

Abstract
Background
In patients with hemodynamically significant pulmonary embolism, physiologic fibrinolysis fails to dissolve thrombi acutely and recombinant tissue plasminogen activator (r-tPA) therapy may be required, despite its bleeding risk. To examine potential mechanisms, we analyzed the expression of key fibrinolytic molecules in experimental pulmonary emboli, assessed the contribution of α2-antiplasmin to fibrinolytic failure and compared the effects of plasminogen activation and α2-antiplasmin-inactivation on experimental thrombus dissolution and bleeding.
Methods
Pulmonary embolism was induced by jugular vein infusion of 125I-fibrin or FITC-fibrin-labeled emboli in anesthetized mice. Thrombus site expression of key fibrinolytic molecules was determined by immunofluorescence staining. The effects of r-tPA and α2-antiplasmin-inactivation on fibrinolysis and bleeding were examined in a humanized model of pulmonary embolism.
Results
The plasminogen activation and plasmin inhibition system assembled at the site of acute pulmonary emboli in vivo. Thrombus dissolution was markedly accelerated in mice with normal α2-antiplasmin levels treated with an α2-antiplasmin-inactivating antibody (p<0.0001). Dissolution of pulmonary emboli by α2-antiplasmin-inactivation alone was comparable to 3 mg/kg r-tPA. Low dose r-tPA alone did not dissolve emboli, but was synergistic with α2-antiplasmin-inactivation, causing more embolus dissolution than clinical dose r-tPA alone (p<0.001) or α2-antiplasmin-inactivation alone (p<0.001). Despite greater thrombus dissolution, α2-antiplasmin-inactivation alone, or in combination with low dose r-tPA, did not lead to fibrinogen degradation, did not cause bleeding (vs. controls) and caused less bleeding than clinical dose r-tPA (p<0.001).
Conclusion
Although the fibrinolytic system assembles at the site of pulmonary emboli, thrombus dissolution is halted by α2-antiplasmin. Inactivation of α2-antiplasmin was comparable to pharmacologic r-tPA for dissolving thrombi. However, α2-antiplasmin-inactivation showed a unique pattern of thrombus specificity, because unlike r-tPA, it did not degrade fibrinogen or enhance experimental bleeding. This suggests that modifying the activity of a key regulator of the fibrinolytic system, like α2-antiplasmin, may have unique therapeutic value in pulmonary embolism.
Keywords: α2-antiplasmin inactivation, fibrinolysis, pulmonary embolism, bleeding, thrombus dissolution
Introduction
Pulmonary embolism (PE) affects millions of patients each year and is a leading cause of hospital mortality1–3. Physiologic or endogenous fibrinolysis usually fails to dissolve pulmonary emboli (PE), which may cause acute obstructive complications such as hypotension and shock. The persistence of thromboemboli also may lead to serious complications like chronic thromboembolic pulmonary hypertension and right ventricular dysfunction4. Anticoagulation is the standard therapy for PE, but anticoagulation only prevents new thrombus formation on pre-existing thromboemboli and does not cause thrombus dissolution5. In patients with massive PE, pharmacologic, recombinant tissue plasminogen activator (r-tPA) therapy is given to rapidly dissolve thromboemboli to enhance hemodynamic function, relieve hypotension and reduce mortality6, 7. However, treatment with r-tPA and other plasminogen activators causes serious bleeding which restricts its use to patients with massive PE who have a high risk of mortality6, 7.
Why endogenous fibrinolysis fails to dissolve acute PE is not well understood. Studies using genetically modified mice and inhibitors have shown that tissue-type (t)8 and urinary-type (u) plasminogen activator (PA)9, which convert plasminogen to the active enzyme plasmin, contribute to the dissolution of experimental pulmonary emboli. In a similar fashion, their inhibitor, plasminogen activator inhibitor-1 (PAI-1) suppresses fibrinolysis10. The plasmin inhibitor α2-antiplasmin11, 12 affects the dissolution of experimental pulmonary emboli and epidemiologic studies in humans identify higher α2-antiplasmin levels as a risk factor for venous thromboembolism13. Whether these components of the fibrinolytic system are expressed at the site of acute pulmonary emboli is unknown. Similarly little information exists about the relative contribution of the endogenous plasminogen activation and plasmin inhibition, and their interactions, to the rate and extent of fibrinolysis in vivo. Finally, it is uncertain whether promoting fibrinolysis by selectively altering plasminogen activation or plasmin inhibition has the same downstream effects on coagulation and bleeding.
We examined the expression of the plasminogen activation and plasmin inhibition system at the site of experimental pulmonary emboli. We compared the effects of plasminogen activation to α2-antiplasmin inactivation on experimental fibrinolysis, fibrinogen levels and bleeding. Experimental α2-antiplasmin inactivation enhances endogenous fibrinolysis to levels comparable to that achieved with higher dose r-tPA, but unlike r-tPA, does not cause fibrinogen destruction or enhance surgical bleeding. These data suggest that plasmin inhibition by α2-antiplasmin is a key rate-limiting step in the acute dissolution of experimental pulmonary emboli.
Methods
Proteins and reagents
Reagents were purchased from the following sources: human α2-antiplasmin (Athens Res. and Tech. GA); human plasmin (Calbiochem); bovine thrombin (Sigma, St. Louis, MO); citrated frozen human plasma (Lampire Biological Laboratories, PA); chimeric α2-antiplasmin inactivating antibody (TS23, Translational Sciences, Memphis, TN); 125I-fibrinogen (Perkin-Elmer, MA); FITC-fibrinogen (Molecular Innovations, MI); r-tPA (Alteplase, Genetech Inc.); all the other reagents if not specified (Sigma, St. Louis, MO).
