

Abstract
Receptor tyrosine kinases (RTKs) have provided molecular targets for the development of novel, prognosis‐improving agents in many cancers; however, resistances to these therapies occur. On the cellular level, one resistance mechanism is attributed to functional RTK redundancies and compensatory cross‐signaling, leading to perception of RTKs as signaling and target networks. To provide a basis for better exploitation of this network in Ewing sarcoma, we generated comprehensive qPCR gene expression profiles of RTKs in Ewing sarcoma cell lines and 21 untreated primary tumors. Key findings confirm broad‐spectrum RTK expressions with potential for signaling redundancy. Profile analyses with regard to patient risk‐group further revealed several individual RTKs of interest. Among them, VEGFR3 and TIE1 showed high‐level expressions and also were suggestive of poor prognosis in localized tumors; underscoring the relevance of angiogenic signaling pathways and tumor‐stroma interactions in Ewing sarcoma. Of note, compared to localized disease, tumors derived from metastatic disease were marked by global high‐level RTK expressions. Nine individual RTKs were significantly over‐expressed, suggesting contributions to molecular mechanisms of metastasis. Of these, ROR1 is being pursued as therapeutic target in leukemias and carcinomas, but un‐characterized in sarcomas. We demonstrate expression of ROR1 and its putative ligand Wnt5a in Ewing sarcomas, and of an active ROR1 protein variant in cell lines. ROR1 silencing impaired cell migration in vitro. Therefore, ROR1 calls for further evaluation as a therapeutic target in metastatic Ewing sarcoma; and described as a pseudo‐kinase with several isoforms, underlines these additional complexities arising in our understanding of RTK signaling networks.
Keywords: Ewing sarcoma, Metastasis, Receptor tyrosine kinase, ROR1, Therapeutic target
Highlights
Comprehensive gene expression profiles provide a baseline reference.
Aspects of patient risk‐groups and metastasis are addressed.
Individual receptor tyrosine kinases (RTKs) of therapeutic interest are revealed.
ROR1 is identified as a potential therapeutic target in metastatic Ewing sarcoma.
ROR1 underlines pseudo‐kinases and isoforms as additional complexities in RTK signaling networks.
Abbreviations
- MSC
mesenchymal stem cell
- qPCR
real-time quantitative PCR
- ROR1
receptor tyrosine kinase-like orphan receptor 1
- RTK
receptor tyrosine kinase, for a listing of RTK abbreviations see Table A.1
1. Introduction
Ewing sarcoma is a rare cancer, but the second most common sarcoma of bone in childhood and adolescence (Bernstein et al., 2006; Gurney et al., 1999). Metastasis at diagnosis is the most significant adverse prognostic factor (Cotterill et al., 2000; Ladenstein et al., 2010; Paulussen et al., 1998). Risk‐adapted and metastasis‐targeted therapeutic strategies therefore are key aims of current translational research.
Given the complex series of events adding to the metastatic process, it is plausible that molecular players and consequently targets should be distinct from those active in localized tumors (Krishnan et al., 2005; Valastyan and Weinberg, 2011). At the same time it remains undefined, whether primary genetic determinants predestine to metastasis, or whether secondary events that generate a metastatic tumor sub‐clone prevail. Several microarray gene expression studies aimed to identify prognostic and/or metastatic gene expression signatures in Ewing sarcoma (Ohali et al., 2004; Schaefer et al., 2008; Volchenboum et al., 2015; Zambelli et al., 2010). So far, while focusing on individual signature genes, validations from a comprehensive gene pathway or gene family perspective are lacking.
Receptor tyrosine kinases (RTKs) and their dysregulation represent key oncogenic and tumor‐maintaining events in many cancers (Gschwind et al., 2004; Lemmon and Schlessinger, 2010). Furthermore, their ligand‐mediated activation and intrinsic kinase activities are well amenable to therapeutic inhibition, making RTKs highly investigated drug targets. Multiple ligand‐blocking antibodies and small molecule kinase inhibitors are in development and clinical application (Gschwind et al., 2004; Jänne et al., 2009). Because oncogenic RTKs are not principally unique to cancer entities (Jänne et al., 2009; Lemmon and Schlessinger, 2010), rare diseases such as Ewing sarcoma can profit from therapeutic advances in more prevalent cancers. This was demonstrated in an advent of pre‐clinical and clinical IGF1R‐targeted investigations in pediatric sarcomas, stimulated by remarkable responses of Ewing sarcoma patients in early phase trials (Olmos et al., 2010; Tolcher et al., 2009).
More recently, distinct RTK functions in the metastatic cellular capacities and consequently targeting potential are reaching focus. RTKs interact with integrin and cadherin cell adhesion molecules to induce and exert pro‐migratory and pro‐invasive signals through intracellular FAK and SRC kinases (Steeg, 2006). In Ewing sarcoma, examples are PDGFR and EGFR signaling identified as metastasis‐associated pathways, and AXL and PTK7 found over‐expressed in a poor prognosis gene signature (Ohali et al., 2004; Schaefer et al., 2008). ERBB4 was shown to interact with E‐cadherin to initiate FAK signaling, and therapeutic SRC inhibition impaired cell migration and invasion (Mendoza‐Naranjo et al., 2013; Shor et al., 2007). RTKs may therefore provide molecular targets also for metastasis‐directed therapeutic strategies.
Despite broad and confirmed molecular bases, resistances to RTK‐targeted therapies occur. Including Ewing sarcoma, where subsequent clinical trials of IGF1R‐targeted antibodies demonstrated no more than modest activity, and concluded that additional markers predictive of benefit were required (Juergens et al., 2011; Pappo et al., 2011). As one resistance mechanism to RTK‐targeted strategies, signaling redundancies, cross‐talk, and mutual compensation of signaling input have been identified (Jänne et al., 2009; Lemmon and Schlessinger, 2010; Stommel et al., 2007). In glioblastoma and lung cancer, resistance to EGFR‐targeted agents was conferred by redundant IGF1R downstream signaling, as well as by compensatory MET amplification, maintaining ERBB3 signaling input (Chakravarti et al., 2002; Engelman et al., 2007; Jun et al., 2014). Vice versa, resistance of pediatric rhabdomyosarcoma (RMS) cell lines to IGF1R inhibitors was attributed to over‐expression of EGFR and MET receptors. In keeping, in both RMS and Ewing sarcoma cell lines, IGF1R inhibitor sensitivity was restored by simultaneous silencing of RON, a MET family RTK (F. Huang et al., 2010; Potratz et al., 2010). RTK signaling hence emerges as an interdependent cellular network of therapeutic relevance.
As this introduction points out, the family of RTKs makes for prime candidates of therapeutic targets, including metastasis‐directed targets; with the draw‐back of our to date insufficient understanding of confounding RTK network interactions. Objective of this work therefore was to provide a basis for systematic exploration of the RTK network in Ewing sarcoma. Comprehensive RTK gene expression profiles of Ewing sarcoma cell lines and untreated primary tumors were generated. To incorporate metastasis‐specific aspects, profiles were characterized with respect to tumor origin from localized or metastatic disease.
2. Methods
2.1. Tumor samples and clinical data
21 samples of untreated Ewing sarcoma primary tumors were available. Diagnosis was based on standard histopathologic criteria and on EWS‐FLI1 or EWS‐ERG fusion transcripts. All patients were enrolled in clinical trials of the German Cooperative Ewing Sarcoma Study Group of the German Society of Pediatric Hematology and Oncology (GPOH), 19 patients into EURO‐E.W.I.N.G.99 and one patient each into CESS86 and EICESS92, respectively. Trials were in compliance with the Declaration of Helsinki, with approval of concerned ethics committees and patient's written informed consent obtained before registration of patients. In this cohort, all patients with metastatic disease presented with metastasis not restricted to the lung, but to bone/bone marrow or other sites, defining them as high‐risk R3 patients. In four cases diagnostic staging showed more than five bone metastases. In these cases tissue samples were from the diagnostic biopsy site. Clinical survival data were available for 20 of 21 patients and survival by age and by (high‐risk) metastatic status as key prognostic factors reflected published cohorts (Figure A.1)(Ladenstein et al., 2010, 2014, 1998, 2008). For risk‐group assignment and further characteristics see Table 1.
Table 1.
Patient characteristics.
| No. | Study | Sex | Age [years] | Disease extension | Tumor volume [ml] | Tumor site | Histologic response | Risk group | Event | EFS [month] | OS [month] | Status |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | EE99 | f | 13 | Localized | >200 | Axial | Good | R1 | 98.5 | 98.5 | CR | |
| 2 | CESS86 | m | 17 | Localized | NA | Axial | NA | NA | NA | NA | NA | |
| 3 | EICESS92 | m | 12 | Localized | <100 | Limb | Poor | SR | 83.9 | 83.9 | CR | |
| 4 | EE99 | f | 9 | Localized | <200 | Axial | NA | R1 | 1st loc, 2nd met | 33.5 | 73.8 | DOD |
| 5 | EE99 | f | 6 | Localized | <200 | Axial | Poor | R2loc | 91.7 | 91.7 | CR | |
| 6 | EE99 | m | 0.75 | Localized | <200 | Axial | Poor | R2loc | 99.9 | 99.9 | CR | |
| 7 | EE99 | m | 8 | Localized | <200 | Axial | NA | R1 | 90.9 | 90.9 | CR | |
| 8 | EE99 | m | 8 | Localized | <200 | Axial | NA | R1 | 59.9 | 59.9 | CR | |
| 9 | EE99 | f | 10 | Localized | <200 | Axial | Good | R1 | Combined | 19.1 | 24.0 | DOD |
| 10 | EE99 | f | 25 | Localized | <200 | Limb | Good | R1 | 59.0 | 59.0 | CR | |
| 11 | EE99 | m | 12 | Localized | <200 | Limb | Poor | R2loc | No CR | 12.4 | 21.7 | DOD |
| 12 | EE99 | m | 18 | Localized | >200 | Limb | Good | R1 | 11.3 | 11.3 | CR | |
| 13 | EE99 | m | 9 | Localized | >200 | Limb | Good | R1 | 63.8 | 63.8 | CR | |
| 14 | EE99 | m | 1.4 | Localized | <200 | Axial | NA | R1 | 52.6 | 52.6 | CR | |
| 15 | EE99 | m | 15 | Localized | >200 | Axial | Good | R1 | No CR | 12.2 | 15.4 | DOD |
| 16 | EE99 | f | 14 | Metastatic: bone, lung, + | >200 | Multifocal (>5) | NA | R3 | No CR | 15.0 | 36.1 | DOD |
| 17 | EE99 | m | 12 | Metastatic: bone, BM, lung, + | >200 | Multifocal (>5) | NA | R3 | No CR | 9.7 | 13.0 | DOD |
| 18 | EE99 | m | 13 | Metastatic: bone, + | <200 | Axial | NA | R3 | No CR | 12.6 | 22.1 | DOD |
| 19 | EE99 | f | 6 | Metastatic: bone | <200 | Multifocal (>5) | NA | R3 | No CR | 8.9 | 10.8 | DOD |
| 20 | EE99 | f | 19 | Metastatic: bone, BM | NA | Multifocal (>5) | NA | R3 | No CR | 5.6 | 7.1 | DOD |
| 21 | EE99 | m | 14 | Metastatic: lung, + | NA | Limb | Poor | R3 | Combined | 13.4 | 15.3 | DOD |
Sex: f = female, m = male. Disease extension: BM = bone marrow, + = other sites. Histologic response: good=<10% viable tumor cells in surgical specimen, poor=>10% viable tumor cells in surgical specimen, NA = not available due to early surgery or no surgery of the primary tumor. ‐groups EICESS92: SR = standard‐risk with localized disease and tumor volume <100 ml, HR = localized disease and tumor volume >100 ml or metastases. Risk‐groups EE99 (= EURO‐E.W.I.N.G.99): R1 = standard‐risk with localized disease and good histologic response to chemotherapy or tumor volume <200 ml if no surgery or surgery previous to chemotherapy, R2loc = high‐risk with localized disease and poor histologic response to chemotherapy or tumor volume >200 ml if no surgery or surgery previous to chemotherapy, R2pulm = high‐risk with metastases restricted to the lung, R3 = patients with metastases other than pulmonary. Event: loc = local relapse, met = distant metastasis, combined = combined local and distant relapse. CR = complete remission. EFS = event‐free survival. OS = overall‐survival. Status: CR = alive in complete remission, DOD = death of disease. NA = not available.
