

ABSTRACT
Bacterial lipoproteins are embedded in the cell membrane of both Gram-positive and Gram-negative bacteria, where they serve numerous functions central to cell envelope physiology. Lipoproteins are tethered to the membrane by an N-acyl-S-(mono/di)-acyl-glyceryl-cysteine anchor that is variously acylated depending on the genus. In several low-GC, Gram-positive firmicutes, a monoacyl-glyceryl-cysteine with an N-terminal fatty acid (known as the lyso form) has been reported, though how it is formed is unknown. Here, through an intergenic complementation rescue assay in Escherichia coli, we report the identification of a common orthologous transmembrane protein in both Enterococcus faecalis and Bacillus cereus that is capable of forming lyso-form lipoproteins. When deleted from the native host, lipoproteins remain diacylated with a free N terminus, as maturation to the N-acylated lyso form is abolished. Evidence is presented suggesting that the previously unknown gene product functions through a novel intramolecular transacylation mechanism, transferring a fatty acid from the diacylglycerol moiety to the α-amino group of the lipidated cysteine. As such, the discovered gene has been named lipoprotein intramolecular transacylase (lit), to differentiate it from the gene for the intermolecular N-acyltransferase (lnt) involved in triacyl lipoprotein biosynthesis in Gram-negative organisms.
IMPORTANCE This study identifies a new enzyme, conserved among low-GC, Gram-positive bacteria, that is involved in bacterial lipoprotein biosynthesis and synthesizes lyso-form lipoproteins. Its discovery is an essential first step in determining the physiological role of N-terminal lipoprotein acylation in Gram-positive bacteria and how these modifications impact bacterial cell envelope function.
KEYWORDS: firmicutes, acyl transferase, lipoproteins
INTRODUCTION
Lipoproteins are bacterial cell membrane components common throughout Gram-negative and Gram-positive bacteria, constituting 2 to 5% of all cellular proteins (1–5). Lipoproteins invariably contain a signature acylated N-terminal cysteine residue that tethers an otherwise soluble globular protein domain to the membrane surface. Positioned at the membrane-environment interface, lipoproteins play critical roles in ion and nutrient capture, solute transport, cell adhesion, and assembly of protein complexes and as protein-folding chaperones.
All lipoproteins are initially translated as precursors containing an N-terminal signal peptide harboring a conserved amino acid lipobox motif sequence preceding an invariant cysteine residue (6, 7). Once transported to the outer face of the membrane, prolipoprotein diacylglycerol transferase (Lgt) attaches a diacylglycerol residue from a phospholipid donor through a thioether bond (8). Signal peptidase II (Lsp) then cleaves the leader peptide immediately upstream from the cysteine to liberate the α-amino group (9). In Escherichia coli and other Gram-negative bacteria, biosynthesis is completed by an apolipoprotein N-acyltransferase (Lnt), which transfers a fatty acid from a membrane phospholipid to form an amide linkage (10). While both Lgt and Lsp are highly conserved across genera, lnt sequence orthologs are confined to Gram-negative bacteria and mycobacteria, which also make triacylated lipoproteins (5, 11). However, lipoprotein N-terminal modification in other Gram-positive bacteria still occurs and, in low-GC firmicutes in particular, is more complex and varied than previously appreciated (2). In addition to the conventional unmodified N-terminal and N-acylated forms, Kurokawa et al. discovered three novel lipoprotein forms: the peptidyl form, the N-acetyl form, and the lyso form (12). Lyso-form lipoproteins contain an N-acyl chain but differ in having a monoacyl-glyceryl group (akin to lysophospholipids) attached to a cysteine. Lyso-form lipoproteins are found in several Gram-positive firmicutes, including Enterococcus faecalis and Bacillus cereus (12). It is currently unclear how the mature lyso-form lipoprotein is made, but in E. faecalis, this likely occurs after canonical processing by Lgt and Lsp (13).
For the mechanism to make the final N-acyl-S-monoacyl-glyceryl-cysteine structure that is characteristic of lyso-form lipoproteins, two theories have been proposed: O-deacylation of the conventional triacyl form in tandem with N-terminal acylation via an Lnt-like reaction or intramolecular transacylation from the S-diacylglyceryl group to the α-amino group of cysteine (12). However, there are no apparent lnt sequence orthologs in firmicute genomes, nor are there any known examples of enzyme-catalyzed lipoprotein intramolecular transacylation (5). The general function of lipoprotein aminoacylation among firmicutes also remains to be seen. In Gram-negative organisms, Lnt-catalyzed lipoprotein N-acylation is necessary for efficient localization of outer membrane (OM) lipoproteins and the ensuing cell division (14–16). This cellular role for N-acylation is not applicable to lyso-form lipoproteins, which are found in cell envelopes lacking an OM. Therefore, the identification of the enzyme that N-terminally acylates lyso-form lipoproteins is a necessary first step, not just in determining how these lipoproteins are made but in elucidating the physiological role of N-acylation in Gram-positive bacteria as well. Using cross-complementation of an Lnt depletion strain of E. coli as a selection strategy and coupling this with mass spectrometry (MS) analyses of lipoproteins, we report herein the identification of a single gene capable of forming lyso-form lipoproteins in two low-GC firmicute hosts: E. faecalis and B. cereus.
RESULTS
Intergenic lipoprotein N-acylation cross-complementation screen design.
In contrast to the essential role of Lnt in E. coli, lipoproteins are nonessential in many firmicutes (3, 5), and there are no known phenotypes attributable to the N-terminal acylation state that could be used for direct selection in native hosts. We hypothesized that as long as lipoproteins were N-acylated, whether in triacylated or lyso form, they would be compatible substrates for the E. coli localization of lipoproteins (Lol) export machinery, since triacyl and lyso-form lipoproteins both share an α-amino-linked acyl chain (4). Fukuda et al. have extensively characterized the substrate specificity of Lol transport and have shown N-acylation to be a critical component in substrate recognition (16). Following acylation, the LolCDE complex extracts lipoproteins from the inner membrane (IM) and transfers them to LolA (Fig. 1A), which shuttles lipoproteins across the periplasm (17). Lipoproteins are then transferred to LolB, itself a lipoprotein, which subsequently inserts the acylated N-terminal anchor of OM-destined lipoprotein into the membrane bilayer (18). These data, when coupled with previous studies showing the cross recognition of E. coli lipoprotein substrates by firmicute lipoprotein biosynthetic proteins when heterologously expressed (19, 20), suggested that an intergenic complementation screen to find the gene(s) responsible for lyso-form lipoprotein N-acylation would be feasible. To address potentially incompatible acyl chain substrate specificities between the putative firmicute lyso-form N-acyl lipoprotein transferase and the acyl chain composition available in E. coli, we built two separate genomic DNA libraries of small inserts (3 to 5 kb) from both B. cereus and E. faecalis. While B. cereus elaborates mainly saturated branched-chain fatty acids, the complement of fatty acids in E. faecalis more closely matches that of E. coli in being even-numbered, linear acyl chains and containing unsaturation units (21). High-quality libraries were built in the multiple-copy-number plasmid pUC19 using genomic DNA randomly sheered by NEBNext double-stranded DNA (dsDNA) Fragmentase, allowing for at least 3-fold genomic coverage per transformation reaction, with more than 90% of library plasmids harboring the firmicute genomic DNA insert.
