

Abstract
Escherichia coli Endonuclease III (EndoIII) and MutY are DNA glycosylases that contain [4Fe4S] clusters and that serve to maintain the integrity of the genome after oxidative stress. Electrochemical studies on highly-oriented pyrolytic graphite (HOPG) revealed that DNA binding by EndoIII leads to a large negative shift in midpoint potential of the cluster, consistent with stabilization of the oxidized [4Fe4S]3+ form. However, the smooth, hydrophobic HOPG surface is non-ideal for working with proteins in the absence of DNA. In this work, we use thin film voltammetry on a pyrolytic graphite edge electrode to overcome these limitations. Improved adsorption leads to substantial signals for both EndoIII and MutY in the absence of DNA, and a large negative potential shift is retained with DNA present. In contrast, the EndoIII mutants E200K, Y205H, and K208E, which provide electrostatic perturbations in the vicinity of the cluster, all show DNA-free potentials within error of wild type; similarly, the presence of negatively charged poly-L glutamate does not lead to a significant potential shift. Overall, binding to the DNA polyanion is the dominant effect in tuning the redox potential of the [4Fe4S] cluster, helping to explain why all DNA-binding proteins with [4Fe4S] clusters studied to date have similar DNA-bound potentials.
Graphical abstract
INTRODUCTION
E. coli endonuclease III (EndoIII) is a DNA glycosylase that excises oxidized pyrimidines from DNA, functioning as part of the base excision repair (BER) pathway in order to maintain the integrity of the genome (1). EndoIII contains a [4Fe4S]2+ cluster that is relatively insensitive to reduction and oxidation in solution (2); as a result, it was initially proposed that the cluster served only a structural role within the protein. MutY is another E. coli BER glycosylase, homologous to EndoIII, that also contains a [4Fe4S]2+ cluster (3). MutY, found in organisms from bacteria to man, is involved in the repair of oxoG:A mismatches (4); in humans, inherited defects in MUTYH are associated with a familial form of colon cancer known as MUTYH-associated polyposis (MAP) and many MAP-associated variants are localized near the [4Fe4S] cluster (4). Furthermore, in the case of MutY, it has been shown that the cluster is not required for folding or stability (3), or direct participation in the intrinsic glycosidic bond hydrolysis catalysis (5), making the widespread presence of conserved, non-catalytic [4Fe4S] clusters difficult to explain.
Notably, the earliest studies with EndoIII and MutY looked only at free protein in solution, neglecting the effect of DNA binding on redox potential. Experiments carried out on DNA-modified electrodes have demonstrated that, in both EndoIII and MutY, the cluster undergoes a negative shift in potential associated with binding to the DNA polyanion and is activated toward reversible redox activity (6). In these experiments, DNA monolayers were formed on gold electrodes, and, upon addition of EndoIII or MutY, a reversible signal with a midpoint potential ranging from 60–95 mV versus NHE was observed. Importantly, the introduction of just a single mismatch or abasic site into DNA led to signal attenuation, showing that electron transfer between the protein and the electrode was through the π-stacked base pairs in a process known as DNA-mediated charge transport (DNA CT) (7). In this process, charge is funneled from the electrode surface through the π-stack of the DNA bases to reach the redox probe (a protein in this case); the only requirement is that the probe must be electronically coupled to the DNA π-stack. Remarkably, the sensitivity to base stacking observed with EndoIII and MutY was comparable to that obtained using small molecules such as Nile blue or methylene blue that intercalate directly into the base stack.
The expanded potential window of highly-oriented pyrolytic graphite (HOPG) and the ability to form pyrene-modified DNA films on the surface made it possible to directly compare the potential of proteins in the presence and absence of DNA (8). Experiments with EndoIII revealed that DNA binding shifts the reduction potential of the [4Fe4S]3+/2+ couple by −200 mV to favor oxidation. Thermodynamically, this shift corresponded to a large (~3 orders of magnitude) increase in the DNA binding affinity of the oxidized form of the protein. Crystal structures of EndoIII and MutY with and without DNA do not show any significant structural change upon DNA binding (9–12), so this dramatic result was attributed to a combination of electrostatic effects resulting from the negatively charged DNA backbone and decreased solvent accessibility of the cluster in DNA-bound protein, which is in agreement with the known sensitivity of [4Fe4S] clusters to their local environment (13). By demonstrating that DNA binding brought the redox potential of EndoIII into a biologically relevant window, this result served to explain the previously observed redox insensitivity of free EndoIII and provided evidence in favor of a redox role for the DNA-bound protein cluster.
Since these experiments were carried out, a wide range of DNA processing enzymes have been revealed to contain [4Fe4S] clusters with properties similar to EndoIII and MutY. These include the Archaeoglobus fulgidus uracil DNA glycosylase (UDG), archaeal and eukaryotic versions of the nucleotide excision repair helicase XPD and the E. coli R-loop maturation helicase DinG (14), all of which were found to have similar DNA-bound potentials (~80 mV versus NHE) as measured on DNA-modified gold electrodes (6,7). The similar DNA-bound midpoint potentials and picosecond kinetics of DNA CT together suggested that DNA CT could provide a means for these enzymes to localize efficiently to the vicinity of their target lesions (15). Indeed, experiments carried out both in vitro and in vivo have led to the development of a model for DNA repair in which two [4Fe4S] cluster proteins use DNA CT to communicate with each other over long molecular distances via electron transfer self-exchange reactions (7, 15). As evidenced through the potential shift, DNA binding activates the proteins toward oxidation to the [4Fe4S]3+ state (8). When the DNA intervening between the two proteins is undamaged, the self-exchange reaction can proceed efficiently, with the result that one of the DNA-bound proteins is reduced and its affinity for DNA lowered. This protein is then free to diffuse to another region of the genome. However, in the case of an intervening mismatch or lesion that impairs CT by disrupting π-stacking, this self-exchange reaction is inhibited. Both proteins then remain bound to the DNA in the vicinity of the lesion, significantly reducing the range over which the slower processes of diffusion must occur and facilitating repair of a relatively large genome on a biologically relevant time scale (15).