Pulmonary embolism
Animal studies were approved by the Institutional Animal Care and Use Committee. The dissolution of experimental pulmonary emboli, fibrinogen degradation and bleeding were examined in a humanized model, in which adult male and female α2-antiplasmin−/− mice (KOMP, UC Davis, CA) on a C57Bl/6 background were supplemented with physiologic amounts of human α2-antiplasmin. To prepare plasma clots, pooled fresh frozen human plasma (5 μl) was mixed with trace amount of 125I-fibrinogen (~5000 cpm), 0.25 U/ml (NIH units) bovine thrombin and 100 mM CaCl2 in a total clot volume of 12.5 μl. After overnight incubation at 37°C, the clots were compressed, washed thoroughly with saline and cut into 20 pieces prior to embolization. Mice were anesthetized by ketamine, xylazine and atropine and kept on continuous anesthesia during the surgery. An anterior midline incision was made in the neck and the left jugular vein was exposed. A small incision was made in the jugular vein and a non-occlusive PE-10 catheter was inserted. Human α2-antiplasmin (4.7 mg/kg) was promptly infused intravenously to achieve physiologic levels. Then clots in saline were embolized into the lungs through the PE-10 catheter. After 30 min, various treatment agents such as r-tPA (0.1–3.0 mg/kg), human α2-antiplasmin inactivating antibody (α2-antiplasmin-I; 10 mg/kg) and or a combination of r-tPA + α2-antiplasmin-I were given intravenously. Tail bleeding was measured after PE as described below. After 4 h PE, the mice were euthanized, blood was collected for plasma preparation from cardiac puncture and the heart and the lung tissue were harvested for further study.
Immunohistology studies of PE were performed in wild-type, α2-antiplasmin+/+ mice (C57Bl/6, Jackson Labs) and human clots were labeled with FITC-fibrinogen instead of 125I-fibrinogen as described. Four hours after PE, the mice were euthanized, the lungs were perfused with saline and were isolated for immunofluorescence staining as described below.
Measurement of thrombus and clot dissolution
To assess thrombus dissolution in vivo, the radioactive content of the clots in the catheter prior to embolization was measured in a gamma counter before clot injection. The total radioactivity of the injected thrombus was determined by subtracting the radioactivity remaining in the injection tubing after embolization. After 4 h the heart and lungs were isolated and the total remaining pulmonary and vascular radioactivity was measured by gamma scintillation counting. The percent thrombus dissolution = 100% × (total thrombus cpm − residual thrombus cpm)/(total thrombus cpm).
To measure clot dissolution in vitro, clots were formed by mixing together in a 5 ml Sarstedt tube, 20 μl of human plasma with or without α2-antiplasmin-inactivating antibody (1.4 μg, 0.25 μl, 100 nM final), trace amounts of 125I-fibrinogen (0.3 μl, ~9000 cpm), 10 μl solution of calcium (25 mM) and thrombin (2.5 U/ml) at 37 °C for 1 h. After counting each tube to determine the amount of cpm per clot, the tubes were placed on ice. Tissue plasminogen activator (r-tPA, Activase, Genentech, South San Francisco, CA) was added to each tube (70 μl at a concentration of 0.2 nM) including no r-tPA as control. Then 10 μl supernatant was counted to determine baseline counts. Tubes were incubated at 37 °C and 10 μl of the supernatant was removed at 3 h to measure the release of 125I-fibrin degradation products by gamma counting in 10 μl samples. The percentage of clot dissolution was determined by dividing the counts per minute of released 125I-fibrin degradation products by the original 125I-fibrin associated with the clot and multiplying by 100%.
Tail bleeding and hemoglobin assays
Tail artery and vein bleeding were measured by tail tip amputation method about 2.5 h (mean 2.3 ± 0.4 h) after thromboembolism14. In anesthetized mice, tails were pre-warmed for 5 min in 3 ml of saline (pre-warmed) at 37°C. A distal 2 mm segment of the tail was amputated and bleeding was monitored for 30 min after re-immersing the tail in 0.9% warm saline tube on the heating pad at an angle of 45°. Total bleeding time was measured as summation of bleeding and re-bleeding time recorded for 30 min. Total blood in saline was centrifuged and the pellet was dissolved in 3 ml of Drabkin’s reagent and measured according to manufacturer’s instructions (Sigma, St. Louis, MO).
Immunofluorescence staining and analysis
Lungs were inflated by tracheal infusion of OCT embedding medium (Fisher Scientific, TX), harvested and embedded in OCT medium. Lung cryosections (8 micron) were cut on slides, air dried, fixed in chilled acetone for 10 min and again dried for 15 min. After washing in PBS, sections were blocked with 10% normal donkey serum for 1 h and incubated with primary antibody in 2% normal donkey serum for 90 min at room temperature. After PBS washings, sections were probed with fluorophore-conjugated donkey secondary antibodies as we have described previously15. The primary antibodies include rabbit anti-mouse against tPA (ASMTPA-GF; Molecular Innovations), uPA (urokinase plasminogen activator) (ASMUPA-GF-HT; Molecular Innovations) and PAI-1 (IASMPAI-GF; Innovative Research), rabbit anti-mouse plasminogen (Abcam), goat anti-mouse α2-antiplasmin (AF1239; R&D Systems), and rat anti-mouse Ly6G (clone1A8, #127602; Biolegend). Alexa Fluor 555 conjugated donkey anti-rabbit, donkey anti-goat and goat anti-rat were used as secondary antibodies. DAPI containing Vectashield hardset mounting media (Vector Laboratories, CA) was used to stain cell nuclei. Slides were scanned for fluorescence images with Aperio image fluorescence scanner (AperioScanScope, Vista, CA). Images were taken at 20× magnification (100 μm) using ImageScope software to examine the expression levels of tPA, uPA, plasminogen, PAI-1 and α2-antiplasmin at the thromboembolus site as well as in distal pulmonary arteries without thrombus. The area of interest in pulmonary arteries (in 100 μm images) with and without thromboemboli were selected and total positive fluorescence area was measured using Image Pro Plus 6.2 and analyzed in Graph Pad Prism 5.2.
Fibrinogen assay
Blood samples from the mouse heart were collected in sodium citrate/EDTA to prepare platelet poor plasma. Total fibrinogen was quantitatively determined by sodium sulfite method16 in the presence of aprotinin (0.4 TIU/ml, Sigma-Aldrich) as we have described previously17.
