments (Fig. 2 and Table 2b, respectively). Serum ACE and TNF‐α levels were also elevated significantly for the skin phenotype and trended towards elevation for the combined phenotype compared to the lung phenotype (Fig. 3c,d, respectively).
Figure 3.
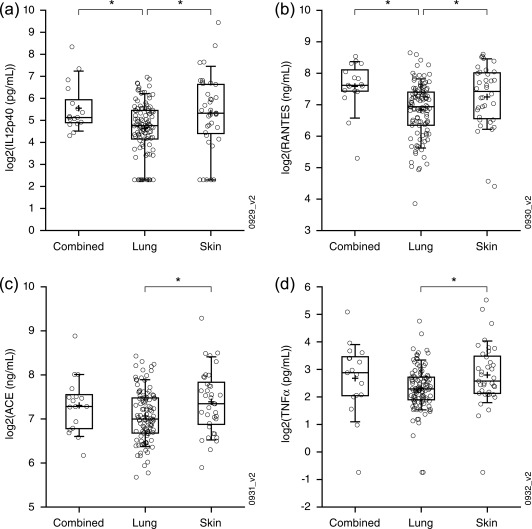
Serum biomarkers. (a–d) Interleukin (IL)−12p40 (a), regulated upon activation, normal T cell expressed and secreted protein (RANTES) (b), ACE (b) and tumour necrosis factor (TNF)‐α (d) levels from serum collected from sarcoidosis patients of the indicated phenotype were compared. Indicated significant differences (*) were those reaching a P‐value of less than 0·05 as determined by Student's t‐test.
Gene set variation analysis
In order to avoid the biases of pathway enrichment analysis inherent to utilizing a knowledge base, we next employed GSVA to compute enrichment scores using our sarcoidosis gene signatures (Table 1) and compared enrichment scores between phenotypes (Table 3a). This analysis indicates which gene signatures represent biological processes that may differ among phenotypes. We observed that the signature comprised of the up‐regulated genes in the combined phenotype compared to healthy controls was enriched increasingly in the combined phenotype compared to the lung phenotype and the skin phenotype (Table 3a, ‘Combined versus controls – up’). Given that the combined phenotype represents more extensive organ involvement compared to the other two phenotypes, this observation suggests that the underlying molecular dysregulation may change for increased organ involvement and that the specific processes involved may be described by this gene signature. To investigate further the involved biology, we performed pathway enrichment on the up‐regulated genes, the down‐regulated genes and all genes from the combined phenotype signature (Fig. 4a). We found that the up‐regulated genes were enriched significantly for multiple signalling pathways that were not enriched in the down‐regulated genes, including IFN signalling, complement system and IL‐15 production.
Table 3.
(a) Baseline gene set variation analysis (GSVA)
| Gene signature | Directionality | Combined versus lung | Skin versus lung | Combined versus skin |
|---|---|---|---|---|
| Lung versus controls | Up | |||
| Lung versus controls | Down | |||
| Skin versus controls | Up | +0·32 | ||
| Skin versus controls | Down | |||
| Combined versus controls | Up | +0·35 | +0·28 | |
| Combined versus controls | Down | |||
| Core | Up | +0·30 | ||
| Core | Down |
| (b) Lung GSVA | ||||
|---|---|---|---|---|
| Gene signature | Directionality | Placebo | Ustekinumab | Golimumab |
| Lung versus controls | Up | –(0·15) | –(0·19) | |
| Lung versus controls | Down | +0·31 | ||
| Skin versus controls | Up | |||
| Skin versus controls | Down | +0·26 | ||
| Combined versus controls | Up | |||
| Combined versus controls | Down | +0·28 | ||
| Core | Up | –(0·16) | ||
| Core | Down | +0·27 | ||
| (c) Combined GSVA | ||||
|---|---|---|---|---|
| Gene signature | Directionality | Placebo | Ustekinumab | Golimumab |
| Lung versus controls | Up | –(0·35) | ||
| Lung versus controls | Down | +0·41 | ||
| Skin versus controls | Up | |||
| Skin versus controls | Down | |||
| Combined versus controls | Up | |||
| Combined versus controls | Down | +0·38 | ||
| Core | Up | |||
| Core | Down | |||
Enrichment scores for each sample were first computed using gene set variation analysis (GSVA), the signatures presented in Table 1 and the core sarcoidosis signature (core). Enrichment scores with GSVA consider directionality, thus each signature was split into up‐ and down‐regulated (compared to control) probesets (Directionality column). Numbers indicate the change in enrichment between the indicated phenotypes with bold + and –(x.x) indicating positive and negative changes, respectively.