2.2. Cell lines
Ewing sarcoma cell lines and Rh30 alveolar rhabdomyosarcoma cells were from the institutional cell line bank. A673 cell line derivates with stable shRNA silencing of EWS‐FLI1 (EWS‐FLI1‐off) or ERG control (EWS‐FLI1‐on) were kindly provided by Prof. S. Lessnick (Huntsman Cancer Centre, University of Utah) and previously described (Smith et al., 2006). HeLa cervix carcinoma, CAPAN‐1 pancreatic adenocarcinoma, HL60 myeloid leukemia and 697 acute lymphoblastic leukemia (ALL) cell lines were from ATCC (Manassas, VA). All lines were cultured in RPMI1640 supplemented with 10% fetal bovine serum (FBS) (Life Technologies, Darmstadt, Germany) and regularly tested to be free of mycoplasma contamination.
MSC cultures were derived from tumor‐negative bone marrow aspirates of five Ewing sarcoma, two rhabdomyosarcoma, and two leukemia patients. Mononuclear cells were isolated from bone marrow by Ficoll density gradient centrifugation and cultured in uncoated tissue culture flasks in DMEM medium (high glucose and pyruvate) supplemented with 10% FBS, 2 mM l‐glutamin (Life Technologies, Darmstadt, Germany) and 3 ng/ml recombinant human basic fibroblast growth factor (PeproTech, Rocky Hill, NJ). Adherent MSC were selected by repetitive medium changes. Osteogenic and adipogenic differentiation potential was confirmed as previously described (Ern et al., 2010). Healthy‐donor peripheral blood mononuclear cells (PBMC) were isolated accordingly and directly processed for studies. Fibroblasts from skin biopsies were kindly provided by Prof. H. Omran (University Children's Hospital Münster). They were cultured in DMEM medium supplemented with FBS and 2 mM l‐glutamine (Life Technologies, Darmstadt, Germany). All primary cells were collected and utilized following informed consent and approval by ethical committees.
2.3. RNA isolation and cDNA preparation
Tumor RNA was extracted as described (Schaefer et al., 2008). Cell line RNA was extracted using the Qiagen RNeasy Mini Kit (Qiagen, Hilden, Germany). MSC RNA was harvested after 1–3 passages (14–30 days). A total of 2 μg of RNA were reverse‐transcribed using random hexamers and M‐MLV reverse transcriptase according to the manufacturer's protocol (Promega, Madison, WI).
2.4. Primers and probes
For listings of RTK families and abbreviations see Table A.1. RTK primers and probes were previously described and validated (Eurogentec, Seraing, Belgium) (Table A.2) (Müller‐Tidow et al., 2004). For Wnt5a analyses the A&B TaqMan Gene Expression Assay was used (Applied Biosystems, Foster City, CA).
2.5. Real‐time quantitative reverse transcription PCR
High‐throughput qPCR analysis was done in 384‐well plate format using the Tecan Genesis RP150 automated pipetting system (Männedorf, Switzerland) and an ABIPrism 7900 HT Sequence Detection System (Perkin–Elmer/Applied Biosystems, Foster City, CA). PCR reactions were performed in duplicate and contained 250 nM of each primer and probe in a final volume of 12 μl. Real‐time PCR conditions were 50 °C/2 min, 95 °C/10 min, followed by 40 cycles of 95 °C/15 s and 60 °C/1 min cDNA concentrations of cell line samples were adjusted to Ct values of GAPDH housekeeping control gene to ensure equal PCR amplification efficiencies. Relative gene expression levels were calculated by ΔΔCt‐method compared to GAPDH and a calibrator sample containing cDNA of HeLa, HL60, and TC32 cells. cDNA concentrations of tumor samples were highly unequal. Therefore, concentrations remained un‐adjusted and relative gene expressions were calculated based on standard curves generated by serial dilutions of the calibrator cDNA using SDS 2.1 software (Applied Biosystems, Foster City, CA) and normalized to GAPDH.
2.6. Western blotting
Procedures and buffers were previously described (Potratz et al., 2010). Antibodies were: ROR1 goat polyclonal (1:250; R&D Systems, Minneapolis, MN); PARP rabbit polyclonal; phospho‐Ser473‐AKT rabbit polyclonal (both 1:100; Cell Signaling; Beverly, MA); DVL3 mouse monoclonal (1:750); actin goat polyclonal antibody (1:1000; both Santa Cruz Biotechnology, Santa Cruz, CA). Secondary HRP‐antibodies were from Santa Cruz Biotechnology (anti‐goat, 1:3000), BD Pharmingen (anti‐rabbit, 1:5000) (Franklin Lakes, NJ), and Cell Signaling (anti‐mouse, 1:5000). Densitometric analyses were performed using ImageJ software (version 1.49v) (Schneider et al., 2012).
2.7. Flow cytometry
Cells were stained with 0.4 μg of ROR1 polyclonal antibody (R&D Systems, Minneapolis, MN) or IgG isotype control (Jackson Immunoresearch, Baltimore, PA) for 30 min at 4 °C. After washing, 0.75 μg of FITC‐conjugated secondary antibody (rabbit anti‐goat IgG; Jackson Immunoreseach, Baltimore, PA) were added and incubated for 10 min at room temperature. Following additional washes, stained cells were fixed in PBS containing 1% paraformaldehyde (PFA) and analyzed on a FACSCantoII flow cytometer (BD Bioscience, Franklin Lakes, NJ) using FACS Diva software.
2.8. siRNA transfection
Several independent ROR1 siRNAs were evaluated. Hs_ROR1_5 FlexiTube siRNA (Qiagen, Hilden, Germany) was most efficient and used in these studies. As negative control, AllStars Negative Control siRNA (Qiagen, Hilden, Germany) was used. Reverse transfection of 85 or 100 nM siRNA used 7.5 μl Lipofectamine RNAiMAX reagent (LifeTechnologies, Darmstadt, Germany) according to the manufacturer's protocol, but in RPMI medium and a transfection volume of 1 ml.
2.9. Wound‐healing assay
In 24‐well plates, cells were allowed to adhere for 24 h before setting a scratch. Cell migration was followed by bright field microscopy and photographed at time points indicated.
2.10. Migration assay
Cells were starved in FBS‐free medium for 12 h before 6 × 104 cells were seeded into ThinCert™ chambers (8 μm filter; Greiner Bio‐One, Frickenhausen, Germany) in a 24‐well plate. Recombinant Wnt5a was from R&D Systems (Minneapolis, MN). After 48 h cells on the upper surface of the filter were scraped off and cells migrated to the lower surface were fixed in 4% PFA for 10 min. Membranes were washed in PBS and DAPI‐stained for 10 min. Following additional PBS washes, membranes were mounted onto microscopy slides and migrated cells were counted in 5 fields per membrane at 100× magnification.
2.11. Compounds and viability assay
BMS‐536924 IGF1R inhibitor and D4476 casein kinase inhibitor were from SelleckChem (Houston, TX). Viability assays were performed in 96‐well plates using WST‐1 assay according to the manufacturer's protocol (Roche Applied Science, Indianapolis, IN).
2.12. Bioinformatics and statistics
Analysis and visualization of PCR data concerning RTK gene expression characteristics was performed on the R project for statistical computing (R versions up to 3.1.1. http://www.r‐project.org/). The workflow was based on standard R functions. Definitions of high‐ and low‐expression groups were established on the base‐2 logarithm of expression values. Analysis of differential gene expression was based on t‐test and multiple testing correction according to Benjamini–Hochberg (FDR) (Benjamini and Hochberg, 1995). Kaplan–Meier and COX survival analysis was performed according to the ‘survival’ package in R. Cluster analysis was of type hierarchical, average linkage utilizing the Pearson correlation as a proximity measure. Chi‐square test and Fisher test were applied to test for differences between defined risk‐groups. Both tests gave concordant results. Chi‐square test results are given throughout. Analysis of differential ROR1 gene expression and functional effects was based on t‐test. The significance level was 5 percent in all cases.
3. Results
3.1. RTK gene expression profiles of Ewing sarcoma cell lines
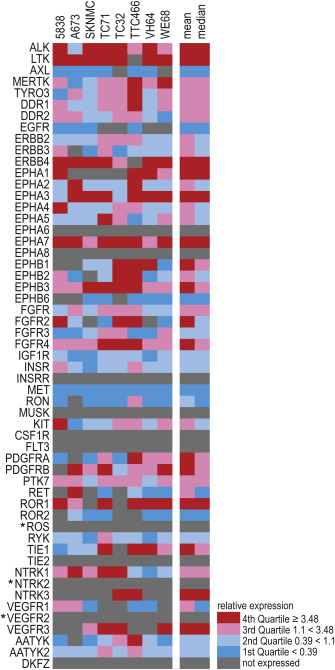
Gene expression analyses comprised 55 of 58 RTKs described in the human genome at the time of primer design, not included were three proposed RTKs of unknown cDNA sequence (EPHB4, EPHX, AATYK3) (Robinson et al., 2000). For listings of RTK families and abbreviations see Table A.1. Eight Ewing sarcoma cell lines and a calibrator sample of broad‐spectrum gene expression containing cDNAs of cancer cell lines from all three germ layers, represented by HeLa, HL60 and TC32, were analyzed. RTKs EPHA6, EPHA8, INSRR, CSF1R, FLT3, ROS1, TIE2, NTRK2, VEGFR2 and DKFZ were not detected in the calibrator sample or in at least three Ewing sarcoma cell lines, which may have served as alternative calibrator (Figure A.2). Given the limited amount of Ewing sarcoma tumor samples available, subsequent analyses were restricted to the 45 RTKs with validated expression in calibrator and cell lines.
3.2. RTK gene expression profiles of Ewing sarcomas
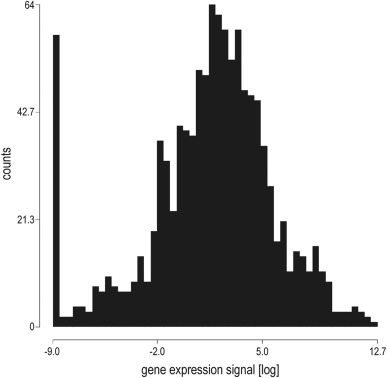
21 Ewing sarcomas were analyzed. All samples were untreated primary tumors. For patients and tumor characteristics see Table 1. Assuming Ewing sarcoma to derive from a mesenchymal precursor cell according to current perception (Toomey et al., 2010), four mesenchymal stem cell (MSC) cultures were analyzed as control. The 5% of expression signals below a near‐normal distribution were defined as not expressed (Figure A.3). RTK expression patterns are depicted as a heatmap in Figure 1A.
Figure 1.