While DNA binding is clearly of critical importance to the redox activity of these enzymes, it is not clear that it represents the only way to modulate the potential. It was recently reported that carboxylic acid monolayers had a similar activating effect as DNA, although, in contrast with the above model, they identified the relevant couple as the [4Fe4S]2+/+ rather than the [4Fe4S]3+/2+ couple (16). With respect to the latter point, the high potential of the reversible DNA-bound signal on both gold and HOPG, EPR spectroscopy of oxidized DNA bound EndoIII and MutY, and the observation of both couples in the expected potential regimes on HOPG support the original [4Fe4S]3+/2+ assignment (6,8,15). Furthermore, this assignment is in agreement with the known potential ranges accessed by the [4Fe4S]3+/2+ couple of HiPIPs (17). Regardless of redox couple assignment, the possibility of activation by other molecules remains an interesting point deserving further investigation.
In addition to other molecules, charged amino acid residues near the cluster might also be expected to affect the potential. This was explored in a recent study in which several EndoIII mutants, E200K, Y205H, and K208E, were prepared and extensively characterized on DNA-modified gold electrodes; although these residues are located within 5 Å of the cluster, all of the mutants had indistinguishable DNA-bound midpoint potentials (18). Overall, these observations suggested that DNA binding was the dominant environmental effect in modulating potential, but the narrow accessible potential window on gold prevented further investigation.
In this work, we used direct electrochemistry on carbon electrodes to address the capacity of DNA and other polyanions to activate [4Fe4S] proteins for redox activity and to assess the ability of local electrostatics to shift the potential of EndoIII in the absence of DNA. Because the hydrophobic surface of HOPG is unsuitable for protein adsorption and difficult to prepare (8, 19–20), we turned to the rougher, more hydrophilic pyrolytic graphite edge (PGE) electrode for these experiments, using the technique of thin film voltammetry to immobilize proteins in a stable layer on the electrode surface (21–24). To enhance signal sizes, we also included single-walled carbon nanotubes (CNT) when possible, taking advantage of their high conductivity and the additional 3-dimensional surface area they can provide for protein binding (21). In summary, this platform provided an ideal and reliable way to improve our understanding of the factors important to tuning the potential of DNA processing enzymes containing [4Fe4S] clusters.
MATERIALS and METHODS
EndoIII Overexpression and Purification
WT E. coli EndoIII was overexpressed in BL21star-(DE3)pLysS cells containing a pET11-ubiquitin-His6-nth construct and purified as detailed previously (18), with the exception that the final buffer contained 10% rather than 20% glycerol (20 mM sodium phosphate, pH 7.5, 0.5 mM EDTA, 150 mM NaCl, 10% glycerol). For electrochemical experiments, glycerol was removed from the protein solution using a HiPrep 26/10 desalting column (GE Healthcare) equilibrated with a buffer consisting of 20 mM sodium phosphate, pH 7.5, 0.5 mM EDTA, 150 mM NaCl. Following buffer exchange, the protein was concentrated in two steps. First, 10,000 molecular weight cutoff (MWCO) Amicon Ultra 15 mL centrifugation filter units (Millipore) were used to concentrate each protein solution to a total volume of 1 mL or less. Samples were then transferred to 10,000 MWCO Amicon Ultra 0.5 mL centrifugation filter units (Millipore) and concentrated until the initially yellow protein solutions were very dark in color (approximately 300 μL final volume from 6 L of bacterial culture). Protein purity was confirmed by SDS-PAGE. Immediately following concentration of the sample, the [4Fe4S] cluster loading ratio was calculated by dividing the total [4Fe4S] cluster concentration as determined from the UV-visible absorbance spectrum using ε410 = 17,000 M−1cm−1 by the total protein concentration as measured in a Bradford assay; typical cluster loading ratios for WT EndoIII were 70–75%.
MutY Overexpression and Purification
MBP (Maltose Binding Protein)-MutY fusion protein was expressed and purified using a slightly modified version of a previously reported protocol (25). Modifications to the protocol included changes in “buffer A” to a resuspension buffer (20 mM sodium phosphate, pH 7.5, 200 mM NaCl, 1 mM EDTA, 1 mM DTT, 1 mM PMSF, and 10% glycerol) and use of an amylose column to eliminate the necessity of a streptomycin sulfate and ammonium sulfate precipitation. During the amylose preparation, the sample was washed with amylose wash buffer (20 mM sodium phosphate, pH 7.5, 200 mM NaCl, 1mM EDTA pH 8) and eluted in amylose elutant buffer (20 mM sodium phosphate, pH 7.5, 200 mM NaCl, 1 mM EDTA pH 8, 10 mM maltose). The resultant fractions were concentrated using an ultrafiltration cell with a 10,000 MWCO filter with stirring at 4°C. Protein was then diluted 10-fold in heparin buffer A (20 mM sodium phosphate, pH 7.5, 1 mM EDTA, 5% glycerol in water), applied to a Pharmacia Hi-trap heparin column on an AKTApurifier FPLC system, and eluted using a 10% linear gradient in heparin buffer A to 100% heparin buffer B (20mM sodium phosphate, pH 7.5, 1mM EDTA, 5% glycerol, and 1 M NaCl in water). MBP-MutY eluted at 450 mM NaCl (45% heparin buffer B). Purity of protein samples was confirmed via 12% SDS page stained with SYPRO orange. The [4Fe4S] cluster loading was determined using the UV-visible absorbance at 410 nm (ε410 = 17,000 M−1cm−1) and at 280 nm (ε280 = 143,240 M−1cm−1); samples were typically 65–75% loaded.
DNA preparation
DNA strands for EndoIII experiments were purchased from Integrated DNA Technologies, with sequences as follows:
20-mer: 5′-GTG AGC TAA CGT GTC AGT AC-3′
Complement: 5′-GTA CTG ACA CGT TAG CTC AC-3′
Single-stranded DNA oligomers (5 μmol) were resuspended in MilliQ water and purified by ethanol precipitation. Briefly, 1000 μL of cold 200 proof ethanol and 50 μL of 3 M NaCl were added to 100 μL single-stranded DNA in water and vortexed; DNA solutions were then frozen in liquid nitrogen for rapid precipitation and spun at 16,000 RCF (25 minutes) to form a pellet which was then re-dissolved in EndoIII storage buffer (20 mM sodium phosphate, pH 7.5, 0.5 mM EDTA, 150 mM NaCl). Single-stranded DNA was quantified by UV-vis using ε260 values calculated using the Integrated DNA Technologies oligo analyzer tool; these were 197,800 M−1cm−1 for the 20-mer strand and 190,200 M−1cm−1 for its complement. Equimolar amounts of each strand were then annealed by incubation at 90°C for 5 minutes followed by slow cooling to ambient temperature.