A general linear model was used to perform the paired comparison of enrichment scores at week 28 to those at week 0 within each treatment group and separately for the lung phenotype. Numbers indicate the change in enrichment between weeks 28 and 0 bold + and –(x.x) indicating positive and negative changes, respectively.
A general linear model was used to perform the paired comparison of enrichment scores at week 28 to those at week 0 within each treatment group and separately for the combined phenotype. Numbers indicate the change in enrichment between weeks 28 and 0 with bold + and –(x.x) indicating positive and negative changes, respectively.
Figure 4.
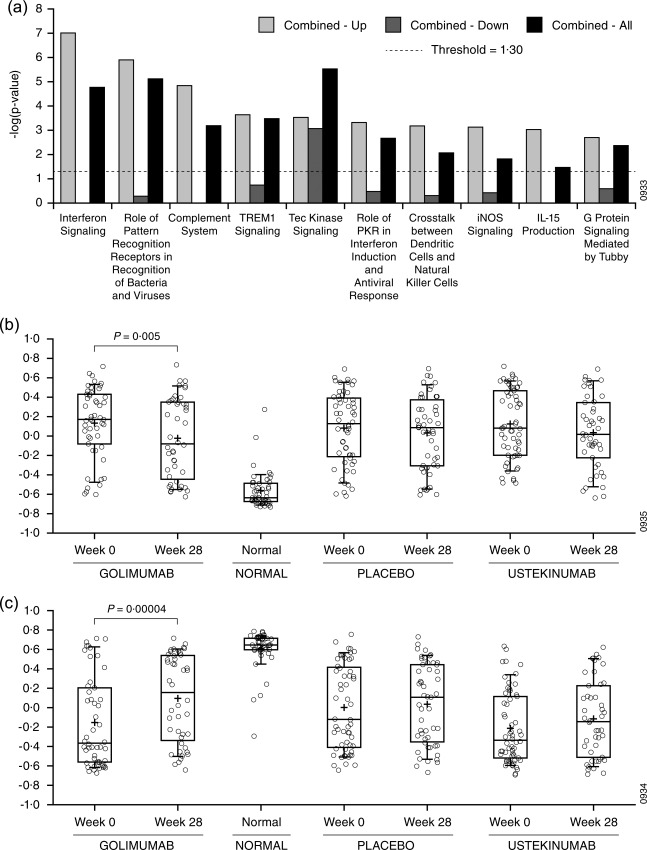
Gene set variation analysis of whole blood microarray results. (a) Probesets included in the combined phenotype versus controls signature (Combined – All) were analysed with Ingenuity Pathway Analysis. Up‐ and down‐regulated genes in this signature we also each analysed separately (Combined – Up and Combined – Down, respectively). Bar heights indicate the significance of enrichment for a given signature from Ingenuity's knowledge base. Enrichment scores less than the value indicated by the dashed line were not considered significant. (b,c) GSVA was performed for the up‐regulated (b) and down‐regulated (c) probesets in the core sarcoidosis signature. Enrichment scores are shown for weeks 28 and week 0 separated into treatment groups for sarcoidosis subjects and for controls. Changes satisfying P < 0·01 were considered significant.
We next explored the effect of study drug treatments on signature enrichments scores. While placebo had no significant effect on gene expression, ustekinumab and golimumab treatment resulted in changes in enrichment scores towards values associated with the healthy controls for the lung phenotype and the combined phenotype (Tables 3b,c, respectively). Treatment effects were not detected for the skin phenotype. Interestingly, ustekinumab treatment tended to trend selectively towards correcting genes that were up‐regulated at baseline compared to healthy controls, while golimumab treatment tended to correct genes that were down‐regulated. Pooling phenotypes and focusing on enrichment with the core sarcoidosis signature demonstrated that golimumab moved enrichment scores significantly for both the up‐ and down‐regulated genes (Fig. 4b,c, respectively) towards normal while treatment with placebo or ustekinumab did not. This is consistent with the clinical observation that golimumab trended towards clinical efficacy for the combined and skin phenotypes pooled 8.