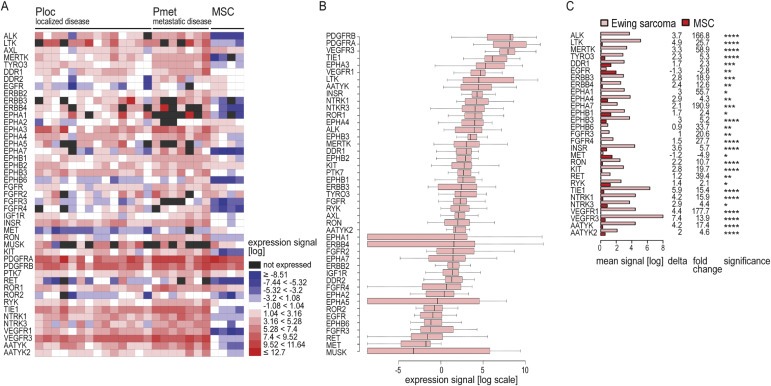
Receptor tyrosine kinase gene expression profiles of Ewing sarcomas (A) Heatmap representation of gene expressions based on log expression signal in 15 Ewing sarcoma primary tumors from localized disease (Ploc), 6 from metastatic disease (Pmet), and 4 MSC cultures. Black boxes: the 5% of expression signals (log) below a near‐normal distribution (Figure A.3) were defined as not expressed. (B) Boxplot representation of gene expressions (log). (C) Genes differentially expressed in Ewing sarcomas and MSC. Depicted are mean expression signals (log); delta, describing the difference in mean expression signals (log) of Ewing sarcomas minus MSC cultures; and fold change of mean expression signals (log) in Ewing sarcomas relative to MSC cultures. Significance: **** p < 0.0001, *** p < 0.001, ** p < 0.01, * p < 0.05.
While cluster analysis separated MSC from Ewing sarcomas with generally higher RTK expression levels, no additional clusters of tumor samples or RTKs were identified. All RTK families were represented and no predominant families were evident. Instead, often several RTK family members were expressed, underlining the potential for functional redundancy within and across RTK families. All 45 RTKs were expressed in seven of 21 tumors. ERBB4, EPHA1, EPHA5 and MUSK were expressed in ≤15 samples and showed most variable expression levels in boxplot analysis (Figure 1B). Highest median expression levels were observed for PDGFRB, PDGFRA and VEGFR3, followed by TIE1 and EPHA3. Interestingly, PDGFRA and PDGFRB were not over‐expressed, but rather expressed at similar levels compared to MSC (Figure 1A and C). Differential expressions between tumors and control cultures were observed in 29 RTKs (Figure 1C). Of these, 27 were over‐expressed in Ewing sarcomas, with strongest differences in VEGFR3, TIE1 and LTK. In contrast, EGFR and MET were under‐expressed in tumors compared to MSC. Together with EPHB6, FGFR3, RET and MUSK, they showed lowest median expression levels (Figure 1B).
3.3. Distribution of RTK expression levels across patient risk‐groups
The clinical data available with our samples allowed for analysis of RTK expressions with respect to patient risk‐groups, which were assigned according to clinical study (Table 1). Patients with localized disease were summarized as Ploc (n = 15) and patients with metastatic disease as Pmet (n = 6).
First, to better compare expression levels across the 45 RTKs, we defined high‐level expression as a signal greater than or equal to the median of all RTK signals. By this definition, high‐level expression was most frequent for TIE1, VEGFR3, INSR, and PDGFRA with 21 and 20 samples, respectively, while ROR2, MET and EGFR showed no high‐level expressions (Figure 2A (left panel)). Distribution of high‐level expressions between localized and metastatic disease groups is shown in Figure 2A (right panel).
Figure 2.
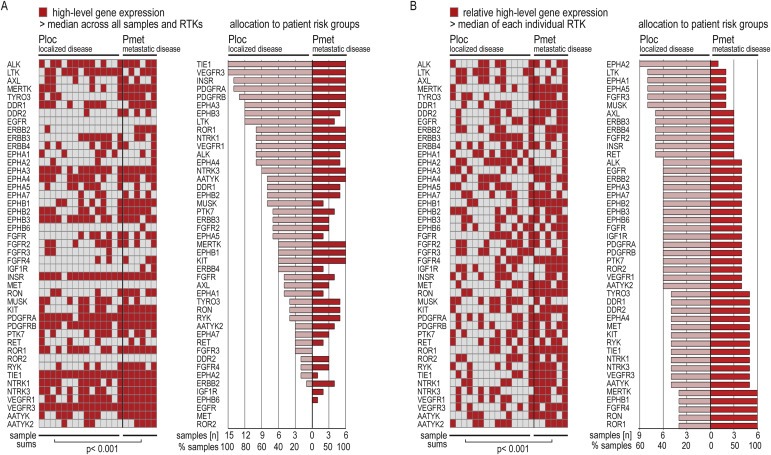
Distribution of RTK expression levels across patient risk‐groups (A) High‐level gene expressions across all RTKs. Left panel: Heatmap representation. Right panel: Allocation to tumors from localized (Ploc) and metastatic (Pmet) disease, sorted by frequency in Ploc. (B) Relative high‐level expressions within individual RTKs. Left: Heatmap representation. Right panel: Allocation to Ploc and Pmet, sorted by frequency in Ploc. For high‐level definitions see 3.3.
Second, to assess how expression levels within individual RTKs distributed between risk‐groups, we defined signals greater than or equal to the median of 21 tumor samples per individual RTK as relative high‐level expressions (Figure 2B (left panel)). By this definition, LTK, EPHA1, EPHA2, EPHA5, FGFR3 and MUSK showed relative high‐level expression predominantly in localized disease. In contrast, ROR1, RON, FGFR4, EPHB1 and MERTK were high‐level expressed in all of the six tumors from metastatic disease (Figure 2B (right panel)).
In both analyses, column sums per samples confirmed significantly more frequent high‐level RTK expressions in tumors from metastatic versus localized disease (left panels of Figure 2A and B). Thus, these expression‐based analyses suggest that, while distinct RTKs may predominate in localized versus metastatic disease, a global high‐level RTK expression marks metastatic tumors; underlining contribution of RTKs metastatic mechanisms and thus potential as metastasis‐directed targets.
3.4. RTK expression levels and prognosis in localized disease
Because we found that high‐level RTK expression was linked to metastatic disease with poor prognosis, we questioned whether this was also of prognostic impact within the localized disease group. We assigned relative high‐level expressions of individual RTKs within the 14 samples from localized disease with clinical follow‐up (Figure 3A) and performed Kaplan–Meier survival analysis. Survival of high‐level versus low‐level expressing patients was significantly different for ERBB3, EPHA7, RYK, TIE1 and VEGFR3. High‐level expression was associated with shortened survival in all cases (Figure 3B). The coarseness of plots reflects limited power of this analysis due to small sample size and only four events in this cohort. While acknowledging no more than indicative character for this analysis, these findings together with the above data (3.3) support a prognostic impact of individual RTKs.
Figure 3.
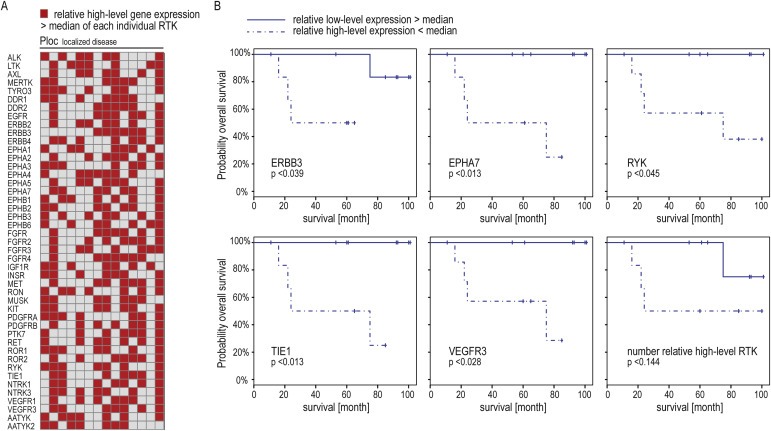
Prognostic RTKs in localized tumors (A) Heatmap representation of relative high‐level expressions within individual RTKs in 14 localized tumors. (B) Kaplan–Meier survival curves based on median expressions as cutoff. Bottom right: survival impact of global high‐level expression. Cutoff was median sum of relative high‐level expressions.
We also aimed to assess whether global relative high‐level RTK expression was of impact within this group. The definition was based on sample sums of relative high‐level RTK expressions greater or less than the median of 22. As expected, global high‐level expressers fared worse than patients with less frequent high‐level expressions, but this was not significant (Figure 3B (bottom right panel)).
3.5. RTKs over‐expressed in tumors from metastatic disease
Since global high‐level RTK expression was linked to metastatic disease and distribution analyses indicated individual RTKs with predominant high‐level expression in metastatic tumors (1, 2), we tested for RTKs with significantly different expression levels between risk‐groups. This revealed nine RTKs, all over‐expressed in tumors from metastatic versus localized disease (Figure 4). Expression levels also constituted over‐expression compared to MSC cultures in all RTKs but ERBB2. With ROR1, RON, EPHB1 and MERTK, the analysis confirmed RTKs suggest by expression level distributions (Figure 2; 3.3). Of note, RYK also was among RTKs with potential prognostic impact in localized disease (Figure 3; 3.4).
Figure 4.
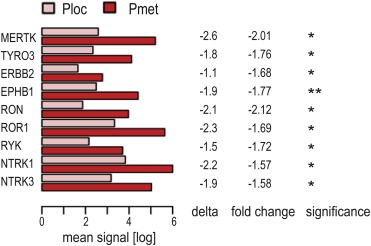
RTKs with significantly different expression between risk‐groups. Genes differentially expressed in primary tumors from localized (Ploc) and metastatic (Pmet) Ewing sarcoma. Depicted are mean expression signals (log); delta, describing the difference in mean expression signals (log) in Ploc minus Pmet tumors; and fold change of mean expression signals (log) in Ploc relative to Pmet tumors. Significance: ** p < 0.01, * p < 0.05.
3.6. ROR1 is expressed in Ewing sarcoma cell lines
Of the nine RTKs over‐expressed in tumors from metastatic disease, we chose to investigate ROR1 (receptor tyrosine kinase‐like orphan receptor 1) further; prompted by an increasing translational interest in ROR1 as targetable oncogene in leukemias and metastatic carcinomas (Bicocca et al., 2012; Cui et al., 2013; Zhang et al., 2012a). ROR1 gene and protein expressions were validated at variable levels in Ewing sarcoma cell lines, comparable to or higher than the 697 acute lymphoblastic leukemia (ALL) cell line included as positive control (Bicocca et al., 2012) (Figure 5A). Western blotting protein bands recapitulated described isoforms of 105, 115, 130 and 260 kDa (Hojjat‐Farsangi et al., 2013; Kaucká et al., 2011). The 130 kDa variant, previously assigned to mature, functional, cell surface‐located ROR1 (Hojjat‐Farsangi et al., 2013), correlated with ROR1 FACS‐expression analyses, with positive populations in 5838, A673, SKNMC and TC71 cells, but <5% positive cells in Rh30 and TC32, supporting presence of functional ROR1 in Ewing sarcoma (Figure 5B). Of note, the CAPAN‐1 cell line, included as a negative control based on published literature (Zhang et al., 2012b), here did show low‐level ROR1 expressions, greater than Rh30 and TC32. Given ROR1 gene expression in MSC (Figure 1A), we evaluated five additional MSC cultures. Gene expression was detectable in all, but 130 kDa ROR1 protein was restricted to two cultures. Fibroblasts, included to represent a normal tissue, also expressed 130 kDa ROR1, while PBMC (peripheral blood mononuclear cells) were negative.
Figure 5.