For MutY experiments, DNA substrates containing oxoG (8-oxo-guanine) or FA (2′-fluoro-adenine) were synthesized at the University of Utah DNA and Peptide Synthesis Core Facility and unmodified strands were ordered from Integrated DNA Technologies. The following DNA duplexes were used:
15-mer: 5′-GGA GCC AXG AGC TCC-3
15-mer Complement: 3′-CCT CGG TYC TCG AGG-5′
30-mer: 5′-CGA TCA TGG AGC CAC XAG CTC CCG TTA CAG-3′
30-mer Complement: 3′-GCT AGT ACC TCG GTG YTC GAG GGC AAT GTC-5′
X = G or oxoG and Y = C, FA, or A
Oligonucleotides containing the central oxoG or FA were deprotected and cleaved from the column by incubation in NH4OH; 2-mercaptoethanol was added into oxoG samples to prevent oxidation. The cleaved DNA substrates were dissolved in H2O, filtered with a 0.2 μm filter, and HPLC purified using a Beckman Gold Nouveau system with a Waters AP1DEAE 8HR column; a 10–100% gradient of 90:10 H2O/acetonitrile with 2 M NH4Ac was used in purification. Isolated fractions were dried down and de-salted using SEP-PAK cartridges, and DNA integrity was confirmed using MALDI-MS. All DNA substrates were stored dried in the −20°C freezer prior to annealing.
Electrochemistry
All electrochemical experiments were performed on an edge-plane pyrolytic graphite electrode (Pine Research Instrumentation) with a geometric surface area of 0.196 cm2. To generate a rough surface suitable for protein binding, the electrode was abraded with 400 grit sandpaper and cleaned by sonication for 1 minute each in ethanol and water. After sonication, the absence of electroactive impurities was verified by scanning in EndoIII storage buffer (20 mM sodium phosphate, 0.5 mM EDTA, 150 mM NaCl, pH 7.5) or MutY storage buffer (20 mM sodium phosphate, 1 mM EDTA, 150 mM NaCl, pH 7.6) as appropriate.
Single-walled carbon nanotubes (CNTs) were found to enhance greatly the signal size of adsorbed protein, so they were included in the formation of all thin films unless otherwise noted. Protein thin films were formed from several (typically 3–6) alternate layers of 10 μL single-walled carbon nanotubes (CNT) in water (0.25 mg/mL) and 10 μL EndoIII (150 μM in storage buffer) or MutY (50 μM) in a 1:1 mix with aqueous CNTs. Each layer was gently dried under an argon gun, and the process was repeated until the surface was coated by a viscous film, which was then secured with 5% Nafion in water (diluted from 10% in water as purchased) to prevent dispersal (21). For experiments including DNA, CNTs generally hindered electrochemical signals, so these films were formed in their absence. Poly-L glutamate (MW 50–100 kDa) was used to assess the effects of a negatively charged non-substrate on potential. Although Nafion also carries a negative charge at the pH values used, it was applied only to the top of a multilayer film to form a binding layer, minimizing interactions with the electroactive protein; in contrast, poly-L glutamate and DNA were incorporated directly into the thin film with protein to maximize any possible interactions.
After thin film formation, 50 μL of EndoIII or MutY storage buffer was pipetted on top of the film and an Ag/AgCl reference in 3 M NaCl and Pt auxiliary electrode were submerged in the resulting droplet. Reduction potential, current, and charge measurements were then taken by cyclic voltammetry (CV), square wave voltammetry (SQWV) and differential pulse voltammetry (DPV); all experiments were conducted at ambient temperature (20 °C). Electroactive area was determined by plotting the scan rate dependence of the CV current generated by 1 mM [Ru(NH3)6]Cl3 in storage buffer and applying the Randles-Sevcik equation (26),
| (1) |
Ip is the peak current in amperes, F is Faraday’s constant (96485 C·mol−1), R is the universal gas constant (8.314 J·(mol·K)−1), T is temperature in K, n is the number of electrons transferred per CV peak, A is electrode area in cm2, D is the diffusion coefficient in cm2·s−1 (9.0 × 10−6 for [Ru(NH3)6]3+; 27), C° is bulk protein concentration in mol·cm−3, and ν is the scan rate in V·s−1. Potentials were converted to NHE by adding 0.212 V to the value measured by Ag/AgCl, using the value of 0.209 mV value at 25 C given by the supplier, BASi®, and applying a temperature correction (28). To prevent leakage of NaCl into the buffer and subsequent wandering of the reference potential, the glass frit of the electrode was immersed in a gel loading pipet tip containing 3 M NaCl with 4% dissolved agarose, and dried in this mix overnight. CNTs, 10% aqueous Nafion, poly-L glutamate, and [Ru(NH3)6]Cl3 were purchased from Sigma-Aldrich, while the Ag/AgCl reference electrode in 3 M NaCl was purchased from BASi®.
RESULTS
Direct Electrochemistry of WT EndoIII and MutY
To examine variations in EndoIII potentials with various substitutions or binding partners, EndoIII thin films anchored to the surface by Nafion were prepared on a PGE electrode. In the absence of DNA, a quasi-reversible signal was observed by CV (Figure 1). The signal under these conditions was relatively small (3 ± 1 μC reductive peak, −4 ± 1 μC oxidative peak), and the reductive peak partially overlapped the much larger wave of oxygen reduction, making it more challenging to quantify. By adding 0.25 mg/mL CNTs to form a protein/CNT/Nafion thin film, the peak areas increased by an order of magnitude to reach 16 ± 3 μC and −18 ± 5 μC reductive and oxidative peaks, respectively, while the peak potentials remained unaltered from those without the CNTs at 74 ± 20 mV and 162 ± 18 mV (all potentials vs NHE).