Discussion
Our analysis of peripheral transcriptomics and serum proteins revealed pronounced molecular differences between the three sarcoidosis phenotypes designated in the clinical trial from which the samples were collected. Multiple computational approaches to analysing the transcriptomic data suggested an increase in IFN signalling in the combined phenotype compared to the other phenotypes (Fig. 2 and Table 3a; recall that the ‘Combined versus controls – up’ signature in Table 3a was shown to enrich for IFN signalling in Fig. 4a). Consistent with this observation, IL‐12p40 and RANTES were increased in the combined phenotype compared to the lung phenotype (Fig. 3a,b, respectively). The skin phenotype was also enriched significantly in IFN signalling, but only by one computational approach (Fig. 2), and was also characterized by increased serum IL‐12p40 and RANTES compared to the lung phenotype (Fig. 3a,b, respectively). We also note that the skin phenotype was characterized by a significant increase in ACE and TNF‐α compared to the lung phenotype, while the combined phenotype trended in the same direction. Taken together, we hypothesize that the combined phenotype represents the most molecularly dysregulated phenotype and the lung phenotype represents the least molecularly dysregulated phenotype.
Enrichment of multiple signalling pathways, including TCR signalling and IL‐15 signalling, were consistent with the notion that the combined phenotype represents the most molecularly dysregulated of the three phenotypes. Increased TCR signalling enrichment for the combined phenotype compared to the other two phenotypes (Fig. 2) is consistent with worsening lymphopenia, which may relate to disease severity 15. In our study, we also note increased enrichment for IL‐15 signalling for the combined phenotype compared to the other phenotypes (Fig. 2). Interestingly, the IL‐15 signalling pathway has been implicated previously in sarcoidosis. Muro and co‐workers reported an increase in IL‐15‐positive cells in tissue from sarcoidosis subjects compared to healthy controls 19. Agostini and co‐workers demonstrated increased proliferation of bronchoalveolar lavage (BAL) T cells from sarcoidosis subjects in the presence of IL‐15 or IL‐2, but not TNF‐α 20. Closer examination of the genes driving enrichment in our study revealed that while there was a mix of directionality, both IL‐15 and IL‐15 receptor were up‐regulated for the combined phenotype in a trend that was less pronounced for the skin phenotype and absent for the lung phenotype (Table 4). These results suggest that IL‐15 signalling may be associated with the extent of organ involvement in sarcoidosis.
Table 4.
Interleukin (IL)−15 signalling
| Gene symbol | Combined (n = 17) | Skin (n = 39) | Lung (n = 108) |
|---|---|---|---|
| AKT3 | –(1·9) | –(1·7) | –(1·9) |
| ATM | –(1·6) | –(1·5) | –(1·5) |
| BCL2 | –(1·8) | –(1·7) | |
| BCL2L1 | +1·6 | ||
| IL15 | +1·6 | ||
| IL15RA | +1·6 | +1·5 | |
| JAK2 | +1·7 | ||
| LCK | –(1·5) | ||
| PIK3C2B | –(1·5) | –(1·6) | |
| PLCG1 | –(1·6) | –(1·6) | |
| RRAS2 | –(1·5) | –(1·6) |
Genes that overlapped between the indicated signature from Table 1 and the IL‐15 signalling signature from Ingenuity's knowledge base are shown. The fold‐change for comparison of sarcoidosis to control are shown for each gene and phenotype. Data showing bold + and –(x.x) indicate up‐ and down‐regulation compared to control, respectively, and blank cells indicate a gene that did not meet statistical significance for that comparison. The number of sarcoidosis subjects per group from Table 1 is also shown.
Taken together, these data suggest that molecular dysregulation may be related to the extent of disease, given that the combined phenotype represents the greatest extent of organ involvement in this cohort. Considering this hypothesis, our analysis suggests that increased organ involvement is characterized by two features: (1) increased dysregulation of existing pathogenic processes, supported by the ability of the core sarcoidosis signature to differentiate the combined phenotype from the lung phenotype (Table 3a), and (2) the emergence of new pathogenic processes, supported by the observation of enrichment of signalling pathways in the combined phenotype or skin phenotype that are not enriched in the lung phenotype (e.g. IFN signalling, Fig. 2).
Our hypothesis for variable contributions of multiple signalling pathways across extents of organ involvement in sarcoidosis is consistent with the clinical heterogeneity seen in this disease 21, 22. These findings, coupled with variation in the efficacy of TNF‐α blockade among different sarcoidosis cohorts 8, 9, 23, support the possibility that different forms of sarcoidosis may require different therapeutic interventions due to variation in causal molecular processes. In the clinical trial from which the present samples were collected, the skin phenotype was the only phenotype in which TNF‐α blockade with golimumab trended towards effective. This raises the possibility that some forms of sarcoidosis are more TNF‐α‐driven, which is consistent with the observation that TNF‐α levels were increased for the skin phenotype compared to the lung phenotype (Fig. 3d). This possibility is also consistent with a second trial of TNF‐α blockade but instead with infliximab in chronic pulmonary sarcoidosis, where greater efficacy of infliximab was demonstrated in patients with elevated serum TNF‐α protein levels 9. In addition, a subset analysis of the same study revealed statistically significant clinical improvement in those patients with specific forms of skin involvement 24, which is also consistent with our hypothesis that this phenotype may be TNF‐α‐driven.