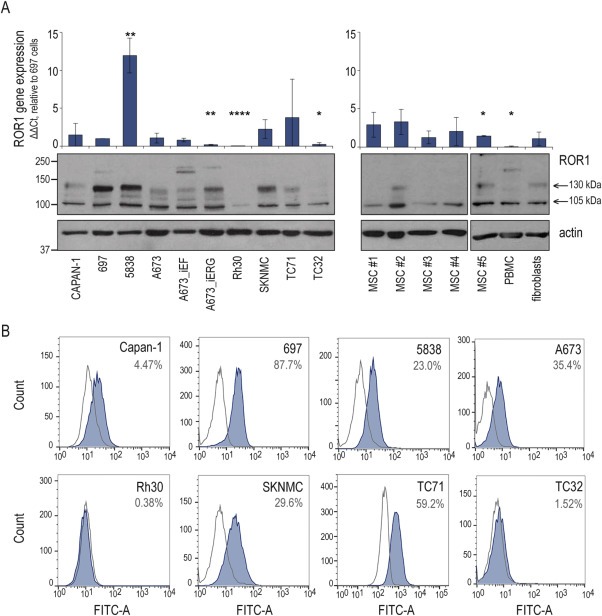
ROR1 expression in Ewing sarcoma (A) Upper panels: ROR1 gene expression. qPCR DDCt‐method relative to GAPDH and to 697 ALL positive control cells. Graphs and error bars show mean and standard deviation of at least three independent experiments. Significance: **** p<0.0001, *** p<0.001, ** p<0.01, * p<0.05. Lower panels: Western blot of ROR1 protein expression, actin as loading control. (B) FACS analysis of ROR1 cell surface expression (blue graph) and IgG control (open curve). Percentages are ROR1 positive cells. Representative of three experiments.
3.7. ROR1 silencing impairs Ewing sarcoma cell survival and migration
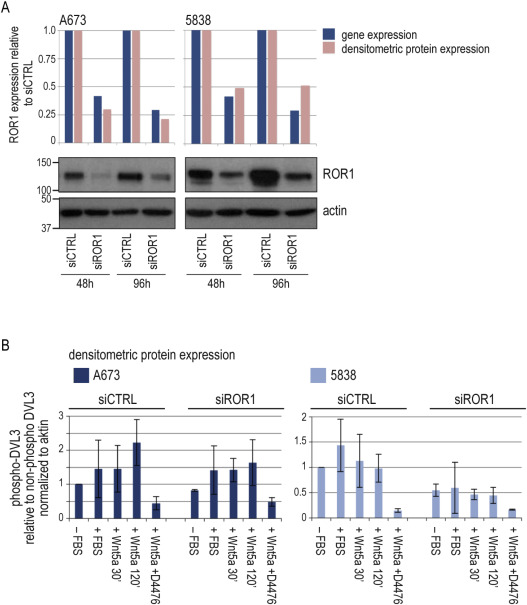
To address ROR1 cellular function in Ewing sarcoma, we utilized siRNA knockdown in A673 and 5838 cells (Figure A.4A). Loss of ROR1 impaired cell survival as shown by relative cell number and cell morphology (Figure 6A). Also, in serum‐starved conditions, preexisting apoptotic PARP cleavage appeared enhanced by ROR1 silencing. FBS‐induced AKT survival signaling was mitigated (Figure 6B). Both wound healing and migration assays, showed impaired cellular migration of ROR1 knockdown cells compared to control in A673 and 5838 cells (not statistically significant) (Figure 6C and D). ROR1 function in the cellular RTK network must be expected to be complex and beyond the scope of this first characterization. Still, with an absolute lack of data on ROR1 within the Ewing sarcoma RTK network, but a paramount role of IGF1R signaling, we tested for ROR1 influence on IGF1R co‐targeting. A dose‐response assay of the IGF1R tyrosine kinase inhibitor BMS‐536924 revealed A673 and 5838 sensitivities as previously described (Potratz et al., 2010), but no sensitizing effect of concomitant ROR1 siRNA silencing in this setting (Figure 6E).
Figure 6.
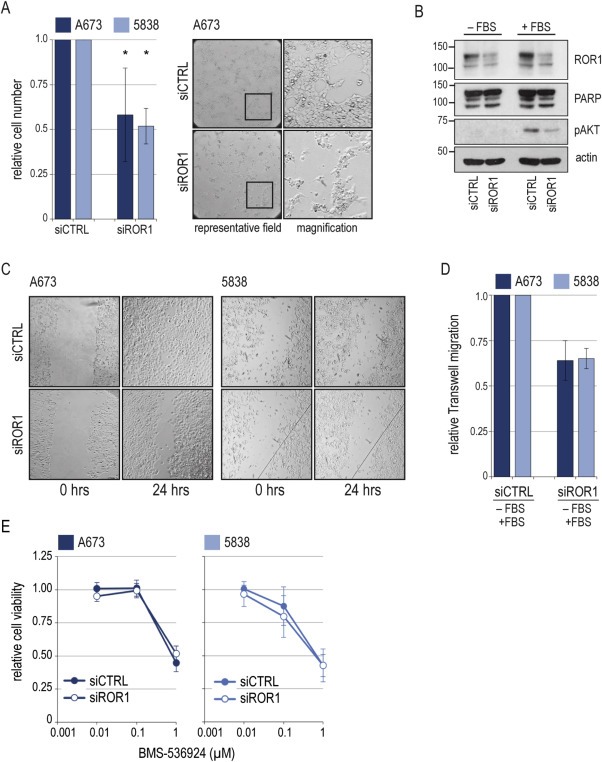
Functional effects of ROR1 silencing A673 and 5838 cells were transfected with siRNA targeting ROR1 (siROR1) or non‐silencing control (siCTRL). (A) Left panel: Viable cells were counted by trypan blue exclusion 96 h after transfection. Graphs and error bars represent mean and standard deviation (SD) of three independent experiments. Significance: * p < 0.05. Right panel: Representative brightfield microscopies of cell morphology. (B) Western blot of signaling effects. 96 h after transfection, cells were serum‐starved for 24 h and stimulated with 10% FBS for 30 min. Actin as loading control. (C) Wound‐healing assays. Wound set 96 h after transfection. (representative of three independent experiments). (D) Migration assays were performed 48 h after siRNA transfection. Insert medium was FBS‐free, migration gradient was 10% FBS. Graphs and error bars represent mean and SD of two independent experiments in triplicate. (E) Dose‐response assay of BMS‐536924 IGF1R tyrosine kinase inhibitor. Cells were treated as of 36 h after transfection for 72 h. Graphs are relative to non‐BMS‐treated control. Mean and SD of two independent transfections with two independent drug treatments each.
3.8. Wnt5a is a potential ROR1 ligand in Ewing sarcoma cells
As one mechanism of ROR1 activation, Wnt5a ligand binding has been demonstrated in chronic lymphocytic leukemia (CLL) and solid cancers (Fukuda et al., 2008; Hojjat‐Farsangi et al., 2014). Wnt5a expression analysis in Ewing sarcomas found mean expression signals of both ROR1 and Wnt5a ∼10‐fold higher in tumors from metastatic versus localized disease, and cell lines showed a trend towards correlating expression (Figure 7A). In the relative low‐level expressing A673 cell line, Wnt5a expression increased following ROR1 knockdown in the absence of serum, suggesting a regulatory loop (Figure 7B). Also, A673 cells showed chemotactic migration towards Wnt5a, which was impaired by ROR1 silencing (Figure 7C). On the signaling level, Wnt5a did not affect AKT signaling in A673 or 5838 cells in the tested conditions (Figure 7D). But, recent findings in CLL of a Wnt5a‐ROR1 mediated phosphorylation of Dishevelled (DVL) family proteins (Janovska et al., 2015) prompted to test DVL3 as such read‐out in Ewing sarcoma. The casein kinase inhibitor D4476, shown to block Wnt5a‐mediated DVL3 activation, was employed as control (Bryja et al., 2007). Indeed, Wnt5a stimulation of A673 cells resulted in DVL3 phosphorylation, mitigated by ROR1 silencing, supporting a functional Wnt5a‐ROR1 signaling axis in Ewing sarcoma (Figure 7D; mean densitometric signals of additional experiments are shown in A.4B). Interestingly, in the 5838 cell line with ∼10‐fold endogenous Wnt5a expression (Figure 7A), phosphorylated DVL3 dominated, independent of supplemented serum or Wnt5a; and was reduced by ROR1 silencing, but without apparent increase in non‐phosphorylated DVL3 levels. Together, these data reveal a ROR1 contribution to migratory pro‐metastatic capacities of Ewing sarcoma cell lines and provide first evidence of functional Wnt5a‐ROR1 signaling in Ewing sarcoma.
Figure 7.
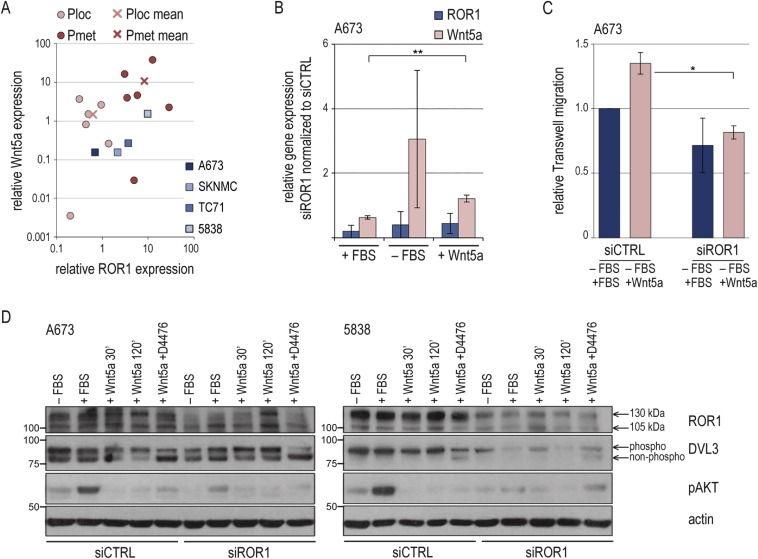
Wnt5a is a potential ROR1 ligand (A) Correlation plot of ROR1 and Wnt5a gene expressions. Wnt5a gene expression was analyzed in cell lines and 12 Ewing sarcoma specimens by qPCR and ΔΔCt‐method compared to GAPDH and MSC cultures (triplicate analysis). In cell lines, markers depict mean of at least three independent measurements. ROR1 data were as in Figures 1 and 5A. (B) 48 h after transfection, cells were serum‐starved for 12 h before supplementing 10% FBS or 400 ng/ml Wnt5a for another 12 h as indicated. ROR1 and Wnt5a gene expressions in siROR1 cells were analyzed by qPCR and ΔΔCt‐method compared to GAPDH and siCTRL (triplicate analysis). Graphs and error bars represent mean and SD of three independent experiments. Significance: ** p < 0.01, * p < 0.05. (C) Migration assays were performed 96 h after siRNA transfection. Insert medium was FBS‐free, migration gradient was 10% FBS or 400 ng/ml Wnt5a. Graphs and error bars represent mean and SD of two independent experiments. (D) Western blot of signaling effects. 48 hrs after transfection cells were serum‐starved for 12 h before stimulation with Wnt5a for 30 min or 120 min (200 ng/ml) in the presence or absence of casein kinase inhibitor D4476 (100 μM). Densitometric analysis of DVL3 is provided in Figure A.4B.
4. Discussion
In this study we provide a first complete gene expression profile focused on the receptor tyrosine kinase (RTK) signaling network in Ewing sarcoma cell lines and tumor samples. The study did not aim to identify a prognostic gene signature, but to provide a complementing baseline for additional investigative angles needed to fully unravel this network. Tumors from both localized and metastatic disease were included to address metastasis‐specific aspects. Both general aspects (4.1) and individual RTKs of interest (4.2) were revealed.