FIG 1.

Lipoprotein maturation in E. coli and complementation strategy. (A) Lipoproteins are sequentially modified by Lgt, Lsp, and Lnt. Once triacylated, lipoproteins are shuttled across the periplasm, where they are inserted into the OM by the Lol export machinery. The Bam complex, catalyzing β-barrel protein assembly and insertion into the membrane, folds LptD in association with LptE, in turn ensuring proper LPS assembly at the outer leaflet of the membrane. The LPS leaflet excludes large hydrophilic molecules, such as vancomycin, from entering the cell. The highly abundant lipoprotein Lpp forms covalent linkages to peptidoglycan through residue K58. (B) The Kanr-TT-araC-PBAD cassette was constructed and inserted directly upstream from lnt by Red-mediated recombination to generate l-arabinose-dependent expression in E. coli. The cassette contained a transcriptional terminator to prevent read-through transcription. (C) Wild-type E. coli strain BW25113 (WT) and PBAD-lnt (strain KA325), Δlpp (strain TXM327), and PBAD-lnt Δlpp (strain KA349) mutants were streaked onto LB agar plates with or without l-arabinose and vancomycin. Plates lacking arabinose contained glucose to suppress basal PBAD expression, while all plates were supplemented with palmitate as a potential source of fatty acids.
In order to identify candidate firmicute proteins able to N-terminally acylate lipoproteins and thus restore OM transport in E. coli, a conditionally lethal mutant of lnt was constructed in E. coli. The Kanr-TT-araC-PBAD cassette was integrated directly upstream from the chromosomal lnt gene (Fig. 1B), and the resulting strain KA325 was indeed dependent on l-arabinose for growth (Fig. 1C). Phenotypic rescue by growth in the absence of the lnt inducer l-arabinose would thus nominate candidate firmicute lipoprotein N-acyltransferase genes.
Vancomycin reduces background growth in lpp null strains.
Despite the high library coverage (see Materials and Methods), the initial attempts to identify rescuing DNA fragments from either B. cereus or E. faecalis were unsuccessful (data not shown). Presumably, this was due to incomplete rescue through low expression, protein misfolding, and/or low affinity for lipoprotein substrates at either the N-acylation or transport stage. Transport is critical to cell viability, as the abundant OM lipoprotein Lpp (Braun's lipoprotein) will continue to be covalently cross-linked to peptidoglycan from the IM, ultimately leading to cell lysis (22). With upwards of 1 million Lpp molecules per cell (23), robust transit to the OM must be maintained to prevent the accumulation of Lpp in the IM and the ensuing Lpp-peptidoglycan adducts from forming. To lower the bar for rescue, we deleted Lpp, which is not essential (24), from our assay strain background in order to facilitate phenotypic rescue. Growth of this strain (KA349), albeit markedly slower than with the lnt inducer l-arabinose, was still observed after a 24-h incubation even without induction (Fig. 1C). This growth may be due to leakiness of the expression construct, residual Lnt activity, or slow but sufficient transit of diacylated lipoproteins to the OM by the Lol export machinery. Regardless, it was necessary to reduce this background growth in order to perform the screen. We reasoned that other lipoprotein-dependent cell processes may be compromised in an lnt-depleted state, such as the OM β-barrel assembly machinery (Bam) and lipopolysaccharide (LPS) transport (Lpt) complexes. The Bam complex, composed of an OM β-barrel and several lipoproteins, folds OM proteins (OMPs) and integrates them into the membrane (4). LptD, an OM β-barrel folded by the Bam complex, can only be folded when associated with its lipoprotein partner LptE (25). Together, these proteins assemble the OM LPS layer in the outer leaflet. Since LPS forms a highly selective permeability barrier that protects the cell from harmful agents (26), a decrease in OM-located Bam, Lpt, and LPS due to retention of lipoprotein components in the IM would enhance susceptibility to normally excluded molecules, such as the large-glycopeptide antibiotic vancomycin (Fig. 1A). Indeed, low levels of vancomycin completely abrogated the growth of the lpp null strain KA349 in the absence of l-arabinose-induced lnt (Fig. 1C).
E. faecalis WMC_RS08810 and B. cereus BC1526 complement Lnt-depleted E. coli.
With the selection assay conditions established, E. faecalis and B. cereus genomic DNA was introduced into KA349 by electroporation and plated on LB agar containing carbenicillin, to select for successful plasmid transformants, glucose, to repress gratuitous expression from the PBAD-lnt construct, vancomycin, to reduce background growth as described above, and palmitate, to provide a potential acyl chain substrate. Plasmids from colonies showing enhanced growth on selection plates were sequenced. Of 16 chosen colonies transformed with E. faecalis genomic DNA, 12 contained overlapping inserts that shared a single intact, unannotated open reading frame (WMC_RS08810) (Fig. 2A). Similarly, seven of eight chosen colonies transformed with B. cereus genomic DNA contained an overlapping insert that included the open reading frame BC1526 (Fig. 2B). The Basic Local Alignment Search Tool (BLAST) search algorithm identified the two genes as orthologs sharing 34% amino acid identity. Both genes are predicted to be membrane proteins containing a domain of unknown function (DUF1461). One plasmid from each strain bearing the candidate lnt rescue gene of interest was chosen for further analysis; these plasmids were designated pEF4 and pBC7.
FIG 2.