α2-antiplasmin activity
The effect of α2-antiplasmin on plasmin activity was assessed by examining the cleavage of a paranitroanilide substrate S2251. Purified α2-antiplasmin (20 μl, 60 nM, Calbiochem) was mixed with no antibody or a purified α2-antiplasmin-inactivating antibody or purified control, anti-nitrophenol monoclonal antibody of the same isotype as α2-antiplasmin-inactivating antibody, (MCA334B AbD Serotec) (20 μl, 60 nM) at room temperature for 10 minutes. Then assay buffer (20 μl, 50 mM Tris HCl, 100 mM NaCl, pH 7.4) and plasmin substrate S2251 (20 μl, S2251, 0.5 mM, Chromogenix) were added to the wells of a microtiter plate. Plasmin (20 μl, 20 nM, Calbiochem) was added to start the reaction. The cleavage of S2251 substrate was monitored at room temperature in a microtiter plate reader (BIO-TEK, Synergy HT) at A405. Experiments were performed in duplicate and the amount of plasmin activity was expressed relative to the amount of plasmin activity in the absence of α2-antiplasmin or any antibody. Plasma α2-antiplasmin activity was measured in a similar fashion in duplicate. Plasma samples were treated with methylamine (0.1 M, Sigma) to prevent interference from α2-macroglobulin18. Plasma samples were incubated with plasmin (100 nM, Haematologic Technologies, Inc.) and the cleavage of S2251 substrate (0.5 mM) in microtiter plate wells was monitored for 20 min. in a microtiter plate reader. Plasma samples were compared to a standard curve in which purified α2-antiplasmin was added in eight different concentrations (0 to 100 nM) to α2-antiplasmin-deficient plasma. Plasma levels of α2-antplasmin antigen were also measured at different dilutions in α2-antiplasmin−/− mice given α2-antiplasmin by ELISA as directed by the manufacturer (Innovative Research, Novi, MI).
Statistical analysis
Statistical analysis was done in Graph Pad Prism 5.2 software (San Diego, CA). Data represent the mean ± standard error. Normally distributed data were analyzed by an unpaired Student’s t-test, a one way or two-way ANOVA using the Neuman–Keuls correction. Data that were not normally distributed were analyzed by a Mann–Whitney test or a one way Kruskal-Wallis analysis using Dunn’s correction. A two-tailed p<0.05 was considered statistically significant.
Results
Pulmonary emboli contain plasminogen activators and plasmin inhibitors
When examined 4 h after thromboembolism, FITC-labeled pulmonary emboli were found adjacent to the tPA-immunostained endothelium in pulmonary arteries (Fig. 1A); tPA was also detected in the non-embolized pulmonary arteries as well as bronchial arteries as previously described19. Endothelial cell expression of tPA was confirmed by double immunostaining with CD31 (Supplementary Data Fig. 1). By comparison to tPA, uPA expression was present not only at the margins of the thrombus but also within the pulmonary embolus itself (Fig 1B and 1D). In the absence of a thromboembolus, uPA expression was identified in pulmonary arteries as well as in extravascular lung tissue and, appeared to be associated with DAPI-stained cells. Neutrophils express uPA and the uPA-receptor; immunostaining showed that neutrophils could be detected at the margins as well as within the body of the pulmonary embolus (Supplementary Data Fig. 2). In the absence of pulmonary emboli, neutrophils were also found in the lung parenchyma and occasionally seen in non-embolized pulmonary vessels. Plasminogen expression was increased nearly 10-fold at the thrombus site when compared to pulmonary arteries without thromboemboli (Fig 1C and D, p<0.001). Plasminogen was also found in a circumferential pattern on the periphery of DAPI-stained cells, suggesting that it may be cell-associated (Supplementary Data Fig. 3)20.
Figure 1. Expression of plasminogen activators and plasminogen at the site of pulmonary emboli.
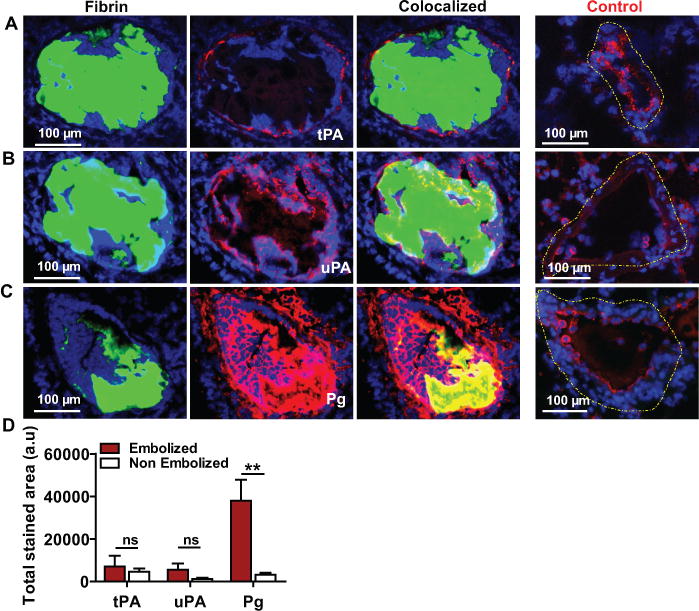
Lung tissue containing FITC-fibrin labeled (green) pulmonary emboli was immunostained (red) to detect the expression of (A) tPA, (B) uPA and (C) plasminogen (Pg) (D) The total immune-stained area (arbitrary units; a.u) for each protein was measured in a 20× (100 μm) image of an embolized vs non embolized (Control; dashed yellow outline) pulmonary artery in the lungs. n=3, mean ± SEM. **p<0.01; ns, non-significant.
There was significant enhancement of α2-antiplasmin staining within the pulmonary emboli by comparison to the negligible staining in the pulmonary vasculature without thromboemboli (Fig. 2A and C, p<0.01). PAI-1 expression was also detected within the pulmonary emboli but, vascular wall expression at the site of the acute pulmonary embolus did not appear to be increased (Figs. 2B,C, p ns) in comparison to non-embolized pulmonary arteries. Taken together, these data indicate that important components of the fibrinolytic system are present at the site of acute pulmonary emboli.
Figure 2. Expression of fibrinolytic inhibitors at the site of pulmonary emboli.