Figure 1.

Representative CVs from EndoIII and MutY thin-films on a PGE electrode. A thin film containing only 75 μM EndoIII capped with Nafion gave a quasi-reversible signal with reductive and oxidative peaks centered, respectively, at 74 ± 11 and 162 ± 20 mV versus NHE (center; red trace). Notably, the addition of CNTs substantially amplified the signal, simplifying quantification (center; blue trace). Similarly, 25 μM MutY in the presence of CNTs yielded a signal with reductive and oxidative peaks at 100 ± 9.0 and 162 ± 3.0 mV versus NHE (bottom). CV scans were taken at a scan rate of 100 mV/s.
The addition of EndoIII and CNTs was associated with a large increase in the capacitance; much of this increase was due to CNTs, as seen in CNT/Nafion thin films, but the protein itself certainly contributed (Figure S1). Notably, the high conductivity of CNTs amplifies redox events at the surface; a CNT/Nafion film shows reversible peaks around 200 mV vs NHE and −80 mV vs NHE, both of which show no splitting and are likely attributable to the reversible reduction of surface oxides on the edge plane and even on the CNTs themselves (Figure S1; 29–31). Indeed, the 200 mV peaks were invariably present, although smaller, in buffer alone and the −80 mV peaks varied in size based upon the freshness of the CNT suspension applied, consistent with this assertion. The presence of protein on the surface markedly suppressed both of these peaks, and the EndoIII signal differed from the background both by its potential, which was essentially identical to that measured in the absence of CNTs, and in the occurrence of peak splitting; the latter suggested a slower process, in agreement with reports of other proteins adsorbed on carbon (21).
WT MutY thin films were prepared just as with EndoIII, although the stock concentrations were somewhat lower (~50 μM for MutY compared to ~150 μM for EndoIII). MutY displayed a quasi-reversible signal similar to EndoIII on CNT/Nafion thin films, with CV peak potentials centered at 100 ± 9 mV for the reductive peak and 162 ± 3 mV for the oxidative peak (Figure 1). Notably, the potentials were within error of the values obtained for EndoIII. The respective peak areas were 2.3 ± 0.3 μC and −3.4 ± 0.1 μC, about an order of magnitude smaller than EndoIII and indicative of lower surface coverage.
For both EndoIII and MutY, the current exhibited a linear dependence on the scan rate (Figure S2), confirming that the protein was adsorbed to the electrode surface rather than diffusing in from solution; this relationship was present whether or not CNTs were included. Surface coverage was initially determined simply by converting the total CV peak charge at a scan rate of 100 mV/s into pmol using Faraday’s constant and dividing by the geometric surface area of the electrode. Because the PGE surface is uneven, the geometric surface area can underestimate the electroactive area by a factor as large as 104 (32). Indeed, using the geometric area of the electrode (0.196 cm2) gave a surface coverage of 550 ± 300 pmol/cm2 for 75 μM EndoIII stock, over 10 times larger than reported for ferredoxin thin films on PGE (40 pmol/cm2; 32) and over 100 times larger than CNT/Nafion/protein thin films on glassy carbon (2–6 pmol/cm2; 21).
By taking the scan rate dependence of the current for [Ru(NH3)6]3+ in EndoIII storage buffer and applying the Randles-Sevcik equation (Equation 1), the electroactive surface area was determined to be 1.0 cm2, about 5 times larger than the simple geometric area. When this correction was applied to a thin film formed from 75 μM EndoIII stock, a value of 108 ± 60 pmol/cm2 was obtained, which is still high but much closer to previously published results on PGE (32). Applying the same correction to films formed from 25 μM MutY stock gave a coverage of 29 ± 6 pmol/cm2, around 25% of that measured for 75 μM EndoIII. To facilitate a more direct comparison, surface coverage on thin films formed with 25 μM EndoIII was measured to be 51 ± 8 pmol/cm2, indicating that MutY adsorption was absolutely less extensive than EndoIII. This result is not surprising, given that unmodified MutY is 39 kDa while EndoIII is only 24 kDa; the 42 kDa N-terminal MBP tag on MutY would only enhance this issue.
Adsorption of proteins to the electrode surface made it possible to estimate electron transfer rate (kET) and transfer coefficient (α) values using the Laviron method for diffusionless systems (Figure S2; 33), where α is a measure of transition state symmetry, taking on values between 0 and 1. The MutY signal was too small to measure the currents at high scan rates, but this analysis could be carried out for EndoIII. In the case of EndoIII, we obtained a kET of 3 ± 0.6 s−1 and α values of 0.4 and 0.6 for the reductive and oxidative peaks, respectively. Assuming that electron transfer is the only reaction taking place on the electrode, the values of α imply a quasi-reversible system. Importantly, electron transfer rates are similar to those reported for other redox-active enzymes/proteins adsorbed to carbon electrodes in the presence of CNTs and Nafion (21).
Electrochemistry of WT EndoIII and MutY in the presence of DNA
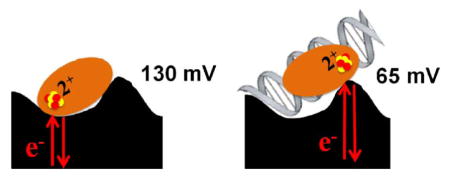
Having found thin films on PGE to facilitate the direct electrochemistry of free [4Fe4S] proteins, we proceeded to see if the DNA-directed potential shift observed on HOPG could be replicated. With this aim in mind, a solution containing EndoIII and 20-mer duplex DNA in a 1:1 ratio in storage buffer was prepared, incubated on ice for 30 minutes and dried to form a thin film on the electrode surface. Inclusion of CNTs in the film resulted in a noisy, widely split (> 100mV) signal that had the same potential as DNA-free protein, indicating that the protein was not DNA-bound. Thus, CNTs were excluded from subsequent experiments with DNA present. Under these conditions, EndoIII showed a very small signal by CV that was much more readily visualized and quantified using square wave voltammetry (SQWV; Figure 2, Table 1). The potential as measured by SQWV was 64 ± 8 mV, representing a negative shift of ~60 mV from the DNA-free value of 130 ± 8 mV. This result is in overall agreement with earlier results supporting the stabilization of the oxidized [4Fe4S]3+ form upon DNA binding, but the shift seen here is markedly smaller (8). This difference can likely be attributed to less than 100% of the protein being DNA bound; free protein likely contributed to the observed peak, leading to an underestimate of the shift. This heterogeneity could be further complicated by the expected random orientation of DNA and protein at the electrode. Indeed, this is a fundamental difference between the HOPG and PGE setups: the signal on HOPG was DNA-mediated, while that on PGE was not.