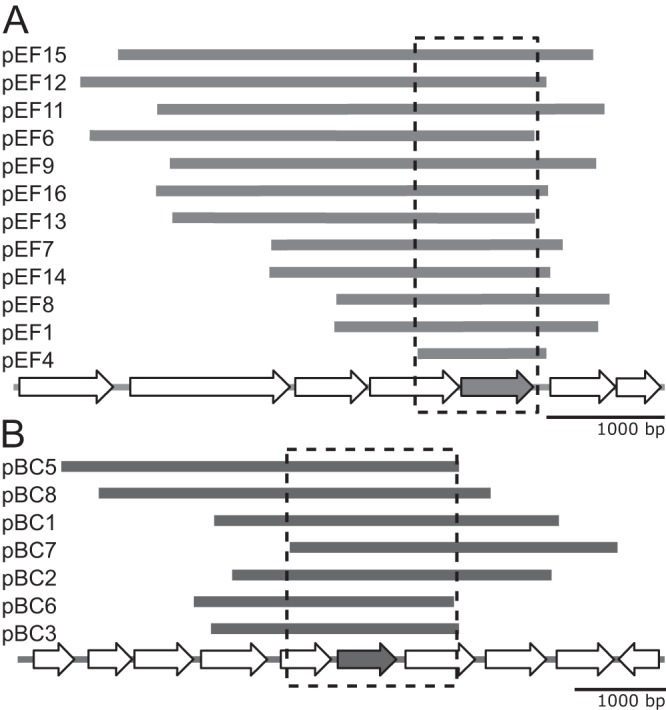
Genetic maps of DNA fragments conveying a viable phenotype in Lnt-depleted E. coli cells. DNA fragments that rescued growth in the Lnt-depleted E. coli mutant strain KA349 (PBAD-lnt Δlpp) were mapped to the E. faecalis genome (A) and the B. cereus genome (B). Fragments are arranged from the top down by size from largest to smallest. The overlapping inserts are boxed in each panel to highlight a single unannotated open reading frame, indicated by the shaded arrow. These genes are E. faecalis WMC_RS08810 and B. cereus BC1526. Plasmids pEF4 and pBC7 were used in subsequent studies.
E. faecalis WMC_RS08810 and B. cereus BC1526 can fully substitute for lnt in E. coli only when lpp is deleted.
To confirm rescue, the growth of PBAD-lnt conditional lpp null strains harboring plasmids (pUC19, pEF4, and pBC7) was monitored with and without l-arabinose (Fig. 3A). While the control strain was dependent on l-arabinose induction of lnt, growth was restored in strains harboring either pEF4 or pBC7 in the absence of l-arabinose, suggesting that pEF4 and pBC7 rescue the lethal phenotype caused by Lnt depletion. To determine whether E. faecalis WMC_RS08810 and B. cereus BC1526 could fully substitute for lnt in E. coli, we performed cotransduction linkage analysis (Fig. 3B). The donor strain TXM541 was constructed with two selectable alleles, chiQ::Apr and lnt::Spt, placed approximately ∼19 kb apart on the chromosome, along with a functional copy of lnt reinserted in a distant chromosomal position. A P1vir lysate was made using TXM541 and transduced into a panel of recipient strains with or without an intact lpp gene and harboring potential rescue plasmids (pUC19, pUC19-lnt, pEF4, and pBC7). Transductants were first plated on medium with apramycin to select for colonies that successfully received the donor DNA fragment of interest and were subsequently patched onto medium with spectinomycin to assess for the lnt::Spt allele. Colonies with coresistance to both apramycin and spectinomycin would indicate that lnt can be functionally replaced in the recipient strain, as it is no longer essential. The observed cotransduction efficiency of the positive control (pUC19-lnt) using P1vir agreed well with the predicted value of 50% (27), while the lnt::Sptr allele could not be established in the pUC19 empty-vector control (Fig. 3B). Cotransduction comparable to that of pUC19-lnt was observed in recipient strains harboring pEF4 and pBC7 and lacking a functional lpp gene. However, cotransduction was not observed in the same recipient strains with an intact lpp gene. This indicates that the firmicute candidate genes cannot rescue cell viability in an lnt null background when lpp is present.
FIG 3.
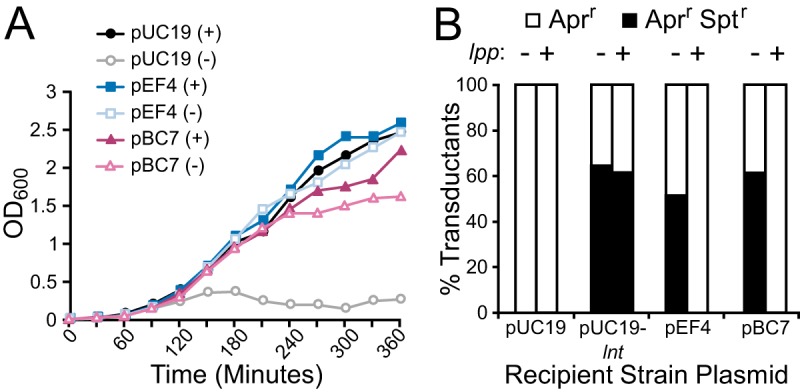
Complementation of lnt depletion by pEF4 and pBC7 in E. coli. (A) The growth of lnt-inducible strains harboring pUC19, pEF4, or pBC7 in an lpp null background grown with (+) or without (−) l-arabinose was measured in LB broth lacking palmitate. (B) The lnt::Sptr- and chiQ::Aprr-linked markers were packaged into P1vir and transduced into recipient strains harboring pUC19, pUC19-lnt, pEF4, or pBC7 in an lpp null (−) or wild-type lpp (+) background. The percentages of transductant colonies resistant to apramycin only (Aprr) or coresistant to both apramycin and spectinomycin (Aprr Sptr) are shown (n = 21 to 60 colonies).
OM lipoprotein localization is stimulated by E. faecalis WMC_RS08810.
We hypothesized that the observed phenotypic rescue was due to restoration of Lol-mediated lipoprotein transport to the OM through α-aminoacylation. To determine the distribution of lipoproteins in the cell envelope, the OM and IM fractions were separated by centrifugation through a discontinuous sucrose gradient. Two distinct samples of strain KA528, featuring the PBAD-lnt construct, were harvested: one was grown with l-arabinose and one without, the latter to serve as the Lnt-depleted sample. The third sample was harvested from KA532 (Δlnt with pEF4), previously constructed by P1vir transduction of the lnt::Sptr allele, so as to monitor WMC_RS08810 activity in the complete absence of Lnt. Chromosomal lpp was deleted in both strains and back complemented with a plasmid bearing a C-terminally Strep-tagged Lpp(K58A)-coding allele (encoding Lpp with a K-to-A change at position 58). The mutation of K58 prevents covalent cross-linking of Lpp to peptidoglycan (22, 28).