4.1. General aspects
Expression profiles represented all RTK families (Robinson et al., 2000), often with more than one family member (Figure 1), and confirm a principal potential for functional redundancy and promiscuity within and across RTK families. Further, expression profiles underline, again, that high‐level expression alone does not define oncogenic or therapeutic significance, but that comparative analyses are required. With the Ewing sarcoma cell‐of‐origin unknown but assigned to a mesenchymal precursor lineage (Riggi et al., 2008; Toomey et al., 2010), we included four MSC cultures as control. Expression patterns were homogenous across these MSC. Of note, compared to this control, Ewing sarcomas' most highly expressed RTKs, platelet‐derived growth factor receptors alpha and beta (PDGFRA/B), were not over‐expressed, but expressed at similar levels in this and published datasets (Figure 1C) (Bozzi et al., 2007; Uren et al., 2003). Together with a lack of activating mutations, this may contribute to the limited clinical efficacy of PDGFR inhibitors observed in Ewing sarcomas (Chugh et al., 2009; Do et al., 2007). Whether in turn in MSC, PDGFR expression may contribute to the specific cell‐of‐origin background that allows for EWS‐FLI1 expression and transformation (Toomey et al., 2010), remains un‐investigated. In any way, the range of RTK expression levels observed in MSC (Figure 1A) points back to the importance of a better definition of the Ewing sarcoma cell‐of‐origin, to clarify such potential RTK contributions.
To compare RTK expression profiles not alone to MSC, but with respect to patient risk‐group, we analyzed tumors from both localized and metastatic disease. Unfortunately, for metastatic disease matched primary and metastasis tissue was not available. Rather, sample numbers were limited to 15 from localized and 6 from metastatic disease, with four events in the localized‐disease group. Of note, all metastatic cases were high‐risk R3 disease and four patients presented with multifocal (>5) bone metastases. Their survival reflected published reports with particularly poor prognosis (Figure A.1B) (Haeusler et al., 2010; Ladenstein et al., 2010; Paulussen et al., 1998). For these samples it cannot be distinguished, whether they represent primary tumors of a multifocal disease subgroup, or more closely reflect biology of tumor metastases. Acknowledging these limitations and an exploratory character, we found that compared to localized disease, tumors from metastatic disease were marked by global high‐level RTK expressions and over‐expression of a subset of RTKs (2, 4). Given the vast evidence on individual RTKs' oncogenic and pro‐metastatic contributions, this is not unexpected, but now confirms these findings from an RTK family perspective. Likewise, though with limited significance due to sample numbers, our family‐focused analyses support a prognostic impact of RTKs in localized disease (Figure 3). Again, a subset of localized tumors defined as global high‐level expressers fared worse, though this did not reach significance. On individual RTK's level, RYK was revealed as potential prognostic factor in localized tumors and at the same time over‐expressed in metastatic tumors. Together, these findings prompt to hypothesize that RTKs may contribute to a molecular continuum between poor‐prognostic localized and metastatic disease, to be further investigated.
4.2. Discussion of selected receptor tyrosine kinases
4.2.1. Angiogenic VEGFR3 and TIE1 receptors
The angiogenic growth factor RTKs vascular endothelial growth factor receptor 1 (VEGFR1) and 3 (VEGFR3), and tyrosine kinase with immunoglobulin‐like and EGF‐like domains 1 (TIE1) stand out in our analyses (1, 2, 3). In Ewing sarcoma, expression, crosstalk to IGF1R signaling, and co‐targeting studies defined VEGFRs as part of the tumor cell's RTK network (Dalal et al., 2005; Kurmasheva et al., 2009). The VEGF targeting antibody bevacizumab is under clinical investigation (www.clinicaltrials.gov). But, VEGFR3 in particular and the TIE signaling system have not been studied. TIE signaling is considered to mediate stabilization and/or remodeling of vessels subsequent to VEGFR driven events and therapeutic VEGFR signaling disruption. The regulatory mechanisms and angiogenic outcome appear context and cell type dependent (J. Huang et al., 2009; Jones et al., 2001; Yun et al., 2013). With TIE ligand antagonists and novel VEGFR inhibitors of distinct targets spectrums in clinical development, TIE1 and VEGFR3 underscore both knowledge gap and therapeutic potential of the angiogenic network in Ewing sarcoma.
As RTKs primarily expressed in endothelial cells, TIE1 and VEGFR3 also emphasize the aspect of stroma content in analytic specimens. Volchenboum et al. have recently shown a role for tumor stroma in determining prognosis (Volchenboum et al., 2015), fitting the potential prognostic impact of VEGFR3 and TIE1 in this study. Yet, specimens of >65% tumor cell content and expression in Ewing sarcoma cell lines (Figure A.2) argue for tumor cell, not alone stroma expression. Of note, rare VEGFR2 and TIE2 expressions in our Ewing sarcoma cell lines led to exclusion from analysis in tumor samples, whereas other authors described VEGFR2 expression (Dalal et al., 2005; Kreuter et al., 2006). Whether tumor cell expression of angiogenic RTKs reflects a common mesenchymal precursor cell background, a tumor‐intrinsic feature, or one regulatory state of tumor‐stroma interactions remains unclear. To solve this, systematic studies on tumor and stroma tissue will be needed.
4.2.2. Ephrin receptors
Several analyses within this study point out ephrin (EPH) receptors (1, 2, 3, 4). EPH receptors are the largest RTK family, and with pro‐angiogenic and pro‐metastatic functions considered as promising therapeutic targets (Pasquale, 2010; Robinson et al., 2000). However in Ewing sarcoma, they remain orphan RTKs. Only EPHA2, here predominant in localized tumors (Figure 2), has been studied and interestingly been shown to promote tumor angiogenesis (Sáinz‐Jaspeado et al., 2013), underscoring that investigations of this complex RTK family in Ewing sarcoma are warranted.
4.2.3. Receptor tyrosine pseudo‐kinases
ERBB3, RYK, and ROR1 have been described as catalytically inactive pseudo‐kinases (Baselga and Swain, 2009; Gentile et al., 2011; Trivier and Ganesan, 2002). Yet, ERBB3 and RYK affected survival in localized disease (Figure 3B), and RYK and ROR1 were among pro‐metastatic gene candidates (Figure 4). For ERBB3, emerging evidence now challenges the classic oncogenic RTK mechanisms, but highlights the pseudo‐kinase ERBB3 as ‘active’ participant in RTK heterodimer complexes with a key role in oncogenic signaling (Baselga and Swain, 2009). Promiscuous formation of ERBB3 heterodimers was shown to mediate resistance to RTK‐targeted agents such IGF1R inhibitors (Baselga and Swain, 2009; Desbois‐Mouthon et al., 2009). In EWS‐ATF1 translocation‐positive clear cell sarcoma, constitutive ERBB3 activation conferred shortened survival (Schaefer et al., 2006), but so far, none of the above RTKs have been investigated in Ewing sarcoma. In our analyses, the most prominent ERBB3 heterodimer‐partner EGFR was only weakly expressed (Figure 1), but with family receptor ERBB2 and ERBB4 over‐expressed in metastatic disease (Figure 4 and (Mendoza‐Naranjo et al., 2013)), principle dimer partners are available. Together these findings underline pseudo‐kinases as an emerging complexity in RTK signaling networks, including Ewing sarcoma.
4.2.4. Receptor tyrosine kinase‐like orphan receptor 1 (ROR1)
ROR1 is an emerging therapeutic target. Very recently, a first publication in pediatric sarcomas demonstrated activity of ROR1‐specific CAR T cells (X. Huang et al., 2015). Our data match their FACS‐based ROR1 expression analyses (Figure 5). Beyond that, we confirm ROR1 gene expression in Ewing sarcomas tumor samples, with over‐expression in poor‐prognostic metastatic disease. Initial functional experiments reveal ROR1 contribution to cellular migration as pro‐metastatic capacity (Figure 6), underscoring ROR1 as a potential therapeutic target in metastatic disease. A first co‐targeting experiment with an IGF1R inhibitor did not reveal synergistic activities in the tested setting, underlining that broader systematic analyses will be required, to position ROR1 within the RTK network and exploit its full therapeutic potential. Beyond this, ROR1 picks up on general issues emerging in RTK signaling, such as isoform and pseudo‐kinase activities:
In chronic lymphocytic leukemia (CLL) certain ROR1 isoforms have been linked to non‐progressive versus progressive disease (105 kDa and 130 kDa isoforms, respectively) (Hojjat‐Farsangi et al., 2013). In carcinomas with undisputed ROR1 contribution to metastasis and aggressive phenotype (Cui et al., 2013; Zhang et al., 2012a), specific investigation and allocation of ROR1 isoforms is lacking. We show that Ewing sarcoma cell lines express protein band sizes correlating with CLL‐described ROR1 isoforms (Figure 5). All cell lines derived from metastatic or relapsed disease presented a 130 kDa band, which was lacking in TC32, the one line in the examined panel originating from a pre‐treatment primary tumor (Whang‐Peng et al., 1986). Given the potential for prognostic impact, this limited evidence calls for a more detailed analysis of ROR1 isoforms in solid cancers.
ROR1 isoforms furthermore gain relevance in the context of therapeutic targeting in a background of normal tissue ROR1 expression. Gene expression microarrays detected ROR1 in diverse tissues (Dave et al., 2012). Western blot analyses however did not reveal bands indicative of cell surface ROR1 protein in 28 adult and 14 pediatric normal tissues (Baskar et al., 2008; Dave et al., 2012). We did observe such bands in two of five MSC and one fibroblast culture, possibly due to culture effects (Figure 5A). PBMC were negative, as described (Bicocca et al., 2012; Hojjat‐Farsangi et al., 2013). Tissue microarray analyses found high proportions of ROR1 expression in various cancers, with negative normal counterparts (Zhang et al., 2012b). Others describe ROR1 staining in ∼half of 32 adult tissues, yet predominantly cytoplasmic and confined to certain cell types (Dave et al., 2012). At this point, subcellular ROR1 location and possible diagnostically confounding isoforms require better understanding; but active, cell surface ROR1 isoforms appear to be restricted to cancers, underscoring suitability as a therapeutic target.
ROR1 (pseudo‐)kinase activities are another open question, which we did not investigate in this study. Activities appear cell type and context dependent: In acute lymphoblastic leukemia, ROR1 activates MAPK signaling to counteract therapeutic efficacy of dasatinib, without evidence of endogenous ROR1 kinase activity (Bicocca et al., 2012). In contrast, ROR1‐mediated activation of SRC and p38 signaling in NSCLC was kinase‐dependent; whereas at the same time ROR1‐sustained EGFR‐ERBB3 dimer signaling was found kinase‐independent (Yamaguchi et al., 2012). As for mechanisms of ROR1 activation, Gentile et al. described ROR1 binding to constitutively active kinases EGFR and ERBB2 in carcinomas; its activation however, selectively depending on MET trans‐activation (Gentile et al., 2011). Activated MET was not detected in all tested Ewing sarcoma cell lines. In Wnt5a, an alternative, RTK‐independent activation mechanism of ROR1 signaling has been demonstrated, but not investigated in sarcomas (Zhang et al., 2012a). In addition to previously published Wnt expression in Ewing sarcoma cell lines (Uren et al., 2004), we now present first evidence of Wnt5a gene expression in tumor samples (Figure 7), and of functional Wnt5a‐ROR1 signaling in Ewing sarcoma; its activation and regulation mechanisms, as well as functional consequences, remaining to be studied.
This cumulative evidence, in the sense of 4.2.3, underscores a role for the pseudo‐kinase ROR1 as ‘active’ participant, signal modifier and/or enhancer in the RTK network of Ewing sarcomas, whose detailed interconnections remain open.