Figure 2.

EndoIII and MutY thin-film voltammetry on a PGE electrode in the presence of DNA. By SQWV, the presence of DNA resulted in a −65 mV shift in the potential of 75 μM EndoIII (center), with a similar result for 25 μM MutY (bottom). Signals were very small due to surface passivation, but they were still readily apparent by SQWV. Unfortunately, CNTs led to inconsistent and unstable signals, likely interfering with DNA binding, so they could not be used to enhance the signals. Thin films were formed from several layers of a pre-mixed 1:1 protein/DNA solution, and were capped with Nafion. SQWV scans were taken at a frequency of 15 Hz with 0.025 V amplitude, and scans were from positive to negative potentials (indicated by the arrow).
Table 1.
Potentials (versus NHE) of SQWV reductive sweeps for WT and mutant EndoIII and WT MutY in the presence and absence of DNA.
| a Enzyme | b ESQWV (mV), − DNA | b ESQWV (mV), + DNA |
|---|---|---|
| WT EndoIII | 130 ± 8 | c 64 ± 8 |
| WT EndoIII + poly-L glutamate | 110 ± 9 | - |
| EndoIII E200K | 130 ± 7 | - |
| EndoIII Y205H | 125 ± 11 | - |
| EndoIII K208E | 141 ± 12 | - |
| WT MutY | d > 130 mV |
c 85 ± 3 c, e 80 ± 6 |
When used, CNTs were added from a 0.25 mg/mL stock, and all experiments used protein storage buffer as the supporting electrolyte (20 mM sodium phosphate, 0.5 mM EDTA, 150 mM NaCl, pH 7.5 for EndoIII, 20 mM sodium phosphate, 1 mM EDTA, 150 mM NaCl, pH 7.6 for MutY).
Each value is the average of 3 or more separate experiments, and error is standard deviation of the mean.
No CNTs present.
The MutY SQWV peaks (Figure 2) were too small and noisy to measure the potential with confidence.
OG:FA MutY substrate trap DNA
Electrochemistry of MutY thin films prepared in the presence and absence of DNA (a 30-mer duplex, in this case) gave similar results to EndoIII. In the absence of DNA and CNTs, the SQWV peak was too small to measure the potential with confidence, but it was in the same region as EndoIII (~130 mV vs NHE; Figure 2); when DNA was present, the potential shifted to 85 ± 3 mV vs NHE (Figure 2, Table 1). In an effort to obtain larger signal sizes by increasing the proportion of DNA-bound protein on the surface, the experiment was repeated with DNA containing an FA:oxoG substrate trap, which mimics the A:oxoG target of MutY but inhibits N-glycosidic bond cleavage due to the electron withdrawing effect of the fluorine at the sugar 2′ position (34). No significant differences were observed, although the potential of 80 ± 6 mV indicated a similar DNA-bound signal, suggesting that any increase in DNA binding affinity afforded by the substrate trap was not sufficient to overcome the combined effects of DNA-free proteins on the surface and surface passivation by the DNA itself. In considering these results, it is important to note that, while a shift could be observed for both proteins, surface passivation by non-tethered DNA blocked a portion of the available electroactive sites and made the signals small and noisy. Taken together, these results demonstrate the limitations of the PGE surface, but, importantly, they confirm that large potential shifts can be observed on the PGE electrode even under suboptimal conditions.
Direct electrochemistry of WT EndoIII in the presence of poly-L glutamate
We next sought to determine if DNA is unique in its ability to shift the potential of the [4Fe4S] cluster by forming thin films with poly-L glutamate (MW 50–100 kDa). Due to the small size of MutY signals, EndoIII was used for this purpose. Like DNA, poly-L glutamate is polyanionic, but it is not a specific target for EndoIII binding. In these experiments, poly-L glutamate was preincubated with EndoIII in a 1:1 ratio and added to thin films just as with DNA. Unlike with DNA, the measured potentials and peak shapes were indistinguishable whether or not CNTs were included. Interestingly, the presence of poly-L glutamate did not result in a significant potential shift by CV, with the reductive and oxidative peaks centered at 81 ± 1 mV and 143 ± 1 mV, respectively (Figure 3). At 110 ± 9 mV, the SQWV potential is, accounting for error, about 10 mV lower than that of free EndoIII, but is still substantially more positive than the DNA-bound potential (Table 1). The CV values are within error of that for free WT EndoIII (Figure 3, Table 1), and, given that EndoIII does not specifically bind to poly-L glutamate, this result should not be too surprising. It is likely that the [4Fe4S] cluster is not exposed to the negative charges of unbound poly-L glutamate to the extent that it would be when the protein is tightly bound to DNA, so no significant shift can be observed. This is also in line with the insusceptibility of free EndoIII to oxidation or reduction originally noted by Cunningham (2). Overall, these results indicate that the potential of the EndoIII [4Fe4S] cluster can be appreciably altered only when the protein binds directly to a polyanion, such that the cluster experiences the full effect of the negative charges. Thus, DNA appears uniquely able to affect the potential of the cluster both by its charges and by its function as a binding substrate for these types of [4Fe4S] proteins.
Figure 3.