Following sample centrifugation and fractionation, the protein abundance in each fraction was measured by using Bradford reagent (Fig. 4A). Fractions containing proteins corresponding to each membrane were pooled, and their volumes normalized. The protein profile of the Lnt-depleted sample differed notably from the profile of the sample from KA528 grown with l-arabinose, in that the density of the IM increased while that of the OM decreased. The profile of the E. faecalis-complemented strain KA532 mirrored that of the Lnt-induced sample. Coomassie blue staining of the PAGE-separated pooled membrane fractions showed enrichment of porins in the OM fraction for each of the three samples (Fig. 4B). To further assess membrane separation, the activity of the IM-associated enzyme NADH oxidase was measured (Fig. 4C). While the IM from KA532 and KA528 grown with l-arabinose contained the majority of the total NADH oxidase activity, nearly equivalent NADH oxidase activities were observed between the OM and IM from the Lnt-depleted sample (Fig. 4C). The blending of OM and IM markers has previously been observed upon Lnt depletion (14, 22). Collectively, the data indicate that WMC_RS08810 can at least partially restore OM biogenesis in the absence of the lnt N-acyltransferase.
FIG 4.
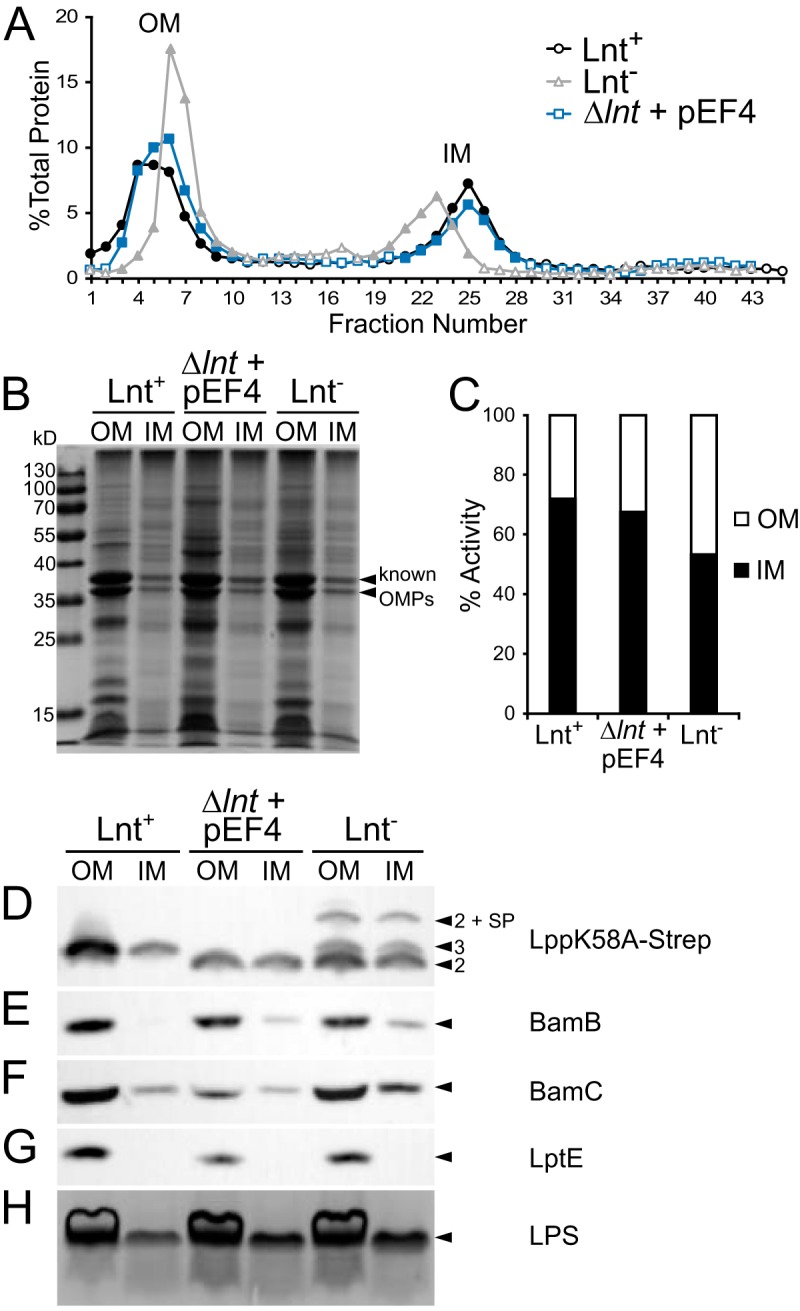
Membrane localization of lipoproteins in strains KA528 and KA532. The total membrane fractions from KA528 (PBAD-lnt) grown in LB with (Lnt+) or without (Lnt−) arabinose and from KA532 (Δlnt with pEF4) were separated by discontinuous sucrose gradient ultracentrifugation. (A) The protein content of the collected fractions was determined by the Bradford assay. OM and IM fractions containing protein were pooled (closed symbols), and their final volumes normalized. (B) Protein profiles of the OM and IM samples were analyzed by SDS-PAGE and Coomassie blue staining. (C) The NADH oxidase activities of the OM and IM samples were measured by following consumption of NADH at 340 nm. (D to G) Samples were immunoblotted with antibodies against Lpp(K58A)–Strep-tag (D), BamB (E), BamC (F), and LptE (G) and detected by HRP chemiluminescence. Lipoproteins having three acyl chains (arrowhead marked “3”), two acyl chains (arrowheads marked “2”), and the signal peptide (SP) are indicated. (H) The LPS profiles of whole-cell samples were detected by SDS-PAGE separation and visualized by silver staining.
To further assess the extent of complementation, the fractions were analyzed by immunoblotting for several key lipoproteins involved in OM biogenesis (Fig. 1A). Immunoblots against Lpp(K58A), BamB, BamC, and LptE revealed lipoprotein enrichment in the OM for all three samples (Fig. 4D to G). The most telling results were seen for Strep-tagged Lpp(K58A) (Fig. 4D), for which distinct bands corresponding to lipoproteins with two and three acyl chains could be observed. The Lnt-depleted sample contained a mixture of triacylated and diacylated Lpp(K58A) in both the OM and IM fractions. Unproteolyzed Lpp with an intact signal peptide was also detected, including in the OM fraction, which is in agreement with the previously observed blending of OM and IM (14, 22). Lpp precursor accumulation in the IM due to the absence of Lnt-catalyzed N-acylation is consistent with slowed Lol transport of diacylated substrates. Unprocessed peptide signal, however, was not observed in the WMC_RS08810-complemented strain even though all Lpp(K58A) signals ran with an electrophoretic mobility consistent with diacylation. The localization of BamB, BamC, and LptE was not appreciably perturbed in any of the samples (Fig. 4E to G). This may reflect the ability of Lol transport to keep pace with de novo diacylglycerol lipoprotein intermediates of low abundance or the inherent substrate preference of the acylation or transport system. Likewise, the amounts and distribution of LPS remained comparable across all samples and are consistent with the proper localization of the Bam/Lpt machinery (Fig. 5H).