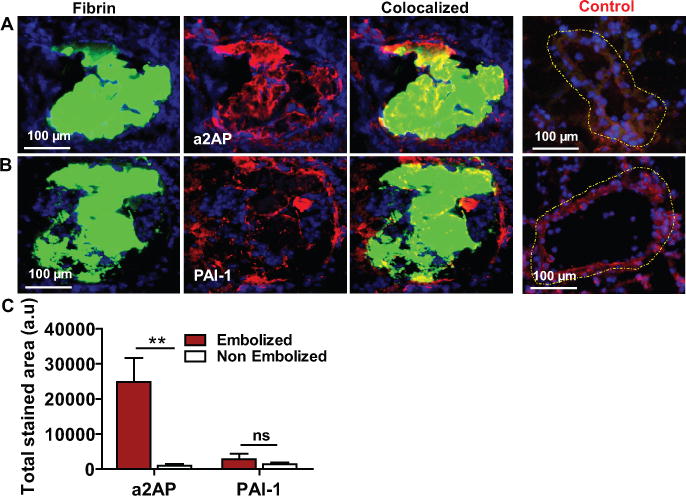
Lung tissue containing FITC-fibrin labeled (green) pulmonary emboli was immunostained (red) to detect the expression of (A) α2-antiplasmin (a2AP) and (B) PAI-1. (C) The total immune-stained area (arbitrary units; a.u) for each protein was measured in a 20× (100 μm) image of embolized vs non-embolized (control; outlined yellow) pulmonary artery in the lungs. n=3, mean ± SEM. **p<0.01; ns, non-significant.
Effect of α2-antiplasmin on endogenous fibrinolysis in pulmonary embolism
Previous studies have shown that in α2-antiplasmin-deficient pulmonary emboli injected into α2-antiplasmin−/− mice dissolve at accelerated rates. However, the extent to which α2-antiplasmin continues to dynamically affect endogenous fibrinolysis in existing pulmonary embolism is unknown. To enhance the translational relevance of these findings for human disease, we created a humanized model of pulmonary embolism in α2-antiplasmin-deficient mice. This allowed us to study the effect of human α2-antiplasmin on the dissolution of human embolized clots. Previous studies have shown that human and mouse α2-antiplasmin react similarly with mouse plasmin21. Physiologic levels of human α2-antiplasmin (mean 58 ± 8 μg/mL, n=4) were achieved in α2-antiplasmin-deficient mice by administration of human α2-antiplasmin, which persisted through the duration of the experiment. When an α2-antiplasmin inactivating monoclonal antibody was administered thirty minutes after embolization to mice with normal α2-antiplasmin levels, it significantly reduced α2-antiplasmin activity by comparison to the control mice (0.11 ± 0.02 vs. 0.92 ± 0.05 uM, p< 0.001), but it but did not significantly affect plasma α2-antiplasmin antigen levels (76 ± 7 ug/ml, n=4, p=0.16). The α2-antiplasmin inactivating antibody enhanced thrombus dissolution (Fig. 3A, p<0.0001) by comparison to control mice with normal α2-antiplasmin levels. In vitro studies confirmed that this antibody neutralized α2-antiplasmin activity in solution (Fig. 3B, C) or bound to fibrin (Supplementary Data Fig. 4), preventing inhibition of plasmin catalytic activity. The α2-antiplasmin-I prevented inhibition of plasmin-mediated fibrinolysis, thereby significantly enhancing the dissolution of human clots (Fig. 3D).
Figure 3. Effect of α2-antiplasmin-inactivation on plasmin activity and the dissolution of pulmonary emboli.
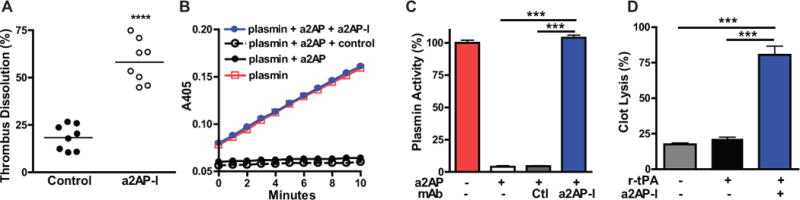
(A) The dissolution of 125I-fibrin-labeled pulmonary emboli was measured in anesthetized α2-antiplasmin (a2AP)−/− mice (N=16) with physiologic levels of α2-antiplasmin (after intravenous supplementation) with or without (control) treatment with an a2AP-inactivating antibody (a2AP-I). The percent thrombus dissolution was assessed by measuring the residual 125I-fibrin radioactivity in the lungs four hours after embolization. ****p<0.0001 vs. control. (B) The activity of plasmin in the presence of no a2AP or a2AP, with or without an a2AP-inactivating (a2AP-I) or a control monoclonal antibody of the same isotype (MCA334B0). Plasmin activity was monitored by cleavage of S2251, a paranitroanilide substrate, at A405. Experiments were performed in duplicate and the amount of plasmin activity was expressed relative to the amount of plasmin activity in the absence of a2AP or any antibody. (C) The effect of no a2AP (Control, Ctl) or a2AP with or without various monoclonal antibodies on plasmin activity, expressed as percentage of the activity of plasmin in the absence of a2AP. ***p<0.001 (D) Effect of an a2AP-I antibody or buffer alone (control) on dissolution of 125I-fibrinogen-labeled human clots in vitro in the presence of trace amounts of r-tPA (0.2 nM) after 3 h incubation at 37 °C. ***p<0.001
Effects of r-tPA and α2-antiplasmin-I on thrombus dissolution
Although tPA was present at the site of the pulmonary emboli, there was minimal thrombus dissolution in control mice (saline treated) indicating that endogenous tPA activity was insufficient to rapidly dissolve the pulmonary embolus (Fig. 4). In this humanized model, low dose r-tPA at 0.1 mg/kg or 0.24 mg/kg did not significantly enhance dissolution of pulmonary emboli by comparison to control mice (Fig. 4). A dose of r-tPA similar to the human clinical dose (1.2 mg/Kg) caused a significant enhancement in thrombus dissolution compared with control mice (36.3±3.9% vs. 16.7± 3.4%, p<0.001). The dissolution of pulmonary emboli was further enhanced by high dose r-tPA (3 mg/Kg) (62.3±3.6% p<0.001). PE dissolution induced by α2-antiplasmin-I (10 mg/Kg) was greater than observed with 1.2 mg/kg of r-tPA (58.3±4.0% vs. 36.3±3.9% p<0.001) and it was comparable to that of high dose r-tPA (62.3±3.6%, 3 mg/kg) (Fig 4).
Figure 4. Effects of pharmacologic r-tPA and α2-antiplasmin inactivation on thrombus dissolution in mice with experimental pulmonary emboli.