Thin-film voltammetry of WT EndoIII with poly-L glutamate and comparison of WT with the mutants E200K, Y205H, and K208E. Unlike DNA, poly-L glutamate caused no significant potential shift even at 6 mM glutamate (top). 75 μM WT and mutant EndoIII in protein/CNT/Nafion thin films had nearly identical potentials, with CV peaks centered around 125 mV versus NHE (center); the similarity is even more apparent by DPV (bottom). Small shifts within the measurement error (10–15 mV) are still possible, but pale in comparison to the effect of DNA. All CVs shown were taken at 100 mV/s, while DPVs were taken at an amplitude of 0.05 V with a 0.5 s pulse period. Poly-L glutamate was pre-incubated with 75 μM EndoIII both with and without CNTs; due to the larger signals, the CVs shown are from films including CNTs.
Direct Electrochemistry of EndoIII mutants
Since EndoIII was observed to undergo a significant potential shift upon DNA binding, but not in the presence of the non-substrate poly-L glutamate, we reasonably assumed that only charges in the immediate vicinity of the cluster could shift the potential. Following this line of reasoning, we sought to determine if altering the charge on amino acid residues near the cluster might have a similarly large effect on the potential. Mutation of nearby residues to give a net positive or negative charge was of further interest because it could, in principle, shift the cluster potential in both positive and negative directions, unlike the unidirectional shift associated with DNA binding.
In order to investigate these possibilities, the following EndoIII point mutants were prepared (18): E200K, Y205H, and K208E. All of these residues are ~5 Å from the cluster, and these mutations span nearly the full range of possible single-charge alterations, going from a single negatively charged residue to a positively charged one (E200K), a neutral residue to a positive residue (Y205H), and a positive residue to a negative one (K208E). Positively charged mutants would be expected to be more repulsive to the [4Fe4S]3+ state, favoring reduction, while negatively charged mutants would be more attractive, stabilizing the oxidized form of the protein. While shifts might be expected, the precise extent cannot be readily predicted; a study on outer-sphere effects in HiPIP [4Fe4S] protein resulted in shifts as large as 150 mV, while surface mutations in certain ferredoxins led to no changes in potential (35, 36).
Surprisingly, all of the mutants exhibited DNA-free potentials on PGE thin films within error of WT (Figure 3, Table 1); this was apparent by CV, SQWV and DPV. However, the standard deviation was relatively high, with the largest value (for WT) around 20 mV. Since conclusions are limited by the largest error among the species investigated, these data are consistent with two possibilities: either there is no change between WT and any of the mutants, or a small shift on the order of 10 mV is present. In either case, it is clear that the effect of changing a single charge, even one in very close proximity to the cluster, is much less dramatic than that of DNA binding.
To confirm the accuracy of potential measurements, thin films formed with 2 mM [Ru(NH3)6]3+ with and without EndoIII were also examined (Figure S3). The [Ru(NH3)6]3+ midpoint potential was consistently ~10 mV vs NHE in the presence and absence of CNTs and Nafion, as well as in the presence and absence of EndoIII. Just as with EndoIII, CNTs did markedly sharpen the peaks and facilitate larger signals, independently confirming their effect on species in a thin film. Overall, these controls verified that the measured potentials were not affected by CNTs or other thin film components, indicating that the observed lack of variation between mutants was due to properties of the proteins themselves. Overall, direct electrochemistry of these EndoIII mutants confirm that DNA binding is a dominating effect relative to single charge reversals in the amino acid sequence (18).
DISCUSSION
In this study, we have used thin film voltammetry on a PGE electrode to measure the potentials of EndoIII and MutY in the presence and absence of DNA, and, in the case of EndoIII, in the presence of poly-L glutamate and with point mutations altering the charged environment near the cluster. Table 1 summarizes all of the results. Notably, the potential shift observed upon DNA binding was smaller than previously reported (8), with the SQWV potentials of DNA-free EndoIII going from 250 ± 30 mV on HOPG to 130 ± 8 mV on PGE. This difference was certainly in large part the result of less than 100% DNA binding on PGE, but the distinct electrode environments on PGE and HOPG may also have played a role (8, 19–20). Indeed, the presence of negatively charged surface oxides on PGE, but not HOPG, would be expected to lower the potential of adsorbed proteins to some extent (29). Even with a lower DNA-free potential, DNA-bound potentials still dropped by 65 mV on PGE, which supports the assertion that DNA binding has a prominent, although not necessarily additive, effect which cannot be duplicated by non-substrate polyanionic molecules such as poly-L glutamate.
The absence of a significant potential shift in the presence of poly-L glutamate does at first appear to be in conflict with the results of the Todorovic group, wherein the redox activation of EndoIII was reported on a mercaptoundecanoic acid monolayer assembled on a gold electrode in the absence of DNA (16). However, this effect was attributed to tight binding to the carboxylic acid film; in contrast, the solvated poly-L glutamate used here would present a very different environment than a thin film of small molecules, and it is unlikely that EndoIII was readily able to bind this non-substrate polymer. While it is reasonable that multiple negative charges could shift the potential, they can only do so if EndoIII is able to bind with some specificity.
The results obtained here for DNA demonstrate that large potential shifts are observable on PGE even under conditions where full DNA binding is unlikely. In contrast to the conditions required to study DNA-binding, the DNA-free EndoIII mutants were studied under conditions that generated very large and readily quantifiable signals; thus, the lack of any apparent change in the potential among these mutants indicates that individual charge alterations near the cluster do not have a significant effect. However, the error in these experiments was relatively high, leaving two interpretations open. First, there may genuinely be no shift. In this case, the conclusion would be that a single charge alteration is insufficient by itself to disrupt the local environment, even for residues ~5 Å from the cluster. Alternatively, because the measurement error was around 20 mV for WT, shifts on the order of 10 mV may have been present but undetectable. Such small shifts would still pale in comparison to those associated with DNA binding, which are larger as independently measured on a PGE electrode (−60 mV for the EndoIII 2+/3+ couple) and on an HOPG electrode (−200 mV; 8). Assuming that the charge mutants of EndoIII do have altered potentials and that the shift is at the upper limit of our error (~10 mV), the effect of DNA binding is at minimum 6 times greater than the effect of a single charge alteration. In either case, it should be noted that, despite the small signals and adverse conditions resulting from DNA in the film, a substantial shift in potential upon DNA binding was still detectable on PGE, while no obvious shift in potential relative to WT was observed for any of the mutants, even though their potentials were determined from very large DNA-free signals under ideal circumstances.