FIG 5.
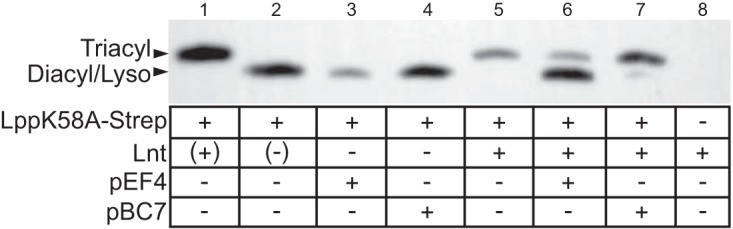
Lipoprotein substrate competition assay. Extracts from lpp null strains harboring various combinations of lipoprotein N-acylating enzymes (+ or −) and Lpp(K58A)–Strep-tagged substrate were separated by SDS-PAGE and immunoblotted with anti-Strep-tag antibody–HRP conjugate. Strain KA528 was grown with (lane 1) and without (lane 2) l-arabinose, while strains KA532 (lane 3), KA583 (lane 4), KA548 (lane 5), KA599 (lane 6), KA600 (lane 7), and TXM327 (lane 8) were grown in LB medium. Parentheses indicate that lnt expression was controlled by adding l-arabinose to the growth medium.
E. coli Lnt and the firmicute candidate proteins compete for a common lipoprotein substrate.
In order to investigate the structure of the lower-molecular-weight bands observed in the membrane-separated WMC_RS08810 sample (Fig. 4D), we designed a substrate competition experiment between Lnt and firmicute candidate N-acyltransferases (Fig. 5). If WMC_RS08810 synthesizes a lyso-form lipoprotein from a diacylglycerol, then Lnt should not be able to convert this pool into mature triacylated lipoprotein, since the α-amino terminus is already modified. Conversely, if WMC_RS08810 induces phenotypic rescue through a mechanism other than lipoprotein aminoacylation, then coexpression of Lnt should convert the entire lipoprotein population into the mature triacylated form with a higher-molecular-weight band. Similarly to WMC_RS08810, Lpp(K58A) from a strain expressing BC1526 (Δlnt mutant carrying pBC7) runs at the same position as diacylglycerol-modified Lpp(K58A) from Lnt-depleted samples (Fig. 5, lanes 3 and 4 versus 2). Cells expressing both Lnt and either firmicute candidate gene show two distinct bands on the gel in comparison to the results for cells expressing Lnt alone (lanes 6 and 7 versus 5), indicative of two noninterconvertible populations of Lpp(K58A). The differences in band intensities between these two strains (lane 6 versus 7) are likely due to enzyme-substrate compatibility, particularly for the acyl chain substrate. As the straight-chained, unsaturated fatty acid composition of E. faecalis more closely matches that of E. coli, WMC_RS08810 may be more efficient in E. coli than BC1526, which normally utilizes branched-chained fatty acids. The observed outcome supports a model whereby the two lipoprotein N-acylating enzymes compete for diacylglycerol-modified Lpp(K58A) substrate, having a free α-amino group. This results in two structures of Lpp(K58A) product depending on which enzyme modifies the lipoprotein: Lnt, forming triacylated lipoprotein, and the firmicute gene product, presumably forming a diacylated lipoprotein, one of which is an N-acyl chain, since this structure cannot be further processed by Lnt. As the total number of acyl chains does not change, N-acylation would have to accompany O-deacylation in order to complete the lyso form. Taken together, these results indicate that lyso-form lipoproteins are synthesized by an intramolecular transacylation mechanism, in which an acyl chain from the diacylglycerol moiety is transferred to the α-amino group of cysteine.
Lpp(K58A) is the lyso form when processed by E. faecalis WMC_RS08810.
To confirm the lyso-form structure, we purified Lpp(K58A)–Strep-tag from E. coli strains KA548 (wild-type lnt) and KA532 (Δlnt with pEF4) and analyzed their N-terminal tryptic peptides by matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS). The MS spectrum of Lpp purified from strain KA548 showed a prominent ion at m/z 1396 (Fig. 6A), corresponding to the mass of conventionally triacylated N-terminal peptide possessing acyl chains totaling 48:1 (with 48 and 1 referring to the number of total carbons and a double bond, respectively). This peak was notably missing from the MS spectrum of Lpp(K58A) purified from strain KA532 (Fig. 6D). Rather, a peak was detected at m/z 1157, equivalent to an N-terminal lipopeptide containing two acyl chains totaling 32:1. To determine the positions of the fatty acids on the lipopeptides, tandem mass spectrometry (MS-MS) analyses were conducted on the m/z 1396 and 1157 parent ions. Both MS-MS spectra share the y series ions corresponding to the SSNAK peptide of Lpp (Fig. 6B and E). In the MS-MS spectrum of m/z 1396, an ion at m/z 1141 corresponds to the product ion that has lost a C16:1 fatty acid (C15H29COOH), and an ion at m/z 1157 corresponds to the loss of a C16:0 fatty acid (C15H30COOH) from either the diacylglycerol or the N terminus. Two additional characteristic fragment ions at m/z 813 and 845 correspond to the N-acyl(C16:0)-dehydroalanyl peptide, generated by the neutral loss of the diacylthioglycerol and the thiolated N-acyl(C16:0) peptide, respectively. Similar N-acylated peptide fragment ions are also evident in the MS-MS spectrum of the m/z 1157 ion from the WMC_RS08810-expressing strain; however, these ions appear at m/z 811 and 843, indicative of a monounsaturated N-terminal fatty acid (C16:1). Additional ions at m/z 901 and 921 correspond to product ions that have lost a C16:0 fatty acid (C15H30COOH) and the N-terminal C16:1 fatty acid (C15H29COOH). Further MS-MS fragment analysis showed that the m/z 1185 parent ion from the WMC_RS08810-expressing strain, which is 28 Da heavier than the m/z 1157 ion due to two additional methylene (CH2) groups, also contained the signature m/z 811 ion, supporting the assignment of an N-acylated parent ion structure (see Fig. S1 in the supplemental material).
FIG 6.

MALDI-TOF MS of E. coli Lpp(K58A) processed by Lnt and of E. faecalis WMC_RS08810. (A, D) Trypsinized Lpp lipopeptides purified from the wild-type lnt strain KA548 (A) and the lnt null, WMC_RS08810-expressing strain KA532 (D) were eluted from nitrocellulose bands and analyzed by MALDI-TOF MS (turquoise traces). Sodium was added to a subset of samples to promote sodiated adduct formation (blue traces). (B, C) The MS-MS spectra of the protonated m/z 1396 (B) and sodiated m/z 1418 (C) parent ions were used to elucidate the triacylated N-terminal structure of Lpp from KA548 (inset in panel A). (E, F) Likewise, the MS-MS spectra of the ions m/z 1157 (E) and 1179 (F) were used to assign the lyso-form N-terminal peptide structure of Lpp from KA532 (inset in panel D). The positions of the O-acyl chains could not be determined on either the triacyl or lyso-form Lpp molecules.