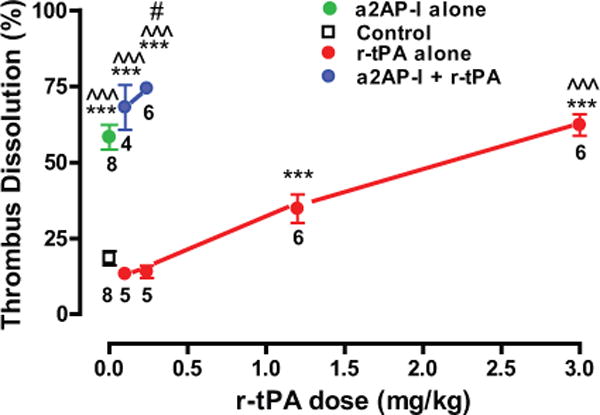
Anesthetized mice were treated thirty minutes after thromboembolism with r-tPA, an α2-antiplasmin-inactivating antibody (a2AP-I) alone, the combination of low dose r-tPA and a2AP-I or nothing (Control) as shown (N=48). Four hours after PE, the experiments were terminated and thrombus dissolution was measured. The number of mice per dose is shown. *** p<0.001 vs. control; ^^^ p<0.001 vs. r-tPA 0.1, 0.24 or 1.2 mg/kg; # p<0.05 vs. r-tPA 3 mg/kg.
To determine whether there was a significant interaction between r-tPA and α2-antiplasmin in the regulation of fibrinolysis, the effects of low dose r-tPA in combination with α2-antiplasmin inactivation were examined. A two-way ANOVA showed that α2-antiplasmin-I + r-tPA significantly enhanced thrombus dissolution vs. r-tPA alone (p<0.0001). The combination of 0.1 mg/kg of r-tPA with α2-antiplasmin-inactivation (10 mg/kg) significantly increased dissolution by comparison to r-tPA 1.2 mg/kg alone (p<0.001) and the same dose of α2-antiplasmin-inactivation alone (p<0.001). The combination of 0.24 mg/kg r-tPA with α2-antiplasmin-inactivation also significantly increased thrombus dissolution vs. α2-antiplasmin-inactivation alone (p<0.01), 1.2 mg/kg r-tPA alone (p<0.001) and 3.0 mg/kg r-tPA alone (p<0.05). This significant enhancement in fibrinolysis is consistent with synergism between the α2-antiplasmin-inactivation and r-tPA in thrombus dissolution.
Effects of r-tPA and α2-antiplasmin on fibrinogen consumption and bleeding
Therapy with r-tPA is associated with consumption of coagulation factors or fibrinolytic system components e.g. fibrinogen degradation22. There was a significant reduction in α2-antiplasmin activity in mice treated with r-tPA (3mg/kg) vs. controls (0.17 ± 0.08 vs. 0.92 ± 0.05 uM, p<0.001), Treatment with r-tPA caused a significant increase in fibrinogen degradation of 25 to 57% in a dose-related fashion by comparison to fibrinogen levels in normal, untreated mice (Fig. 5A). By comparison, α2-antiplasmin-I treatment alone or in combination of α2-antiplasmin-I with low dose r-tPA (0.1 to 0.24 mg/kg) did not show a statistically significant change (≤5%) from normal controls (Fig. 5A). In a two-way analysis of variance, low doses of r-tPA with α2-antiplasmin-I showed significantly less reduction of fibrinogen (p≤0.01) vs. low dose r-tPA alone.
Figure 5. Effects of pharmacologic r-tPA and α2-antiplasmin-inactivation on fibrinogen consumption and bleeding dissolution in mice with experimental PE.
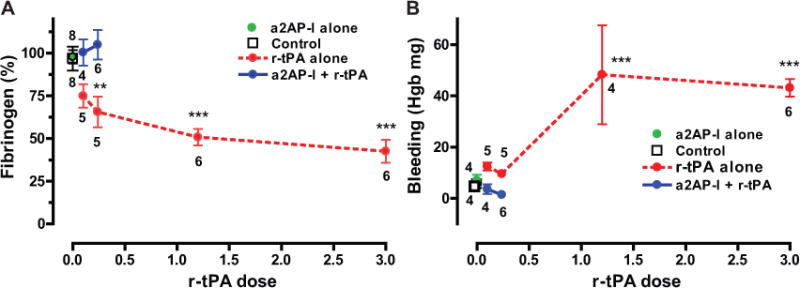
(A) Fibrinogen consumption in experimental groups given an α2-antiplasmin-inactivating antibody (a2AP-I) or r-tPA or no treatment (Control). Fibrinogen levels were measured by the sodium sulfite method and are expressed relative to fibrinogen levels in normal wild-type mice without PE. (B) Tail bleeding was measured as hemoglobin (Hgb) loss by Drabkin’s reagent in the same experimental groups. N per group is shown. **p<0.01, ***p<0.001 vs. controls.
The regulation of endogenous fibrinolysis may affect hemostasis following vessel injury. We compared the effects of α2-antiplasmin-inactivation and r-tPA on hemorrhage using the well-established, tail bleeding method. Bleeding was increased significantly in mice receiving a clinical dose (1.2 mg/kg, p<0.001) or higher dose (3 mg/kg, p<0.001) of r-tPA as compared to control mice (Fig. 5B). Lower dose r-tPA treatment (0.1 or 0.24 mg/kg) did not affect tail bleeding. No increase in bleeding time was observed in mice treated with α2-antiplasmin-I alone (Fig. 5B). Overall, the combination of α2-antiplasmin-I with low dose r-tPA significantly reduced tail bleeding by comparison to low dose r-tPA alone (two-way ANOVA, p<0.0001).
Comparative effects on fibrinolysis and bleeding
Optimal treatment for PE would maximize thrombus dissolution with low bleeding risk (Fig. 6A). Both r-tPA and α2-antiplasmin are in the fibrinolytic pathway, but it is unknown whether they exert the same effects on clot dissolution and bleeding. Low doses of r-tPA were associated with minimal thrombus dissolution and no increases in bleeding risk (Fig. 6B). Higher dose r-tPA (1.2 and 3 mg/kg) increased both thrombus dissolution and bleeding in parallel. In contrast, α2-antiplasmin-inactivation alone, or in combination with low dose r-tPA led to increased thrombus dissolution with low bleeding risk (Fig. 6B).
Figure 6. Comparative effects of plasminogen activation and α2-antiplasmin on thrombus dissolution and associated bleeding.