A similar result obtained with the same mutants on DNA-modified gold electrodes supports the observed lack of potential shifts. In those experiments, the similarity in potential was attributed to the presence of DNA being a dominant effect, with only an increase in current correlated with protein folding stability occurring for K208E and Y205H (18). The smaller accessible window and poor to nonexistent DNA-free signal on gold electrodes prevented the experiments described here from being carried out, but taken along with our results, it appears that the mutants have similar potentials to WT both on and off of DNA.
In summary, these results reveal that, under the same experimental conditions, DNA binding is the dominant factor in tuning the redox potential of the [4Fe4S] cluster. This is supported by the similarity of the DNA-bound midpoint potentials of various repair enzymes (6), which all fall within a range of 60–85 mV vs NHE. In the context of DNA repair, we have previously proposed a model in which these proteins use single-electron transfers to signal to each other across the genome in the search for damage (15). In order for this model to work, the electron transfers must be reversible, necessitating that the proteins involved have approximately equal DNA-bound midpoint potentials. If this were not the case, proteins of lower reduction potential would remain bound at the expense of those of higher potential, and the search process would only be able to proceed by diffusion processes that are too slow to fully account for the time scale of DNA repair (15). This model depends on the large effect of DNA binding to bring potentials into a relevant regime, and our results verify that differences in the protein environment are unlikely to shift the potential sufficiently to affect the reversibility of inter-protein signaling on DNA. Overall, the similar DNA-bound potentials among diverse proteins facilitate not only the DNA damage search, but could also make long-range communication between diverse pathways possible.
Supplementary Material
Acknowledgments
We are grateful to the NIH (GM61077 to JKB, CA06985 to SSD), FAPESP-Brazil (Visiting Associate at Caltech Fellowship 13/10516-4 to FNC) and the Moore Foundation for their support of this research. A.Z. was supported by an NSF fellowship, N.N.N. was supported by a Floyd and Mary Schwall Fellowship for Medical Research, and A.R.A. was supported by the National Institute of Aging of the NIH on a pre-doctoral NRSA (F31AG040954).
Footnotes
Author Contributions
All authors have given approval to the final version of the manuscript.
Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org: Background CV scans, scan rate dependence of protein signals on PGE, [Ru(NH3)6]3+ CVs on PGE.
References
- 1.Kim YJ, Wilson DM., III Overview of Base Excision Repair Biochemistry. Curr Mol Pharmacol. 2012;5:3–13. doi: 10.2174/1874467211205010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cunningham RP, Asahara H, Bank JF, Scholes CP, Salerno JC, Surerus K, Munck E, McCracken J, Peisach J, Emptage MH. Endonuclease III is an iron-sulfur protein. Biochemistry. 1989;28:4450–4455. doi: 10.1021/bi00436a049. [DOI] [PubMed] [Google Scholar]
- 3.Porello SL, Cannon MJ, David SS. A Substrate Recognition Role for the [4Fe4S]2+ Cluster of the DNA Repair Glycosylase MutY. Biochemistry. 1998;37:6465–6475. doi: 10.1021/bi972433t. [DOI] [PubMed] [Google Scholar]
- 4.David SS, O’Shea VL, Kundu S. Base-excision repair of oxidative DNA damage. Nature. 2007;447:941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Woods RD, O’Shea VL, Chu A, Cao S, Richards JL, Horvath MP, David SS. Structure and stereochemistry of the base excision repair glycosylase MutY reveal a mechanism similar to retaining glycosylases. Nucleic Acids Research. 2016;44:801–810. doi: 10.1093/nar/gkv1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boal AK, Yavin E, Lukianova OA, O’Shea VL, David SS, Barton JK. DNA-Bound Redox Activity of DNA Repair Glycosylases Containing [4Fe4S] Clusters. Biochemistry. 2005;44:8397–8407. doi: 10.1021/bi047494n. [DOI] [PubMed] [Google Scholar]
- 7.Arnold AR, Grodick MA, Barton JK. DNA Charge Transport: from Chemical Principles to the Cell. Cell Chemical Biology. 2016;23:183–197. doi: 10.1016/j.chembiol.2015.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gorodetsky AA, Boal AK, Barton JK. Direct Electrochemistry of Endonuclease III in the Presence and Absence of DNA. J Am Chem Soc. 2006;128:12082–12083. doi: 10.1021/ja064784d. [DOI] [PubMed] [Google Scholar]
- 9.Thayer MM, Ahern H, Xing D, Cunningham RP, Tainer JA. Novel DNA binding motifs in the DNA repair enzyme endonuclease III crystal structure. The EMBO J. 1995;14:4108–4120. doi: 10.1002/j.1460-2075.1995.tb00083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fromme JC, Verdine GL. Structure of a trapped endonuclease III-DNA covalent intermediate. The EMBO J. 2003;22:3461–3471. doi: 10.1093/emboj/cdg311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guan Y, Manuel RC, Arvai AS, Parikh SS, Mol CD, Miller JH, Lloyd RS, Tainer JA. MutY catalytic core, mutant and bound adenine structures define specificity for DNA repair enzyme superfamily. Nat Struct Biol. 1998;5:1058–1064. doi: 10.1038/4168. [DOI] [PubMed] [Google Scholar]
- 12.Fromme JC, Banerjee A, Huang SJ, Verdine GL. Structural basis for removal of adenine mispaired with 8-oxoguanine by MutY adenine DNA glycosylase. Nature. 2004;427:652–656. doi: 10.1038/nature02306. [DOI] [PubMed] [Google Scholar]