Both MS-MS spectra of the parent ions m/z 1396 and 1157 showed strong preferential fragmentation to the m/z 506 SSNAK peptide ion, diminishing the diagnostic N-acylated peptide signal intensity. For certain samples, this proved to be problematic and hampered confident structure assignments. Since sodium adducts can improve the signal from triacylglycerides and phospholipids (29), we promoted sodium adduct formation by spiking samples with sodium bicarbonate in order to coerce potentially more informative fragmentation (Fig. 6). While the overall signal intensity declined, sodium adducts were readily formed, as determined by the 22-Da shift in the m/z 1396 and 1157 ions to m/z 1418 and 1179, respectively. Subsequent MS-MS fragmentation of these sodium adducts generated strong N-acylated dehydroalanyl peptide ion signals at m/z 811 and 813 (Fig. 6C and F). Taken together, the absence of the m/z 1396 triacylated peptide peak in the MS spectrum and the prominent N-acylated peptide peaks in the MS-MS spectrum of Lpp(K58A) purified from KA532 confirm that Lpp(K58A) is converted into the lyso form when processed by E. faecalis WMC_RS08810.
Deletion of WMC_RS08810 and BC1526 prevents lyso-form lipoprotein formation in E. faecalis and B. cereus.
To confirm the roles of E. faecalis WMC_RS08810 and B. cereus BC1526 in lyso-lipoprotein formation, the genes were deleted in their native organisms. Native lipoproteins from the wild-type, deletion, and complemented strains were enriched using the Triton X-114 phase partitioning method and separated by SDS-PAGE. The lipoprotein profile and relative abundance were unchanged when either gene was deleted and then back complemented with a plasmid-encoded copy (see Fig. S2 in the supplemental material). This was consistent with the inability of SDS-PAGE to discriminate between diacylglycerol and lyso forms of lipoproteins (Fig. 5). The most abundant lipoproteins from E. faecalis (PnrA) and B. cereus (PrsA), previously characterized by Kurokawa et al. (12), were chosen for further analysis by MALDI-TOF MS-MS.
Based on the proposed intramolecular transacylation mechanism for lipoprotein modification when expressed heterologously in E. coli, deletion of the candidate genes should result in conventional diacylglycerol-modified lipoproteins identical in mass to the lyso form. Indeed, peaks corresponding to the N-terminal peptide (m/z 997 for E. faecalis PnrA and m/z 1335 for B. cereus PrsA) were seen in parent spectra from both wild-type and deletion strains (see Fig. S3 in the supplemental material). The MS-MS spectra contained y series ions corresponding to the N-terminal amino acid sequences GGGK of PnrA (Fig. 7A and B) and GTSSSDK of PrsA (Fig. 7F and E), confirming the lipoprotein identities. However, the parent ions fragmented differently depending on whether the WMC_RS08810 or BC1526 gene was present. Differences in the lipoproteins purified from the wild-type and deletion strains arise from the acylated or deacylated product ions. In the MS-MS spectrum of E. faecalis PnrA from wild-type cells, ions at m/z 715, 733, and 759 correspond to the parent ions having lost a C18:1 fatty acid (C17H33COOH), C18:1 ketene (C16H31CH=C=O), and C16:0 ketene (C14H29CH=C=O), respectively. Two additional characteristic fragment ions observed at m/z 625 and 657 correspond to the N-acyl(C16:0)-dehydroalanyl peptide, resulting from the neutral loss of the monoacyl(C18:1)-thioglycerol, and the thiolated N-acyl(C16:0)-peptide, respectively (Fig. 7A). These peaks are absent from the spectrum of PnrA purified from WMC_RS08810 deletion mutants (Fig. 7B). Rather, this spectrum features ions at m/z 741 and 759, corresponding to the loss of a C16:0 fatty acid (C15H30COOH) and a C16:0 ketene (C14H29CH=C=O), respectively, and an additional ion at m/z 459 whose mass is consistent with the loss of both fatty acids (34:1). Two characteristic peaks at m/z 387 and 419, corresponding to dehydroalanyl and thiolated CGGGK peptides with free α-amino groups, respectively, both support the diacylglycerol lipoprotein structural assignment.
FIG 7.

MALDI-TOF MS-MS of E. faecalis PnrA and B. cereus PrsA. (A to C) MS-MS spectra of the m/z 997 parent ions corresponding to the N-terminal lipopeptides of E. faecalis PnrA purified from the wild-type strain (A), the deletion strain (B), and the plasmid back-complemented strain (C). (F to H) MS-MS spectra of the m/z 1334 parent ion corresponding to the N-terminal lipopeptide of B. cereus PrsA purified from the wild-type strain (F), the deletion strain (G), and the back-complemented strain (H). (D, E, I, J) The elucidated lyso- and diacylglycerol structures are shown for PnrA (D and E) and PrsA (I and J). Images of the SDS-PAGE gel and the parent ion spectra can be found in Fig. S2 and S3, respectively, in the supplemental material.
Similar differences were observed in the MS-MS spectra of B. cereus PrsA purified from wild-type and BC1526 deletion strains (Fig. 7F to H). The MS-MS spectrum of PrsA from the wild type features the N-acyl(C16:0)-dehydroalanyl peptide and thiolated N-acyl(C16:0) peptide peaks at m/z 974 and 1006, respectively, and is consistent with the lyso form (Fig. 7F). These peaks are absent in the MS-MS spectrum of PrsA purified from the deletion strain (Fig. 7G). Instead, two fragment ions at m/z 750 and 782 appeared that correspond to the nonacylated dehydroalanyl and thiolated CGTSSSDK peptides, respectively. Additional ions at m/z 1092 and 822, corresponding to the product ions that have lost a C15:0 fatty acid (C14H28COOH) or both fatty acids (32:0), were also detected. While the exact positions of the O-acyl chains could not be determined for the lyso form of either PnrA or PrsA, back complementation in both E. faecalis and B. cereus deletion strains with plasmids bearing the candidate genes restored lyso-form lipoprotein maturation (Fig. 7C and H).