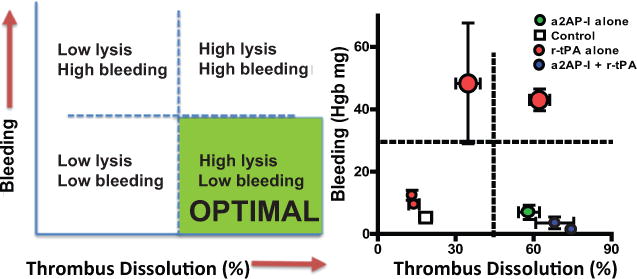
(A) A hypothetical plot of thrombus dissolution and bleeding for potential optimal treatment for PE. (B) Experimental data showing the effects (means ± SE) of r-tPA, α2-antiplasmin-I and the combination of the two agents on thrombus dissolution and bleeding. Higher dose r-tPA (1.2 or 3 mg/kg) is indicated by larger symbols.
Discussion
These experiments with α2-antiplasmin-deficient mice, α2-antiplasmin replacement and α2-antiplasmin-inactivation showed that the plasmin inhibitor, α2-antiplasmin suppressed endogenous fibrinolysis after pulmonary embolism. The magnitude of α2-antiplasmin’s regulatory effect on fibrinolysis was assessed by α2-antiplasmin inactivation and was comparable to the fibrinolysis achieved with pharmacologic doses of r-tPA (3 mg/kg). There was synergism between low dose r-tPA and α2-antiplasmin inactivation indicating that α2-antiplasmin modulated the fibrinolytic effects of r-tPA. While α2-antiplasmin inactivation and higher dose r-tPA were comparably effective in dissolving thrombi, they showed distinctly different effects on fibrinogen degradation and tail bleeding. Fibrinogen levels were significantly reduced by increasing doses of r-tPA, but not by α2-antiplasmin inactivation. Bleeding was enhanced by fibrinolytic doses of r-tPA but not by α2-antiplasmin inactivation.
To our knowledge this is the first demonstration that all of these particular key components of the plasminogen activation and plasmin inhibition systems are expressed at the site of an acute pulmonary embolism. These thrombi also contained neutrophils consistent with an acute inflammatory response. Although increased levels of α2-antiplasmin are associated with an increased risk for human venous thromboembolism by univariate analysis13 and diagnostic agents carrying the N-terminal peptide of α2-antiplasmin can identify developing venous thrombi,23 the presence of α2-antiplasmin in acute PE has not been previously identified.
Three component processes contribute to the regulation endogenous fibrinolysis by plasmin. The first component, plasminogen activation or plasmin generation, is regulated by tPA, uPA and PAI-1 (and other inhibitors). The second component of plasmin inhibition, is mediated by α2-antiplasmin (fast inhibitor) and α2-macroglobulin (slower inhibitor)24. The third component, modification of the plasmin substrate fibrin by activated factor XIII and thrombin activatable fibrinolysis inhibitor (TAFI). Although not investigated in the current study, there is evidence that all of these components contribute to fibrinolysis in experimental pulmonary embolism. Gene deletion studies demonstrate that tPA−/−8 and uPA−/−9 mice show reduced dissolution of experimental pulmonary thrombi/microemboli. PAI-1 (PAI-1−/−)10, TAFI (TAFI−/−)25 or α2-antiplasmin (α2-antiplasmin−/−)11, 12 deficiency or α2-antiplasmin inhibition26 causes enhanced fibrinolytic potential in pulmonary embolism. Studies with specific inhibitors demonstrate that activated factor XIII, through its crosslinking of α2-antiplasmin to fibrin, also regulates fibrinolysis in experimental PE17. Mutch et al. found that PAI-1, TAFI, and α2-antiplasmin had complementary effects on fibrinolysis, although α2-antiplasmin had the dominant role27. In comparative in vivo studies, Dewerchin et al. showed that α2-antiplasmin was more potent than PAI-1 for suppressing dissolution of experimental pulmonary emboli28. This is consistent with our finding that dissolution of experimental pulmonary emboli is restricted by the effects of α2-antiplasmin and not by an inability of the endogenous plasminogen activation system to generate plasmin.
These studies were performed in a humanized model to enhance their relevance for human PE. Still, even meticulous animal models may not fully simulate the complexity of human illnesses. While these studies in pulmonary embolism, and others in ischemic stroke, show that α2-antiplasmin-I is not associated with significant bleeding risk, the safety of this approach for humans will need to be confirmed15, 29. Finally, these studies examined the effects of α2-antiplasmin inactivation on acute PE; its effect on subacute or chronic PE remains unknown.
While equipotent in dissolving pulmonary emboli, α2-antiplasmin inactivation and pharmacologic doses of r-tPA had divergent effects on fibrinogen degradation and bleeding. Low dose r-tPA has minimal effects, but therapeutic doses of r-tPA consume fibrinogen and other coagulation factors, and enhance bleeding30–32. These effects are primarily attributed to the fact that therapeutic doses of r-tPA generate large amounts of plasmin to overcome the effects of α2-antiplasmin and deplete coagulation proteins, which reduces hemostatic potential33. In the presence of massive plasminemia, α2-antiplasmin may have a protective effect by helping to prevent fibrinogen and coagulation factor depletion33. In contrast, α2-antiplasmin inactivation prolongs the half-life of plasmin generated at the site of the fibrin thrombus, but does not interfere with plasmin inactivation in the circulation by α2-macroglobulin and potentially other inhibitors. Even when plasmin generation is slightly enhanced by low dose r-tPA in the presence of α2-antiplasmin inactivation, there is not a signal of enhanced fibrinogen consumption or of bleeding. This is consistent with recent studies of experimental ischemic stroke where α2-antiplasmin inactivation has been shown to reduce bleeding while enhancing dissolution of middle cerebral artery thrombi, reducing brain infarction and increasing survival15,29. This argues that the main effect of α2-antiplasmin inactivation is to enhance local fibrinolysis of thrombi without triggering consumption of coagulation factors.
Despite anticoagulation, inadequate fibrinolysis in patients with acute pulmonary embolism may contribute to acute mortality and longer term disability and death. The present studies suggest that even after the onset of pulmonary embolism, therapeutic modulation of α2-antiplasmin activity may prove an effective strategy to enhance endogenous or physiologic fibrinolysis without significantly increasing the bleeding risk. Initial studies are now underway in an NIH-supported trial to evaluate the safety and biomarker efficacy of α2-antiplasmin in humans.
Supplementary Material
Clinical Perspective.
What is new?
Acute, experimental pulmonary emboli resist dissolution although they contain key components of the fibrinolytic system.