- 13.Stephens PJ, Jollie DR, Warshel A. Protein Control of Redox Potentials of Iron-Sulfur Proteins. Chem Rev. 1996;96:2491–2513. doi: 10.1021/cr950045w. [DOI] [PubMed] [Google Scholar]
- 14.Ren B, Duan X, Ding H. Redox control of the DNA damage-inducible protein DinG helicase activity via its iron-sulfur cluster. J Biol Chem. 2009;284:4829–4835. doi: 10.1074/jbc.M807943200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boal AK, Genereux JC, Sontz PA, Gralnick JA, Newman DK, Barton JK. Redox signaling between DNA repair proteins for efficient lesion detection. Proc Natl Acad Sci USA. 2009;106:15237–15242. doi: 10.1073/pnas.0908059106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moe E, Sezer M, Hildebrandt P, Todorovic S. Surface enhanced vibrational spectroscopic evidence for an alternative DNA-independent redox activation of endonuclease III. ChemComm. 2015;51:3255–3257. doi: 10.1039/c4cc09498k. [DOI] [PubMed] [Google Scholar]
- 17.Dey A, Jenney FE, Jr, Adams MWW, Babini E, Takahashi Y, Fukuyama K, Hodgson KO, Hedman B, Solomon EI. Solvent Tuning of Electrochemical Potentials in the Active Sites of HiPIP Versus Ferredoxin. Science. 2007;318:1464–1468. doi: 10.1126/science.1147753. [DOI] [PubMed] [Google Scholar]
- 18.Pheeney CG, Arnold AR, Grodick MA, Barton JK. Multiplexed Electrochemistry of DNA-Bound Metalloproteins. J Amer Chem Soc. 2013;135:11869–11878. doi: 10.1021/ja4041779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Banks CE, Compton RG. New electrodes for old: from carbon nanotubes to edge plane pyrolytic graphite. Analyst. 2006;131:15–21. doi: 10.1039/b512688f. [DOI] [PubMed] [Google Scholar]
- 20.Armstrong FA, Bond AM, Hill AO, Oliver BN, Psalti ISM. Electrochemistry of Cytochrome c, Plastocyanin, and Ferredoxin at Edge- and Basal-Plane Graphite Electrodes Interpreted via a Model Based on Electron Transfer at Electroactive Sites of Microscopic Dimensions in Size. J Am Chem Soc. 1989;111:9185–9189. [Google Scholar]
- 21.Yin Y, Lu Y, Wu P, Cai C. Direct Electrochemistry of Redox Proteins and Enzymes Promoted by Carbon Nanotubes. Sensors. 2005;5:220–234. [Google Scholar]
- 22.Léger C, Elliott SJ, Hoke KR, Jeuken LJC, Jones AK, Armstrong FA. Enzyme Electrokinetics: Using Protein Film Voltammetry To Investigate Redox Enzymes and Their Mechanisms. Biochemistry. 2003;42:8653–8662. doi: 10.1021/bi034789c. [DOI] [PubMed] [Google Scholar]
- 23.Blanford CF, Armstrong FA. The pyrolytic graphite surface as an enzyme substrate: microscopic and spectroscopic studies. J Solid State Electrochemistry. 2006;10:826–832. [Google Scholar]
- 24.Baffert C, Sybirna K, Ezanno P, Lautier T, Hajj V, Meynial-Salles I, Soucaille P, Bottin H, Léger C. Covalent Attachment of FeFe Hydrogenases to Carbon Electrodes for Direct Electron Transfer. Anal Chem. 2012;84:7999–8005. doi: 10.1021/ac301812s. [DOI] [PubMed] [Google Scholar]
- 25.Chmiel NH, Golinelli MP, Francis AW, David SS. Efficient recognition of substrates and substrate analogs by the adenine glycosylase MutY requires the C-terminal domain. Nucleic Acids Res. 2001;29:553–64. doi: 10.1093/nar/29.2.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kissinger PT, Heineman WR, editors. Laboratory Techniques in Electroanalytical Chemistry. 2. Marcel Dekker; 1996. [Google Scholar]
- 27.Wang Y, Limon-Peterson JG, Compton RG. Measurement of the diffusion coefficients of [Ru(NH3)6]3+ and [Ru(NH3)6]2+ in aqueous solution using microelectrode double potential step potentiometry. J Electroanalytical Chem. 2011;652:13–17. [Google Scholar]
- 28.Greeley RS, Smith WT, Jr, Stoughton RW, Lietzke MH. Electromotive Forces in Aqueous Solutions at Elevated Temperatures. I. The Standard Potential of the Silver-Silver Chloride Electrode. J Phys Chem. 1960;64:652–657. [Google Scholar]
- 29.McCreery RL. Advanced Carbon Electrode Materials for Molecular Electrochemistry. Chem Rev. 2008;108:2646–2687. doi: 10.1021/cr068076m. [DOI] [PubMed] [Google Scholar]
- 30.Blurton KF. An Electrochemical Investigation of Graphite Surfaces. Electrochimica Acta. 1973;18:869–875. [Google Scholar]
- 31.Gooding JJ. Nanostructuring electrodes with carbon nanotubes: A review on electrochemistry and applications for sensing. Electrochimica Acta. 2005;50:3049–3060. [Google Scholar]
- 32.Breton JL, Duff JLC, Butt JN, Armstrong FA, George SJ, Pétillot Y, Forest E, Schäfer G, Thomson AJ. Identification of the iron-sulfur clusters in a ferredoxin from the archaeon Sulfolobus acidocaldarius. Evidence for a reduced [3Fe-4S] cluster with pH-dependent electronic properties. Eur J Biochem. 1995;233:937–946. doi: 10.1111/j.1432-1033.1995.937_3.x. [DOI] [PubMed] [Google Scholar]
- 33.Laviron E. General expression of the linear potential sweep voltammogram in the case of diffusionless electrochemical systems. J Electroanaytical Chem. 1979;101:19–28. [Google Scholar]
- 34.Lee S, Verdine GL. Atomic substitution reveals the structural basis for substrate adenine recognition and removal by adenine DNA glycosylase. Proc Natl Acad Sci USA. 2009;106:18497–18502. doi: 10.1073/pnas.0902908106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Low DW, Hill MG. Backbone-Engineered High-Potential Iron Proteins: Effects of Active-Site Hydrogen Bonding on Reduction Potential. J Amer Chem Soc. 2000;122:11039–11040. [Google Scholar]
- 36.Shen B, Jollies DR, Stout CD, Diller TC, Armstrong FA, Gorst CM, La Mar GN, Stephens PJ, Burgess BK. Azotobacter vinelandii ferredoxin I. Alteration of individual surface charges and the [4Fe-4S]2+/+ cluster reduction potential. J Biol Chem. 1994;269:8564–8575. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.