5. Conclusion
Comprehensive RTK gene expression profiles of Ewing sarcoma cell lines and untreated tumors are presented. Additional analyses with regard to patient risk‐group offer several individual RTKs to be explored as potential therapeutic targets. Here, we emphasize the indicative character of the data. Nonetheless, together they underscore emerging general issues in RTK signaling and challenge our perception of the RTK network in Ewing sarcoma: Angiogenic signaling axes and tumor‐stroma interactions of targeting relevance; EPH receptor signaling demanding better characterization; and receptor tyrosine pseudo‐kinases and isoforms emerging as additional complexities. The latter are only two open issues of interest in ROR1. Initial investigations presented here confirm ROR1 expression and function in metastatic cellular features and identify a Wnt5a‐ROR1 signaling axis, calling for further evaluation of ROR1 as therapeutic target in high‐risk Ewing sarcoma, particularly with ROR1‐targeted agents in development.
Financial support
The studies were supported by Bundesministerium für Bildung und Forschung (BMBF) [01GM0869] to J.P., E.K. and U.D.; the ERA‐Net‐ TRANSCAN consortium PROVABES for personalized translational medicine [01KT1310] to E.K. and U.D.; the Euro Ewing Consortium [602856‐2] and German Cancer Aid [108128] to U.D.; and by the fund “Innovative Medical Research” (IMF) of the University of Münster Medical School [PO211411] to J.P. A.T. received an IMF student scholarship [TI611001]. The funding sources had no involvement in study design; collection, analysis and interpretation of data; in the writing of the manuscript; and in the decision to submit the article for publication.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
The following are the supplementary data related to this article:
Table A.1 List of RTK families, abbreviations and descriptions Classification of receptor tyrosine kinases into families according to Robinson et al. (Robinson et al., 2000). Gene names and description are according to NCBI database (http://www.ncbi.nlm.nih.gov). Alias given where applied in this paper. Proposed RTK of unknown mRNA/cDNA sequence at the time of primer design marked (*) (Robinson et al., 2000).
Table A.2 List of primers and probes Primers and probes as previously described (Müller‐Tidow et al., 2004, 2005). GenBank number as at the time of primer design.
Figure A.1 Kaplan–Meier survival analysis of patient cohort Kaplan–Meier analysis of overall‐survival in 20 Ewing sarcoma patients based on (A) patient age and (B) metastatic status.
{kind=link}
Figure A.2 RTK gene expression profiles of Ewing sarcoma cell lines Relative RTK gene expressions of eight Ewing sarcoma cell lines as calculated by ΔΔCt‐method compared to GAPDH and a calibrator sample. Heatmap depicts fold expression according to quartiles calculated across all samples and RTKs. * = RTKs detected in <3 cell lines and negative in calibrator.
{kind=link}
Figure A.3 Distribution of RTK gene expression signals Distribution of RTK gene expression signals (log) in Ewing sarcomas and MSC controls. The column of 58 signals below a near‐normal distribution was defined as not expressed.
{kind=link}
Figure A.4 Functional effects of ROR1 silencing (A) Time‐course of ROR1 siRNA knockdown. A673 and 5838 cells were transfected with siRNA targeting ROR1 (siROR1) or non‐silencing control (siCTRL). At time points indicated, ROR1 gene expression was analyzed by qPCR by ΔΔCt‐method compared to GAPDH and siCTRL samples (upper panels). In parallel, ROR1 protein expression was assessed by Western blot (lower panels). Bands were analyzed by densitometry (upper panels). (B) Densitometric analysis of DVL3 protein bands. Intensities of DVL3 bands were normalized to corresponding actin loading control. Phospho‐DVL3 is displayed relative to non‐phosphorylated, lower‐band DVL3. Mean and SD of at least two independent experiments as shown in Figure 7D.
{kind=link}
Acknowledgements
We thank Andreas Ranft of the Cooperative Ewing Sarcoma Study group for supplying clinical data, Martina Ahlmann for helpful discussions of the manuscript, and Stephanie Klco‐Brosius for proofreading.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2015.12.009.
Potratz Jenny, Tillmanns Amelie, Berning Philipp, Korsching Eberhard, Schaefer Christiane, Lechtape Birgit, Schleithoff Carolin, Unland Rebekka, Schäfer Karl-Ludwig, Müller-Tidow Carsten, Jürgens Heribert, Dirksen Uta, (2016), Receptor tyrosine kinase gene expression profiles of Ewing sarcomas reveal ROR1 as a potential therapeutic target in metastatic disease, Molecular Oncology, 10, doi: 10.1016/j.molonc.2015.12.009.
References
- Baselga, J. , Swain, S.M. , 2009. Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat. Rev. Cancer. 9, 463–475. [DOI] [PubMed] [Google Scholar]
- Baskar, S. , Kwong, K.Y. , Hofer, T. , Levy, J.M. , Kennedy, M.G. , Lee, E. , Staudt, L.M. , Wilson, W.H. , Wiestner, A. , Rader, C. , 2008. Unique cell surface expression of receptor tyrosine kinase ROR1 in human B-cell chronic lymphocytic leukemia. Clin. Cancer Res. 14, 396–404. [DOI] [PubMed] [Google Scholar]
- Benjamini, Y. , Hochberg, Y. , 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodological). 57, 289–300. [Google Scholar]
- Bernstein, M. , Kovar, H. , Paulussen, M. , Randall, R.L. , Schuck, A. , Teot, L.A. , Juergens, H. , 2006. Ewing's sarcoma family of tumors: current management. Oncologist. 11, 503–519. [DOI] [PubMed] [Google Scholar]
- Bicocca, V.T. , Chang, B.H. , Masouleh, B.K. , Muschen, M. , Loriaux, M.M. , Druker, B.J. , Tyner, J.W. , 2012. Crosstalk between ROR1 and the Pre-B cell receptor promotes survival of t(1;19) acute lymphoblastic leukemia. Cancer Cell. 22, 656–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozzi, F. , Tamborini, E. , Negri, T. , Pastore, E. , Ferrari, A. , Luksch, R. , Casanova, M. , Pierotti, M.A. , Bellani, F.F. , Pilotti, S. , 2007. Evidence for activation of KIT, PDGFRalpha, and PDGFRbeta receptors in the Ewing sarcoma family of tumors. Cancer. 109, 1638–1645. [DOI] [PubMed] [Google Scholar]
- Bryja, V. , Schulte, G. , Rawal, N. , Grahn, A. , Arenas, E. , 2007. Wnt-5a induces Dishevelled phosphorylation and dopaminergic differentiation via a CK1-dependent mechanism. J. Cel. Sci. 120, 586–595. [DOI] [PubMed] [Google Scholar]
- Chakravarti, A. , Loeffler, J.S. , Dyson, N.J. , 2002. Insulin-like growth factor receptor I mediates resistance to anti-epidermal growth factor receptor therapy in primary human glioblastoma cells through continued activation of phosphoinositide 3-kinase signaling. Cancer Res. 62, 200–207. [PubMed] [Google Scholar]
- Chugh, R. , Wathen, J.K. , Maki, R.G. , Benjamin, R.S. , Patel, S.R. , Meyers, P.A. , Myers, P.A. , Priebat, D.A. , Reinke, D.K. , Thomas, D.G. , Keohan, M.L. , Samuels, B.L. , Baker, L.H. , 2009. Phase II multicenter trial of imatinib in 10 histologic subtypes of sarcoma using a Bayesian hierarchical statistical model. J. Clin. Oncol. 27, 3148–3153. [DOI] [PubMed] [Google Scholar]
- Cotterill, S.J. , Ahrens, S. , Paulussen, M. , Jürgens, H.F. , Voûte, P.A. , Gadner, H. , Craft, A.W. , 2000. Prognostic factors in Ewing“s tumor of bone: analysis of 975 patients from the European Intergroup Cooperative Ewing”s Sarcoma Study Group. J. Clin. Oncol. 18, 3108–3114. [DOI] [PubMed] [Google Scholar]
- Cui, B. , Zhang, S. , Chen, L. , Yu, J. , Widhopf, G.F. , Fecteau, J.-F. , Rassenti, L.Z. , Kipps, T.J. , 2013. Targeting ROR1 inhibits epithelial-mesenchymal transition and metastasis. Cancer Res. 73, 3649–3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalal, S. , Berry, A.M. , Cullinane, C.J. , Mangham, D.C. , Grimer, R. , Lewis, I.J. , Johnston, C. , Laurence, V. , Burchill, S.A. , 2005. Vascular endothelial growth factor: a therapeutic target for tumors of the Ewing's sarcoma family. Clin. Cancer Res. 11, 2364–2378. [DOI] [PubMed] [Google Scholar]
- Dave, H. , Anver, M.R. , Butcher, D.O. , Brown, P. , Khan, J. , Wayne, A.S. , Baskar, S. , Rader, C. , 2012. Restricted cell surface expression of receptor tyrosine kinase ROR1 in pediatric B-lineage acute lymphoblastic leukemia suggests targetability with therapeutic monoclonal antibodies. PLoS ONE. 7, e52655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desbois-Mouthon, C. , Baron, A. , Blivet-Van Eggelpoël, M.-J. , Fartoux, L. , Venot, C. , Bladt, F. , Housset, C. , Rosmorduc, O. , 2009. Insulin-like growth factor-1 receptor inhibition induces a resistance mechanism via the epidermal growth factor receptor/HER3/AKT signaling pathway: rational basis for cotargeting insulin-like growth factor-1 receptor and epidermal growth factor receptor in hepatocellular carcinoma. Clin. Cancer Res. 15, 5445–5456. [DOI] [PubMed] [Google Scholar]
- Do, I. , Araujo, E.S. , Kalil, R.K. , Bacchini, P. , Bertoni, F. , Unni, K.K. , Park, Y.-K. , 2007. Protein expression of KIT and gene mutation of c-kit and PDGFRs in Ewing sarcomas. Pathol. Res. Pract. 203, 127–134. [DOI] [PubMed] [Google Scholar]
- Engelman, J.A. , Zejnullahu, K. , Mitsudomi, T. , Song, Y. , Hyland, C. , Park, J.O. , Lindeman, N. , Gale, C.-M. , Zhao, X. , Christensen, J. , Kosaka, T. , Holmes, A.J. , Rogers, A.M. , Cappuzzo, F. , Mok, T. , Lee, C. , Johnson, B.E. , Cantley, L.C. , Jänne, P.A. , 2007. MET amplification leads to Gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 316, 1039–1043. [DOI] [PubMed] [Google Scholar]