Monoclonal antibody inactivation of α2-antiplasmin, an endogenous inhibitor of plasmin, effectively dissolves pulmonary emboli with similar potency to high dose r-tPA.
α2-antiplasmin inactivation synergizes with low dose r-tPA to enhance thrombus dissolution.
Unlike r-tPA, α2-antiplasmin inactivation alone or in combination with low dose r-tPA did not cause fibrinogen degradation or increase bleeding.
What are the clinical implications?
α2-antiplasmin is a dominant regulator that prohibits thrombus dissolution in vivo.
Therapeutic modulation of α2-antiplasmin activity may prove an effective strategy to enhance fibrinolysis without significantly increasing the bleeding risk.
Acknowledgments
The authors appreciate the technical efforts of Nelson Houng.
Source of funding
This work was funded in part by NIH (NINDS NS089707 and by NHLBI HL092750).
Footnotes
Subject codes: Pulmonary embolism, α2-antiplasmin, tissue plasminogen activator, fibrinolysis, bleeding
Disclosures
G.L. Reed is a founder of Translational Sciences.
References
- 1.Goldhaber SZ, Bounameaux H. Pulmonary embolism and deep vein thrombosis. Lancet. 2012;379:1835–1846. doi: 10.1016/S0140-6736(11)61904-1. [DOI] [PubMed] [Google Scholar]
- 2.Laporte S, Mismetti P, Decousus H, Uresandi F, Otero R, Lobo JL, Monreal M, Investigators R Clinical predictors for fatal pulmonary embolism in 15,520 patients with venous thromboembolism: findings from the Registro Informatizado de la Enfermedad TromboEmbolica venosa (RIETE) Registry. Circulation. 2008;117:1711–1716. doi: 10.1161/CIRCULATIONAHA.107.726232. [DOI] [PubMed] [Google Scholar]
- 3.Aujesky D, Jimenez D, Mor MK, Geng M, Fine MJ, Ibrahim SA. Weekend versus weekday admission and mortality after acute pulmonary embolism. Circulation. 2009;119:962–968. doi: 10.1161/CIRCULATIONAHA.108.824292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jaff MR, McMurtry MS, Archer SL, Cushman M, Goldenberg N, Goldhaber SZ, Jenkins JS, Kline JA, Michaels AD, Thistlethwaite P, Vedantham S, White RJ, Zierler BK, American Heart Association Council on Cardiopulmonary CCP, Resuscitation, American Heart Association Council on Peripheral Vascular D, American Heart Association Council on Arteriosclerosis T and Vascular B Management of massive and submassive pulmonary embolism, iliofemoral deep vein thrombosis, and chronic thromboembolic pulmonary hypertension: a scientific statement from the American Heart Association. Circulation. 2011;123:1788–1830. doi: 10.1161/CIR.0b013e318214914f. [DOI] [PubMed] [Google Scholar]
- 5.Kearon C, Akl EA, Comerota AJ, Prandoni P, Bounameaux H, Goldhaber SZ, Nelson ME, Wells PS, Gould MK, Dentali F, Crowther M, Kahn SR, American College of Chest P Antithrombotic therapy for VTE disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141:e419S–494S. doi: 10.1378/chest.11-2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chatterjee S, Chakraborty A, Weinberg I, Kadakia M, Wilensky RL, Sardar P, Kumbhani DJ, Mukherjee D, Jaff MR, Giri J. Thrombolysis for pulmonary embolism and risk of all-cause mortality, major bleeding, and intracranial hemorrhage: a meta-analysis. JAMA. 2014;311:2414–2421. doi: 10.1001/jama.2014.5990. [DOI] [PubMed] [Google Scholar]
- 7.Marti C, John G, Konstantinides S, Combescure C, Sanchez O, Lankeit M, Meyer G, Perrier A. Systemic thrombolytic therapy for acute pulmonary embolism: a systematic review and meta-analysis. Eur Heart J. 2015;36:605–614. doi: 10.1093/eurheartj/ehu218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carmeliet P, Schoonjans L, Kieckens L, Ream B, Degen J, Bronson R, De Vos R, van den Oord JJ, Collen D, Mulligan RC. Physiological consequences of loss of plasminogen activator gene function in mice. Nature. 1994;368:419–424. doi: 10.1038/368419a0. [DOI] [PubMed] [Google Scholar]
- 9.Bdeir K, Murciano JC, Tomaszewski J, Koniaris L, Martinez J, Cines DB, Muzykantov VR, Higazi AA. Urokinase mediates fibrinolysis in the pulmonary microvasculature. Blood. 2000;96:1820–1826. [PubMed] [Google Scholar]
- 10.Carmeliet P, Stassen JM, Schoonjans L, Ream B, van den Oord JJ, De Mol M, Mulligan RC, Collen D. Plasminogen activator inhibitor-1 gene-deficient mice. II. Effects on hemostasis, thrombosis, and thrombolysis. J Clin Invest. 1993;92:2756–2760. doi: 10.1172/JCI116893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matsuno H, Okada K, Ueshima S, Matsuo O, Kozawa O. Alpha2-antiplasmin plays a significant role in acute pulmonary embolism. J Thromb Haemost. 2003;1:1734–1739. doi: 10.1046/j.1538-7836.2003.00252.x. [DOI] [PubMed] [Google Scholar]
- 12.Lijnen HR, Okada K, Matsuo O, Collen D, Dewerchin M. Alpha2-antiplasmin gene deficiency in mice is associated with enhanced fibrinolytic potential without overt bleeding. Blood. 1999;93:2274–2281. [PubMed] [Google Scholar]
- 13.Meltzer ME, Doggen CJ, de Groot PG, Rosendaal FR, Lisman T. The impact of the fibrinolytic system on the risk of venous and arterial thrombosis. Semin Thromb Hemost. 2009;35:468–477. doi: 10.1055/s-0029-1234142. [DOI] [PubMed] [Google Scholar]
- 14.Pleines I, Hagedorn I, Gupta S, May F, Chakarova L, van Hengel J, Offermanns S, Krohne G, Kleinschnitz C, Brakebusch C, Nieswandt B. Megakaryocyte-specific RhoA deficiency causes macrothrombocytopenia and defective platelet activation in hemostasis and thrombosis. Blood. 2012;119:1054–1063. doi: 10.1182/blood-2011-08-372193. [DOI] [PubMed] [Google Scholar]