DISCUSSION
Lipoproteins are highly conserved components of the cell envelopes of bacteria from diverse genera that thus far have been thought to share a biosynthetic pathway (5). The sequential action of diacylglcyerol transferase (Lgt) and the signal peptide II protease (Lsp) is invariant, while those organisms with an OM further process diacylglyceryl lipoproteins by N-acylation (Lnt) (Fig. 1). This modification is important, as the Lol transport machinery preferentially recognizes triacylated lipoprotein substrates and is therefore required for robust translocation rates (14, 16). Since firmicutes lack an OM, it came as no surprise when genomic sequencing failed to identify lnt orthologs. No other enzymes have been implicated in lipoprotein biosynthesis, though lnt has been characterized outside OM-containing bacteria, in mycobacteria and high-GC, Gram-positive organisms (11, 30–32). However, when Kurokawa et al. surveyed the N-terminal structures among a panel of firmicutes, they uncovered novel lipoprotein structural diversity that included N-acylation (12). The discovery of lyso-form lipoproteins and other N-terminally modified forms in low-GC, Gram-positive firmicutes lacking apparent lnt sequence orthologs suggests unique biosynthesis pathways that have been tailored for host organism-specific physiological roles. Here, we have identified one such protein, responsible for lyso-form lipoprotein biosynthesis in E. faecalis and B. cereus.
To identify putative genes involved in lyso-lipoprotein formation, we engineered a selection strain for intergenic complementation (Fig. 1). The essential role of Lnt in the N-acylating substrate for Lol-mediated transport in E. coli, in combination with the reduced burden of lipoprotein processing in an Lpp null background, allowed us to identify two previously unknown candidate genes: E. faecalis WMC_RS08810 and B. cereus BC1526. These genes supported growth in Lnt-deficient E. coli cells, prevented mislocalization of Lpp(K58A), and restored general cell envelope integrity as judged by phenotypic resistance to the cell wall biosynthesis inhibitor vancomycin. Vancomycin is normally unable to cross the OM and inhibit peptidoglycan synthesis unless there are permeability defects. As such, vancomycin is a useful probe of membrane integrity (33, 34) and was critical in reducing background growth in the Lpp null selection strain background. Suppression of Lnt essentiality through loss of Lpp has been observed previously (14, 35). Curiously, the localization of lipoproteins directly involved in OM porin assembly (BamB and BamC), LPS transport (LptE), and LPS levels did not reveal gross mislocalization. It has previously been observed that select OM-targeted lipoproteins remain properly localized in the OM upon Lnt depletion (see Robichon et al. [14] for a detailed discussion). Loss of OM integrity due to Lnt depletion in an Lpp null background may be more complex than simple mislocalization of Bam/Lpt. Regardless, substrate competition experiments, in tandem with MS analyses, confirmed that lyso-form lipoproteins improve cell fitness through restoration of cell envelope integrity. Once more, deletion of either of these genes alone in E. faecalis or B. cereus produced conventional diacylglycerol-modified lipoproteins with free α-amino termini. As lipoprotein N-acylation occurs to be concomitant with O-deacylation, we have named this protein lipoprotein intramolecular transacylase (Lit) to differentiate it from the canonical N-acyl transferase Lnt (Fig. 8).
FIG 8.
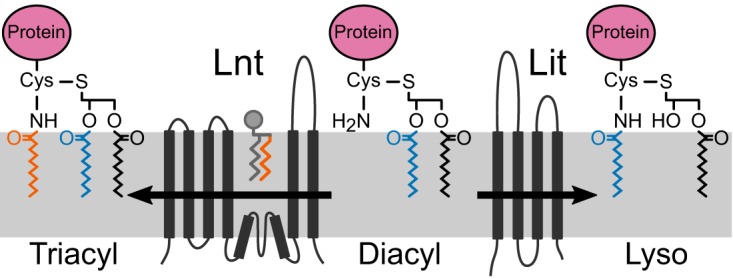
Bacterial lipoprotein N-acylation. In Gram-negative bacteria, Lnt transfers the sn-1 acyl chain (orange) of a membrane phospholipid to form triacylated lipoprotein. In low-GC, Gram-positive firmicute species that make lyso-form lipoproteins, it is proposed that Lit transfers the sn-2 acyl chain (blue) internally from the diacylglycerol moiety to the α-amino group of cysteine. When heterologously expressed in E. coli (Fig. 6), the N-terminal dehydroalanyl fragment ion of Lpp shifts from an Lnt-catalyzed intermolecular sn-1 C16:0 acyl chain transfer (m/z 813, orange acyl chain) to a WMC_RS08810-catalyzed intramolecular sn-2 C16:1 acyl chain transfer (m/z 811, blue acyl chain).
The Lit family of enzymes have no detectable similarity in amino acid sequence or predicted membrane topology to those of Lnt family enzymes (14, 36), as would be predicted for the proposed intramolecular O- to N-transacylation involving the sn-2 fatty acid. At present, a bifunctional enzyme that O-deacylates and adds an N-acyl chain from a neighboring phospholipid cannot be definitively ruled out. This would seem unlikely, however, given the small size (∼220 amino acids with four predicted transmembrane domains) (Fig. 8) (37). While we could not directly determine the exact position of the remaining O-acyl chain on the lyso-form lipoproteins by MS-MS analyses, the fatty acid composition of the membrane phospholipid pool supports an sn-2 origin for the N-terminal acyl chain. In E. coli, de novo phospholipids are synthesized by PlsB and PlsC through sequential addition to the glycerol phosphate backbone of the sn-1 and sn-2 fatty acids, respectively (38, 39). Both enzymes have well-characterized and distinct preferences for acyl chain substrates. PlsB prefers C16:0 and C18:1 fatty acids and typically does not accept C16:1 acyl chains as substrates (40), while PlsC lacks strict substrate specificity and can incorporate saturated or unsaturated fatty acids (38). Interestingly, there was a two-mass-unit difference in the N-acylated lipopeptide fragment ions of Lpp(K58A) when processed by E. faecalis WMC_RS08810 versus E. coli (Fig. 6C and F). Previous studies on acyl chain specificity show that Lnt transfers the sn-1 acyl chain of phospholipid donors, preferring C16:0 saturated acyl chains over C16:1 (41, 42). Indeed, when processed by Lnt, the dominant Lpp fragment ion at m/z 813 contained a C16:0 N-acyl chain (Fig. 6C). As WMC_RS08810-processed Lpp contained a C16:1 N-acyl chain (m/z 811) (Fig. 6F), this suggests that this fatty acid was added to the diacylglycerol donor by PlsC and, thus, that E. faecalis WMC_RS08810 likely catalyzes the transfer of the sn-2 fatty acid to the free α-amino group of the N-terminal cysteine. Mechanistically, two distinct scenarios can be proposed. In the first, O-deacylation through side chain nucleophilic attack of an ester-bound acyl chain forms a covalent enzyme-fatty acid intermediate, which then reacylates to form an amide at the α-amino group of cysteine. Alternatively, the enzyme simply positions the α-amino terminus for direct nucleophilic attack on the ester-linked fatty acid. In either case, the formation of an amide from an ester would be thermodynamically favored and result in net conversion of diacylglycerol lipoprotein precursors into lyso-form lipoproteins. Reconstitution of Lit and in vitro characterization with a defined phospholipid pool representative of the firmicute native host membrane environment will be necessary to confirm acyl chain specificity.