- Ern, C. , Krump-Konvalinkova, V. , Docheva, D. , Schindler, S. , Rossmann, O. , Böcker, W. , Mutschler, W. , Schieker, M. , 2010. Interactions of human endothelial and multipotent mesenchymal stem cells in cocultures. Open Biomed. Eng. J. 4, 190–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda, T. , Chen, L. , Endo, T. , Tang, L. , Lu, D. , Castro, J.E. , Widhopf, G.F. , Rassenti, L.Z. , Cantwell, M.J. , Prussak, C.E. , Carson, D.A. , Kipps, T.J. , 2008. Antisera induced by infusions of autologous Ad-CD154-leukemia B cells identify ROR1 as an oncofetal antigen and receptor for Wnt5a. Proc. Natl. Acad. Sci. U.S.A. 105, 3047–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentile, A. , Lazzari, L. , Benvenuti, S. , Trusolino, L. , Comoglio, P.M. , 2011. Ror1 is a pseudokinase that is crucial for Met-driven tumorigenesis. Cancer Res. 71, 3132–3141. [DOI] [PubMed] [Google Scholar]
- Gschwind, A. , Fischer, O.M. , Ullrich, A. , 2004. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat. Rev. Cancer. 4, 361–370. [DOI] [PubMed] [Google Scholar]
- Gurney, J.G. , Swensen, A.R. , Bulterys, M. , 1999. Malignant bone tumors. In Ries L., Smith M.A., Gurney J.G.(Eds.), Cancer Incidence and Survival Among Children and Adolescents. 99–110. [Google Scholar]
- Haeusler, J. , Ranft, A. , Boelling, T. , Gosheger, G. , Braun-Munzinger, G. , Vieth, V. , Burdach, S. , van den Berg, H. , Juergens, H. , Dirksen, U. , 2010. The value of local treatment in patients with primary, disseminated, multifocal Ewing sarcoma (PDMES). Cancer. 116, 443–450. [DOI] [PubMed] [Google Scholar]
- Hojjat-Farsangi, M. , Khan, A.S. , Daneshmanesh, A.H. , Moshfegh, A. , Sandin, A. , Mansouri, L. , Palma, M. , Lundin, J. , Österborg, A. , Mellstedt, H. , 2013. The tyrosine kinase receptor ROR1 is constitutively phosphorylated in chronic lymphocytic leukemia (CLL) cells. PLoS ONE. 8, e78339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hojjat-Farsangi, M. , Moshfegh, A. , Daneshmanesh, A.H. , Khan, A.S. , Mikaelsson, E. , Österborg, A. , Mellstedt, H. , 2014. The receptor tyrosine kinase ROR1-An oncofetal antigen for targeted cancer therapy. Semin. Cancer Biol. 29C, 21–31. [DOI] [PubMed] [Google Scholar]
- Huang, F. , Hurlburt, W. , Greer, A. , Reeves, K.A. , Hillerman, S. , Chang, H. , Fargnoli, J. , Graf Finckenstein, F. , Gottardis, M.M. , Carboni, J.M. , 2010. Differential mechanisms of acquired resistance to insulin-like growth factor-i receptor antibody therapy or to a small-molecule inhibitor, BMS-754807, in a human rhabdomyosarcoma model. Cancer Res. 70, 7221–7231. [DOI] [PubMed] [Google Scholar]
- Huang, J. , Bae, J.-O. , Tsai, J.P. , Kadenhe-Chiweshe, A. , Papa, J. , Lee, A. , Zeng, S. , Kornfeld, Z.N. , Ullner, P. , Zaghloul, N. , Ioffe, E. , Nandor, S. , Burova, E. , Holash, J. , Thurston, G. , Rudge, J. , Yancopoulos, G.D. , Yamashiro, D.J. , Kandel, J.J. , 2009. Angiopoietin-1/Tie-2 activation contributes to vascular survival and tumor growth during VEGF blockade. Int. J. Oncol. 34, 79–87. [PMC free article] [PubMed] [Google Scholar]
- Huang, X. , Park, H. , Greene, J. , Pao, J. , Mulvey, E. , Zhou, S.X. , Albert, C.M. , Moy, F. , Sachdev, D. , Yee, D. , Rader, C. , Hamby, C.V. , Loeb, D.M. , Cairo, M.S. , Zhou, X. , 2015. IGF1R- and ROR1-specific CAR t cells as a potential therapy for high risk sarcomas. PLoS One. 10, e0133152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janovska, P. , Poppova, L. , Plevova, K. , Plesingerova, H. , Behal, M. , Kaucka, M. , Ovesna, P. , Hlozkova, M. , Borsky, M. , Stehlikova, O. , Brychtova, Y. , Doubek, M. , Machalova, M. , Baskar, S. , Kozubik, A. , Pospisilova, S. , Pavlova, S. , Bryja, V. , 2015. Autocrine signaling by Wnt-5a deregulates chemotaxis of leukemic cells and predicts clinical outcome in chronic lymphocytic leukemia. Clin. Cancer Res. Clincanres. 10.1158/1078-0432.CCR-15-0154 0154.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jänne, P.A. , Gray, N. , Settleman, J. , 2009. Factors underlying sensitivity of cancers to small-molecule kinase inhibitors. Nat. Rev. Drug Discov. 8, 709–723. [DOI] [PubMed] [Google Scholar]
- Jones, N. , Iljin, K. , Dumont, D.J. , Alitalo, K. , 2001. Tie receptors: new modulators of angiogenic and lymphangiogenic responses. Nat. Rev. Mol. Cell Biol. 2, 257–267. [DOI] [PubMed] [Google Scholar]
- Juergens, H. , Daw, N.C. , Geoerger, B. , Ferrari, S. , Villarroel, M. , Aerts, I. , Whelan, J. , Dirksen, U. , Hixon, M.L. , Yin, D. , Wang, T. , Green, S. , Paccagnella, L. , Gualberto, A. , 2011. Preliminary efficacy of the anti-insulin-like growth factor type 1 receptor antibody figitumumab in patients with refractory Ewing sarcoma. J. Clin. Oncol. 29, 4534–4540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun, H.J. , Bronson, R.T. , Charest, A. , 2014. Inhibition of EGFR induces a c-MET-driven stem cell population in glioblastoma. Stem Cells. 32, 338–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaucká, M. , Krejčí, P. , Plevová, K. , Pavlová, S. , Procházková, J. , Janovská, P. , Valnohová, J. , Kozubík, A. , Pospíšilová, S. , Bryja, V. , 2011. Post-translational modifications regulate signalling by Ror1. Acta Physiol. (Oxf). 203, 351–362. [DOI] [PubMed] [Google Scholar]
- Kreuter, M. , Paulussen, M. , Boeckeler, J. , Gerss, J. , Buerger, H. , Liebscher, C. , Kessler, T. , Jurgens, H. , Berdel, W.E. , Mesters, R.M. , 2006. Clinical significance of vascular endothelial growth Factor-A expression in Ewing's sarcoma. Eur. J. Cancer. 42, 1904–1911. [DOI] [PubMed] [Google Scholar]
- Krishnan, K. , Khanna, C. , Helman, L.J. , 2005. The biology of metastases in pediatric sarcomas. Cancer J. 11, 306–313. [DOI] [PubMed] [Google Scholar]
- Kurmasheva, R.T. , Dudkin, L. , Billups, C. , Debelenko, L.V. , Morton, C.L. , Houghton, P.J. , 2009. The insulin-like growth factor-1 receptor-targeting antibody, CP-751,871, suppresses tumor-derived VEGF and synergizes with rapamycin in models of childhood sarcoma. Cancer Res. 69, 7662–7671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladenstein, R. , Pötschger, U. , Le Deley, M.C. , Whelan, J. , Paulussen, M. , Oberlin, O. , van den Berg, H. , Dirksen, U. , Hjorth, L. , Michon, J. , Lewis, I. , Craft, A. , Jurgens, H. , 2010. Primary disseminated multifocal Ewing sarcoma: results of the Euro-EWING 99 trial. J. Clin. Oncol. 28, 3284–3291. [DOI] [PubMed] [Google Scholar]
- Le Deley, M.C. , Paulussen, M. , Lewis, I. , Brennan, B. , Ranft, A. , Whelan, J. , Le Teuff, G. , Michon, J. , Ladenstein, R. , Marec-Bérard, P. , van den Berg, H. , Hjorth, L. , Wheatley, K. , Judson, I. , Juergens, H. , Craft, A. , Oberlin, O. , Dirksen, U. , 2014. Cyclophosphamide compared with ifosfamide in consolidation treatment of standard-risk Ewing sarcoma: results of the randomized noninferiority Euro-EWING99-R1 trial. J. Clin. Oncol. 32, 2440–2448. [DOI] [PubMed] [Google Scholar]
- Lemmon, M.A. , Schlessinger, J. , 2010. Cell signaling by receptor tyrosine kinases. Cell. 141, 1117–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza-Naranjo, A. , El-Naggar, A. , Wai, D.H. , Mistry, P. , Lazic, N. , Ayala, F.R.R. , da Cunha, I.W. , Rodriguez-Viciana, P. , Cheng, H. , Tavares Guerreiro Fregnani, J.H. , Reynolds, P. , Arceci, R.J. , Nicholson, A. , Triche, T.J. , Soares, F.A. , Flanagan, A.M. , Wang, Y.Z. , Strauss, S.J. , Sorensen, P.H. , 2013. ERBB4 confers metastatic capacity in Ewing sarcoma. EMBO Mol. Med. 5, 1019–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller-Tidow, C. , Diederichs, S. , Bulk, E. , Pohle, T. , Steffen, B. , Schwäble, J. , Plewka, S. , Thomas, M. , Metzger, R. , Schneider, P.M. , Brandts, C.H. , Berdel, W.E. , Serve, H. , 2005. Identification of metastasis-associated receptor tyrosine kinases in non-small cell lung cancer. Cancer Res. 65, 1778–1782. [DOI] [PubMed] [Google Scholar]
- Müller-Tidow, C. , Schwäble, J. , Steffen, B. , Tidow, N. , Brandt, B. , Becker, K. , Schulze-Bahr, E. , Halfter, H. , Vogt, U. , Metzger, R. , Schneider, P.M. , Büchner, T. , Brandts, C. , Berdel, W.E. , Serve, H. , 2004. High-throughput analysis of genome-wide receptor tyrosine kinase expression in human cancers identifies potential novel drug targets. Clin. Cancer Res. 10, 1241–1249. [DOI] [PubMed] [Google Scholar]
- Ohali, A. , Avigad, S. , Zaizov, R. , Ophir, R. , Horn-Saban, S. , Cohen, I.J. , Meller, I. , Kollender, Y. , Issakov, J. , Yaniv, I. , 2004. Prediction of high risk Ewing's sarcoma by gene expression profiling. Oncogene. 23, 8997–9006. [DOI] [PubMed] [Google Scholar]
- Olmos, D. , Tan, D.S.W. , Jones, R.L. , Judson, I.R. , 2010. Biological rationale and current clinical experience with anti-insulin-like growth factor 1 receptor monoclonal antibodies in treating sarcoma: twenty years from the bench to the bedside. Cancer J. 16, 183–194. [DOI] [PubMed] [Google Scholar]
- Pappo, A.S. , Patel, S.R. , Crowley, J. , Reinke, D.K. , Kuenkele, K.-P. , Chawla, S.P. , Toner, G.C. , Maki, R.G. , Meyers, P.A. , Chugh, R. , Ganjoo, K.N. , Schuetze, S.M. , Juergens, H. , Leahy, M.G. , Geoerger, B. , Benjamin, R.S. , Helman, L.J. , Baker, L.H. , 2011. R1507, a monoclonal antibody to the insulin-like growth factor 1 receptor, in patients with recurrent or refractory Ewing sarcoma family of tumors: results of a phase II sarcoma Alliance for research through Collaboration study. J. Clin. Oncol. 29, 4541–4547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquale, E.B. , 2010. Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat. Rev. Cancer. 10, 165–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulussen, M. , Ahrens, S. , Burdach, S. , Craft, A. , Dockhorn-Dworniczak, B. , Dunst, J. , Fröhlich, B. , Winkelmann, W. , Zoubek, A. , Jürgens, H. , 1998. Primary metastatic (stage IV) Ewing tumor: survival analysis of 171 patients from the EICESS studies. Eur. Intergroup Coop. Ewing Sarcoma Stud. Ann. Oncol. 9, 275–281. [DOI] [PubMed] [Google Scholar]