- 15.Houng AK, Wang D, Reed GL. Reversing the deleterious effects of alpha2-antiplasmin on tissue plasminogen activator therapy improves outcomes in experimental ischemic stroke. Exp Neurol. 2014;255C:56–62. doi: 10.1016/j.expneurol.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rampling MW, Gaffney PJ. The sulphite precipitation method for fibrinogen measurement; its use on small samples in the presence of fibrinogen degradation products. Clin Chim Acta. 1976;67:43–52. doi: 10.1016/0009-8981(76)90215-1. [DOI] [PubMed] [Google Scholar]
- 17.Reed GL, Houng AK. The contribution of activated factor XIII to fibrinolytic resistance in experimental pulmonary embolism. Circulation. 1999;99:299–304. doi: 10.1161/01.cir.99.2.299. [DOI] [PubMed] [Google Scholar]
- 18.Matsuda T, Ogawara M, Miura R, Seki T, Matsumoto T, Teramura Y, Nakamura K. Selective determination of alpha 2-plasmin inhibitor activity in plasma using chromogenic substrate. Thromb Res. 1984;33:379–388. doi: 10.1016/0049-3848(84)90077-x. [DOI] [PubMed] [Google Scholar]
- 19.Levin EG, Santell L, Osborn KG. The expression of endothelial tissue plasminogen activator in vivo: a function defined by vessel size and anatomic location. J Cell Sci. 1997;110(Pt 2):139–148. doi: 10.1242/jcs.110.2.139. [DOI] [PubMed] [Google Scholar]
- 20.Plow EF, Herren T, Redlitz A, Miles LA, Hoover-Plow JL. The cell biology of the plasminogen system. FASEB J. 1995;9:939–945. doi: 10.1096/fasebj.9.10.7615163. [DOI] [PubMed] [Google Scholar]
- 21.Lijnen HR, van Hoef B, Beelen V, Collen D. Characterization of the murine plasma fibrinolytic system. Eur J Biochem. 1994;224:863–871. doi: 10.1111/j.1432-1033.1994.00863.x. [DOI] [PubMed] [Google Scholar]
- 22.Vaughan DE, Goldhaber SZ, Kim J, Loscalzo J. Recombinant tissue plasminogen activator in patients with pulmonary embolism: correlation of fibrinolytic specificity and efficacy. Circulation. 1987;75:1200–1203. doi: 10.1161/01.cir.75.6.1200. [DOI] [PubMed] [Google Scholar]
- 23.Temme S, Grapentin C, Quast C, Jacoby C, Grandoch M, Ding Z, Owenier C, Mayenfels F, Fischer JW, Schubert R, Schrader J, Flogel U. Noninvasive Imaging of Early Venous Thrombosis by 19F Magnetic Resonance Imaging With Targeted Perfluorocarbon Nanoemulsions. Circulation. 2015;131:1405–1414. doi: 10.1161/CIRCULATIONAHA.114.010962. [DOI] [PubMed] [Google Scholar]
- 24.Harpel PC. Alpha2-plasmin inhibitor and alpha2-macroglobulin-plasmin complexes in plasma. Quantitation by an enzyme-linked differential antibody immunosorbent assay. J Clin Invest. 1981;68:46–55. doi: 10.1172/JCI110253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Swaisgood CM, Schmitt D, Eaton D, Plow EF. In vivo regulation of plasminogen function by plasma carboxypeptidase B. J Clin Invest. 2002;110:1275–1282. doi: 10.1172/JCI15082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Butte AN, Houng AK, Jang IK, Reed GL. Alpha 2-antiplasmin causes thrombi to resist fibrinolysis induced by tissue plasminogen activator in experimental pulmonary embolism. Circulation. 1997;95:1886–1891. doi: 10.1161/01.cir.95.7.1886. [DOI] [PubMed] [Google Scholar]
- 27.Mutch NJ, Thomas L, Moore NR, Lisiak KM, Booth NA. TAFIa, PAI-1 and alpha-antiplasmin: complementary roles in regulating lysis of thrombi and plasma clots. J Thromb Haemost. 2007;5:812–817. doi: 10.1111/j.1538-7836.2007.02430.x. [DOI] [PubMed] [Google Scholar]
- 28.Dewerchin M, Collen D, Lijnen HR. Enhanced fibrinolytic potential in mice with combined homozygous deficiency of alpha2-antiplasmin and PAI-1. Thromb Haemost. 2001;86:640–646. [PubMed] [Google Scholar]
- 29.Reed GL, Houng AK, Wang D. Microvascular thrombosis, fibrinolysis, ischemic injury, and death after cerebral thromboembolism are affected by levels of circulating alpha2-antiplasmin. Arterioscler Thromb Vasc Biol. 2014;34:2586–2593. doi: 10.1161/ATVBAHA.114.304530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Okada K, Lijnen HR, Moreau H, Vanderschueren S, Collen D. Procoagulant properties of intravenous staphylokinase versus tissue-type plasminogen activator. Thromb Haemost. 1996;76:857–859. [PubMed] [Google Scholar]
- 31.Hoffmeister HM, Szabo S, Kastner C, Beyer ME, Helber U, Kazmaier S, Wendel HP, Heller W, Seipel L. Thrombolytic therapy in acute myocardial infarction: comparison of procoagulant effects of streptokinase and alteplase regimens with focus on the kallikrein system and plasmin. Circulation. 1998;98:2527–2533. doi: 10.1161/01.cir.98.23.2527. [DOI] [PubMed] [Google Scholar]
- 32.Rao AK, Pratt C, Berke A, Jaffe A, Ockene I, Schreiber TL, Bell WR, Knatterud G, Robertson TL, Terrin ML. Thrombolysis in Myocardial Infarction (TIMI) Trial–phase I: hemorrhagic manifestations and changes in plasma fibrinogen and the fibrinolytic system in patients treated with recombinant tissue plasminogen activator and streptokinase. J Am Coll Cardiol. 1988;11:1–11. doi: 10.1016/0735-1097(88)90158-1. [DOI] [PubMed] [Google Scholar]
- 33.Leebeek FW, Kluft C, Knot EA, Los P, Cohen AF, Six AJ. Plasmin inhibitors in the prevention of systemic effects during thrombolytic therapy: specific role of the plasminogen-binding form of alpha 2-antiplasmin. J Am Coll Cardiol. 1990;15:1212–1220. doi: 10.1016/s0735-1097(10)80003-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.