Disease driven by IFN signalling was a key assumption for efficacy of ustekinumab in sarcoidosis, but the lung phenotype appeared to be characterized by a lack of enrichment for IFN signalling and reduced IL‐12p40 levels compared to the other phenotypes (Figs 2 and 3a, respectively); however, even phenotypes characterized by enrichment of IFN signalling and increased IL‐12p40 levels (the combined phenotype and the skin phenotype) did not respond to IL‐12p40 blockade with ustekinumab 8. Consistent with this, ustekinumab was less effective at the molecular level compared to golimumab in impacting (1) the core sarcoidosis signature (Fig. 4b,c) and (2) phenotype‐specific signatures (Tables 3b,c). However, ustekinumab was able to partially correct the up‐regulated genes from the core sarcoidosis signature in the lung phenotype (Table 3b), which is notable considering that these genes are enriched for IFN signalling (Fig. 1b). This suggests that ustekinumab may have reduced IFN signalling to some extent, as intended, but the degree of reduction may have been insufficient for clinical response or IFN signalling may not be causal in the disease. Conversely, golimumab appeared to have a greater molecular impact, specifically with respect to the genes that were down‐regulated compared to healthy control at baseline (Tables 3b,c and Fig. 4c). Thus, given that only golimumab trended towards a clinical effect in the trial, we hypothesize that resolution of the down‐regulated genes may be related to clinical response.
A previous study explored sarcoidosis heterogeneity with respect to IFN and TNF‐α 25. In this study, TNF‐α levels differentiated African American sarcoidosis subjects from controls and subjects who had neurological manifestations from those who did not. Conversely, type 1 IFN differentiated European American subjects from controls and subjects with extrapulmonary manifestations from those with only pulmonary manifestations. These results are particularly pertinent in the context of our study, because they are consistent with our hypothesis of relationships between IFN, TNF‐α and the extent of organ involvement in sarcoidosis.
These results suggest that multiple factors may contribute to molecular heterogeneity across sarcoidosis cohorts in a manner that may affect the clinical efficacy of targeted therapeutics. Importantly, this study did not include analysis of samples from diseased tissue. Future studies involving direct sampling of disease tissue will be necessary to assess our findings of pronounced molecular heterogeneity in sarcoidosis and to evaluate the possible role of the IL‐15 pathway in sarcoidosis pathogenesis suggested by our analysis. At a minimum, our data invite speculation that therapeutic interventions in sarcoidosis will be optimized through clinical patient stratification that reflects the molecular heterogeneity of the disease.
Disclosures
C. M., K. L., E. W., R. W., P. S., E. B. and C. B. are or were employees of Janssen Research & Development, LLC at the time of the study and all own(ed) stock in Johnson & Johnson. M. A. J. has acted as a consultant for Biogen, Mallinckrodt, Janssen. He has received grants for his institution from Mallinckrodt and the Foundation for Sarcoidosis Research. R. B. reports grants and personal fees from Janssen Research & Development, LLC during the conduct of the study.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's website.
Fig. S1. Prednisone equivalent dose by phenotype. The average prednisone equivalent doses of corticosteroids at baseline for each sarcoidosis phenotype are shown.
Acknowledgements
We thank the clinical trial participants, without whom this work would not be possible. Support for this study was provided by Janssen Research & Development, LLC.