- European Intergroup Cooperative Ewing's Sarcoma Study-92 Paulussen, M. , Craft, A.W. , Lewis, I. , Hackshaw, A. , Douglas, C. , Dunst, J. , Schuck, A. , Winkelmann, W. , Köhler, G. , Poremba, C. , Zoubek, A. , Ladenstein, R. , van den Berg, H. , Hunold, A. , Cassoni, A. , Spooner, D. , Grimer, R. , Whelan, J. , McTiernan, A. , Jürgens, H. , 2008. Results of the EICESS-92 Study: two randomized trials of Ewing's sarcoma treatment–cyclophosphamide compared with ifosfamide in standard-risk patients and assessment of benefit of etoposide added to standard treatment in high-risk patients. J. Clin. Oncol. 26, 4385–4393. [DOI] [PubMed] [Google Scholar]
- Potratz, J.C. , Saunders, D.N. , Wai, D.H. , Ng, T.L. , McKinney, S.E. , Carboni, J.M. , Gottardis, M.M. , Triche, T.J. , Jurgens, H. , Pollak, M.N. , Aparicio, S.A. , Sorensen, P.H.B. , 2010. Synthetic lethality screens reveal RPS6 and MST1R as modifiers of insulin-like growth factor-1 receptor inhibitor activity in childhood sarcomas. Cancer Res. 70, 8770–8781. [DOI] [PubMed] [Google Scholar]
- Riggi, N. , Suvà, M.-L. , Suvà, D. , Cironi, L. , Provero, P. , Tercier, S. , Joseph, J.-M. , Stehle, J.-C. , Baumer, K. , Kindler, V. , Stamenkovic, I. , 2008. EWS-FLI-1 expression triggers a Ewing's sarcoma initiation program in primary human mesenchymal stem cells. Cancer Res. 68, 2176–2185. [DOI] [PubMed] [Google Scholar]
- Robinson, D.R. , Wu, Y.M. , Lin, S.F. , 2000. The protein tyrosine kinase family of the human genome. Oncogene. 19, 5548–5557. [DOI] [PubMed] [Google Scholar]
- Sáinz-Jaspeado, M. , Huertas-Martinez, J. , Lagares-Tena, L. , Martin Liberal, J. , Mateo-Lozano, S. , de Alava, E. , de Torres, C. , Mora, J. , Del Muro, X.G. , Tirado, O.M. , 2013. EphA2-induced angiogenesis in ewing sarcoma cells works through bFGF production and is dependent on caveolin-1. PLoS ONE. 8, e71449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer, K.-L. , Brachwitz, K. , Braun, Y. , Diallo, R. , Wai, D.H. , Zahn, S. , Schneider, D.T. , Kuhnen, C. , Vollmann, A. , Brockhoff, G. , Gabbert, H.E. , Poremba, C. , 2006. Constitutive activation of neuregulin/ERBB3 signaling pathway in clear cell sarcoma of soft tissue. Neoplasia. 8, 613–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer, K.-L. , Eisenacher, M. , Braun, Y. , Brachwitz, K. , Wai, D.H. , Dirksen, U. , Lanvers-Kaminsky, C. , Juergens, H. , Herrero, D. , Stegmaier, S. , Koscielniak, E. , Eggert, A. , Nathrath, M. , Gosheger, G. , Schneider, D.T. , Bury, C. , Diallo-Danebrock, R. , Ottaviano, L. , Gabbert, H.E. , Poremba, C. , 2008. Microarray analysis of Ewing's sarcoma family of tumours reveals characteristic gene expression signatures associated with metastasis and resistance to chemotherapy. Eur. J. Cancer. 44, 699–709. [DOI] [PubMed] [Google Scholar]
- Schneider, C.A. , Rasband, W.S. , Eliceiri, K.W. , 2012. NIH image to ImageJ: 25 years of image analysis. Nat. Methods. 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shor, A.C. , Keschman, E.A. , Lee, F.Y. , Muro-Cacho, C. , Letson, G.D. , Trent, J.C. , Pledger, W.J. , Jove, R. , 2007. Dasatinib inhibits migration and invasion in diverse human sarcoma cell lines and induces apoptosis in bone sarcoma cells dependent on SRC kinase for survival. Cancer Res. 67, 2800–2808. [DOI] [PubMed] [Google Scholar]
- Smith, R. , Owen, L.A. , Trem, D.J. , Wong, J.S. , Whangbo, J.S. , Golub, T.R. , Lessnick, S.L. , 2006. Expression profiling of EWS/FLI identifies NKX2.2 as a critical target gene in Ewing's sarcoma. Cancer Cell. 9, 405–416. [DOI] [PubMed] [Google Scholar]
- Steeg, P.S. , 2006. Tumor metastasis: mechanistic insights and clinical challenges. Nat. Med. 12, 895–904. [DOI] [PubMed] [Google Scholar]
- Stommel, J.M. , Kimmelman, A.C. , Ying, H. , Nabioullin, R. , Ponugoti, A.H. , Wiedemeyer, R. , Stegh, A.H. , Bradner, J.E. , Ligon, K.L. , Brennan, C. , Chin, L. , DePinho, R.A. , 2007. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science. 318, 287–290. [DOI] [PubMed] [Google Scholar]
- Tolcher, A.W. , Sarantopoulos, J. , Patnaik, A. , Papadopoulos, K. , Lin, C.-C. , Rodon, J. , Murphy, B. , Roth, B. , McCaffery, I. , Gorski, K.S. , Kaiser, B. , Zhu, M. , Deng, H. , Friberg, G. , Puzanov, I. , 2009. Phase I, pharmacokinetic, and pharmacodynamic study of AMG 479, a fully human monoclonal antibody to insulin-like growth factor receptor 1. J. Clin. Oncol. 27, 5800–5807. [DOI] [PubMed] [Google Scholar]
- Toomey, E.C. , Schiffman, J.D. , Lessnick, S.L. , 2010. Recent advances in the molecular pathogenesis of Ewing's sarcoma. Oncogene. 29, 4504–4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivier, E. , Ganesan, T.S. , 2002. RYK, a catalytically inactive receptor tyrosine kinase, associates with EphB2 and EphB3 but does not interact with AF-6. J. Biol. Chem. 277, 23037–23043. [DOI] [PubMed] [Google Scholar]
- Uren, A. , Merchant, M.S. , Sun, C.J. , Vitolo, M.I. , Sun, Y. , Tsokos, M. , Illei, P.B. , Ladanyi, M. , Passaniti, A. , Mackall, C. , Toretsky, J.A. , 2003. Beta-platelet-derived growth factor receptor mediates motility and growth of Ewing's sarcoma cells. Oncogene. 22, 2334–2342. [DOI] [PubMed] [Google Scholar]
- Uren, A. , Wolf, V. , Sun, Y.-F. , Azari, A. , Rubin, J.S. , Toretsky, J.A. , 2004. Wnt/Frizzled signaling in Ewing sarcoma. Pediatr. Blood Cancer. 43, 243–249. [DOI] [PubMed] [Google Scholar]
- Valastyan, S. , Weinberg, R.A. , 2011. Tumor metastasis: molecular insights and evolving paradigms. Cell. 147, 275–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volchenboum, S.L. , Andrade, J. , Huang, L. , Barkauskas, D.A. , Krailo, M. , Womer, R.B. , Ranft, A. , Potratz, J. , Dirksen, U. , Triche, T.J. , Lawlor, E.R. , 2015. Gene expression profiling of Ewing sarcoma tumours reveals the prognostic importance of tumour-stromal interactions: a report from the Children's oncology Group. J. Path: Clin. Res. 1, (2) 83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whang-Peng, J. , Triche, T.J. , Knutsen, T. , Miser, J. , Kao-Shan, S. , Tsai, S. , Israel, M.A. , 1986. Cytogenetic characterization of selected small round cell tumors of childhood. Cancer Genet. Cytogenet. 21, 185–208. [DOI] [PubMed] [Google Scholar]
- Yamaguchi, T. , Yanagisawa, K. , Sugiyama, R. , Hosono, Y. , Shimada, Y. , Arima, C. , Kato, S. , Tomida, S. , Suzuki, M. , Osada, H. , Takahashi, T. , 2012. NKX2-1/TITF1/TTF-1-Induced ROR1 is required to sustain EGFR survival signaling in lung adenocarcinoma. Cancer Cell. 21, 348–361. [DOI] [PubMed] [Google Scholar]
- Yun, J.-H. , Lee, H.M. , Lee, E.H. , Park, J.-W. , Cho, C.-H. , 2013. Hypoxia reduces endothelial Ang1-induced Tie2 activity in a Tie1-dependent manner. Biochem. Biophys. Res. Commun. 436, 691–697. [DOI] [PubMed] [Google Scholar]
- Zambelli, D. , Zuntini, M. , Nardi, F. , Manara, M.C. , Serra, M. , Landuzzi, L. , Lollini, P.-L. , Ferrari, S. , Alberghini, M. , Llombart-Bosch, A. , Piccolo, E. , Iacobelli, S. , Picci, P. , Scotlandi, K. , 2010. Biological indicators of prognosis in Ewing's sarcoma: an emerging role for lectin galactoside-binding soluble 3 binding protein (LGALS3BP). Int. J. Cancer. 126, 41–52. [DOI] [PubMed] [Google Scholar]
- Zhang, S. , Chen, L. , Cui, B. , Chuang, H.-Y. , Yu, J. , Wang-Rodriguez, J. , Tang, L. , Chen, G. , Basak, G.W. , Kipps, T.J. , 2012. ROR1 is expressed in human breast cancer and associated with enhanced tumor-cell growth. PLoS One. 7, e31127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, S. , Chen, L. , Wang-Rodriguez, J. , Zhang, L. , Cui, B. , Frankel, W. , Wu, R. , Kipps, T.J. , 2012. The onco-embryonic antigen ROR1 is expressed by a variety of human cancers. Am. J. Pathol. 181, 1903–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Table A.1 List of RTK families, abbreviations and descriptions Classification of receptor tyrosine kinases into families according to Robinson et al. (Robinson et al., 2000). Gene names and description are according to NCBI database (http://www.ncbi.nlm.nih.gov). Alias given where applied in this paper. Proposed RTK of unknown mRNA/cDNA sequence at the time of primer design marked (*) (Robinson et al., 2000).
Table A.2 List of primers and probes Primers and probes as previously described (Müller‐Tidow et al., 2004, 2005). GenBank number as at the time of primer design.
Figure A.1 Kaplan–Meier survival analysis of patient cohort Kaplan–Meier analysis of overall‐survival in 20 Ewing sarcoma patients based on (A) patient age and (B) metastatic status.
Figure A.2 RTK gene expression profiles of Ewing sarcoma cell lines Relative RTK gene expressions of eight Ewing sarcoma cell lines as calculated by ΔΔCt‐method compared to GAPDH and a calibrator sample. Heatmap depicts fold expression according to quartiles calculated across all samples and RTKs. * = RTKs detected in <3 cell lines and negative in calibrator.
Figure A.3 Distribution of RTK gene expression signals Distribution of RTK gene expression signals (log) in Ewing sarcomas and MSC controls. The column of 58 signals below a near‐normal distribution was defined as not expressed.
Figure A.4 Functional effects of ROR1 silencing (A) Time‐course of ROR1 siRNA knockdown. A673 and 5838 cells were transfected with siRNA targeting ROR1 (siROR1) or non‐silencing control (siCTRL). At time points indicated, ROR1 gene expression was analyzed by qPCR by ΔΔCt‐method compared to GAPDH and siCTRL samples (upper panels). In parallel, ROR1 protein expression was assessed by Western blot (lower panels). Bands were analyzed by densitometry (upper panels). (B) Densitometric analysis of DVL3 protein bands. Intensities of DVL3 bands were normalized to corresponding actin loading control. Phospho‐DVL3 is displayed relative to non‐phosphorylated, lower‐band DVL3. Mean and SD of at least two independent experiments as shown in Figure 7D.