References
- 1. Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. N Engl J Med 2007; 357:2153–65. [DOI] [PubMed] [Google Scholar]
- 2. Adrianto I, Lin CP, Hale JJ et al Genome‐wide association study of African and European Americans implicates multiple shared and ethnic specific loci in sarcoidosis susceptibility. PLOS ONE 2012; 7:e43907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baughman RP, Culver DA, Judson MA. A concise review of pulmonary sarcoidosis. Am J Respir Crit Care Med 2011; 183:573–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bloom CI, Graham CM, Berry MP et al Transcriptional blood signatures distinguish pulmonary tuberculosis, pulmonary sarcoidosis, pneumonias and lung cancers. PLOS ONE 2013; 8:e70630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Maertzdorf J, Weiner J III, Mollenkopf HJ et al Common patterns and disease‐related signatures in tuberculosis and sarcoidosis. Proc Natl Acad Sci USA 2012; 109:7853–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhou T, Zhang W, Sweiss NJ et al Peripheral blood gene expression as a novel genomic biomarker in complicated sarcoidosis. PLOS ONE 2012; 7:e44818.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Judson MA, Marchell RM, Mascelli M et al Molecular profiling and gene expression analysis in cutaneous sarcoidosis: the role of interleukin‐12, interleukin‐23, and the T‐helper 17 pathway. J Am Acad Dermatol 2012; 66:901–10, 910.e1–2. [DOI] [PubMed] [Google Scholar]
- 8. Judson MA, Baughman RP, Costabel U et al Safety and efficacy of ustekinumab or golimumab in patients with chronic sarcoidosis. Eur Respir J 2014; 44:1296–307. [DOI] [PubMed] [Google Scholar]
- 9. Baughman RP, Drent M, Kavuru M et al Infliximab therapy in patients with chronic sarcoidosis and pulmonary involvement. Am J Respir Crit Care Med 2006; 174:795–802. [DOI] [PubMed] [Google Scholar]
- 10. Baughman RP, Bradley DA, Lower EE. Infliximab in chronic ocular inflammation. Int J Clin Pharmacol Ther 2005; 43:7–11. [DOI] [PubMed] [Google Scholar]
- 11. Petrek M, Kolek V, Szotkowska J, du Bois RM. CC and C chemokine expression in pulmonary sarcoidosis. Eur Respir J 2002; 20:1206–12. [DOI] [PubMed] [Google Scholar]
- 12. Sofen H, Smith S, Matheson RT et al Guselkumab (an IL‐23‐specific mAb) demonstrates clinical and molecular response in patients with moderate‐to‐severe psoriasis. J Allergy Clin Immunol 2014; 133:1032–40. [DOI] [PubMed] [Google Scholar]
- 13. Irizarry RA, Hobbs B, Collin F et al Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003; 4:249–64. [DOI] [PubMed] [Google Scholar]
- 14. Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA‐seq data. BMC Bioinformatics 2013; 14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sweiss NJ, Salloum R, Gandhi S et al Significant CD4, CD8, and CD19 lymphopenia in peripheral blood of sarcoidosis patients correlates with severe disease manifestations. PLOS ONE 2010; 5:e9088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Woodruff PG, Boushey HA, Dolganov GM et al Genome‐wide profiling identifies epithelial cell genes associated with asthma and with treatment response to corticosteroids. Proc Natl Acad Sci USA 2007; 104:15858–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Binder EB. The role of FKBP5, a co‐chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology 2009; 34(Suppl 1):S1861–95. [DOI] [PubMed] [Google Scholar]
- 18. Stechschulte LA, Sanchez ER. FKBP51‐a selective modulator of glucocorticoid and androgen sensitivity. Curr Opin Pharmacol 2011; 11:332–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Muro S, Taha R, Tsicopoulos A et al Expression of IL‐15 in inflammatory pulmonary diseases. J Allergy Clin Immunol 2001; 108:970–5. [DOI] [PubMed] [Google Scholar]
- 20. Agostini C, Trentin L, Facco M et al Role of IL‐15, IL‐2, and their receptors in the development of T cell alveolitis in pulmonary sarcoidosis. J Immunol 1996; 157:910–8. [PubMed] [Google Scholar]
- 21. Baughman RP, Teirstein AS, Judson MA et al Clinical characteristics of patients in a case control study of sarcoidosis. Am J Respir Crit Care Med 2001; 164:1885–9. [DOI] [PubMed] [Google Scholar]
- 22. Costabel U, Hunninghake GW. ATS/ERS/WASOG statement on sarcoidosis. Sarcoidosis Statement Committee. American Thoracic Society. European Respiratory Society. World Association for Sarcoidosis and Other Granulomatous Disorders. Eur Respir J 1999; 14:735–7. [DOI] [PubMed] [Google Scholar]
- 23. Rossman MD, Newman LS, Baughman RP et al A double‐blinded, randomized, placebo‐controlled trial of infliximab in subjects with active pulmonary sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2006; 23:201–8. [PubMed] [Google Scholar]
- 24. Baughman RP, Judson MA, Lower EE et al Infliximab for chronic cutaneous sarcoidosis: a subset analysis from a double‐blind randomized clinical trial. Sarcoidosis Vasc Diffuse Lung Dis 2016; 32:289–95. [PubMed] [Google Scholar]
- 25. Sweiss NJ, Zhang W, Franek BS et al Linkage of type I interferon activity and TNF‐alpha levels in serum with sarcoidosis manifestations and ancestry. PLOS ONE 2011; 6:e29126. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's website.
Fig. S1. Prednisone equivalent dose by phenotype. The average prednisone equivalent doses of corticosteroids at baseline for each sarcoidosis phenotype are shown.