The physiological role of lyso-form lipoproteins and the greater role of N-terminal lipoprotein modifications in Gram-positive bacteria in general remain unknown. In contrast to the case for Gram-negative bacteria, enzymes involved in lipoprotein biosynthesis are not essential in Gram-positive organisms (3, 5). Indeed, mutants with deletions of WMC_RS08810 and BC1526 were readily constructed in E. faecalis and B. cereus without any apparent phenotypes. Lit orthologs are present in Lactobacillus bulgaricus and Streptococcus sanguinis, both also known to make lyso-form lipoproteins (12), suggesting that Lit is conserved among organisms that make the lyso form. Conversely, Lit is absent from firmicute species that are known to not make the lyso form, including Staphylococcus aureus strain RN4220, which makes triacylated lipoproteins, Listeria monocytogenes strain ATCC 15313, which has diacylglycerol-modified lipoproteins, and Bacillus subtilis strain 168, which elaborates N-acetylated lipoproteins (12, 43, 44). Lit is not present in any other phylum of bacteria, suggesting that the gene has been independently acquired in firmicutes at the species level as a possible “postedit” to diacylglycerol-modified lipoproteins. Consistent with a relatively recent acquisition, there is no genomic synteny between WMC_RS08810 and BC1526. While the selective pressure operative for N-acylation is currently unknown, the identification of the Lit enzyme family provides opportunities for further study.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The E. coli strains used are derivatives of the reference strain BW25113 and were grown in LB-Miller medium at 37°C with agitation. Cultures were supplemented with 0.2% l-arabinose when necessary. Antibiotic markers were selected with carbenicillin (50 or 100 μg/ml), kanamycin (30 μg/ml), spectinomycin (25 or 50 μg/ml), apramycin (100 μg/ml), and chloramphenicol (25 μg/ml) unless otherwise noted. E. faecalis and B. cereus strains are derivatives of reference strains ATCC 19433 and ATCC 14579, respectively. All strains were grown in tryptic soy broth (TSB) at 37°C and selected with erythromycin (10 μg/ml) or tetracycline (5 μg/ml) when appropriate. E. faecalis was grown with nisin (100 ng/ml) when appropriate. The strains used in this study are listed in Table 1.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid(s) | Relevant genotype/phenotypea | Source |
|---|---|---|
| E. coli strainsb | ||
| BW25113 | E. coli K-12 wild type; Δ(araD araB)567 ΔlacZ4787(::rrnB-3) λ− rph-1 Δ(rhaD-rhaB)568 hsdR514 | CGSC7636c |
| TXM315 | gut::Kanr-rrnB TT-araC-PBAD-lnt | This study |
| TXM327 | lpp::Chlr | This study |
| KA325 | ybeX-(Kanr-rrnB TT-araC-PBAD)-lnt | This study |
| KA349 | TXM327 ybeX-(Kanr-rrnB TT-araC-PBAD)-lnt | This study |
| KA472 | TXM315 lnt::Sptr | This study |
| KA486 | TXM327(pEF4) | This study |
| KA489 | TXM327(pBC7) | This study |
| KA494 | TXM327(pUC19) | This study |
| KA495 | TXM327(pUC19-lnt) | This study |
| KA503 | pUC19 | This study |
| KA504 | pUC19-lnt | This study |
| KA505 | pEF4 | This study |
| KA528 | KA349(pKA524) | This study |
| KA532 | KA486 with lnt::Sptr by transduction and pKA522 | This study |
| TXM537 | KA325(pEF4) | This study |
| TXM538 | KA325(pUC19) | This study |
| TXM539 | KA349(pEF4) | This study |
| TXM540 | KA349(pUC19) | This study |
| TXM541 | KA472 chiQ::Aprr | This study |
| KA548 | TXM327(pKA522) | This study |
| KA555 | pBC7 | This study |
| KA583 | KA489 with lnt::Sptr by transduction and pKA522 | This study |
| KA597 | KA325(pBC7) | This study |
| KA598 | KA349(pBC7) | This study |
| KA599 | TXM327(pEF4, pKA522) | This study |
| KA600 | TXM327(pBC7, pKA522) | This study |
| Firmicutes strains | ||
| TXM465 | E. faecalis ATCC 19433 | Sigma |
| KA477 | B. cereus ATCC 14579 | American Type Culture Collection |
| KA543 | TXM465 ΔWMC_RS08810 | This study |
| KA546 | KA477 ΔBC1526 | This study |
| KA637 | KA546(pKA637) | This study |
| KA666 | KA543(pKA635) | This study |
| Plasmids | ||
| pUC19, pCL25, and pKFC | General cloning vectors; Carr | |
| pEF4 | pUC19 bearing E. faecalis genomic DNA; Carr | This study |
| pBC7 | pUC19 bearing B. cereus genomic DNA; Carr | This study |
| pKA495 | pUC19-lnt; Carr | This study |
| pKA522 | lpp(K58A)-Strep; Kanr | This study |
| pKA524 | lpp(K58A)-Strep; Sptr | This study |
| pKA635 | pMSP3535-WMC_RS08810; Eryr | This study |
| pKA637 | pSP-BC1526-hp; Tetr | This study |
Resistance phenotypes: Carr, carbenicillin; Kanr, kanamycin; Sptr, spectinomycin; Chlr, chloramphenicol; Eryr, erythromycin; Tetr, tetracycline; Aprr, apramycin.
All E. coli strains are derivatives of BW25113.
Strain CGSC7636 at the Coli Genetic Stock Center (CGSC).
Construction of deletion strains, the PBAD-lnt cassette, and plasmids.
All gene deletions in E. coli strains were constructed by using the Red recombinase system and transduced into recipient strains by P1vir when appropriate (45). The primers used in this study are listed in Table S1 in the supplemental material.
The PBAD-lnt cassette was constructed similarly to the previously described method (14), with some modifications. DNA fragments comprising the kanamycin resistance gene (Kanr), the rrnB transcription terminator (TT), the araC gene, and the l-arabinose-inducible PBAD promoter were PCR amplified and assembled into the pUC19 vector using the In-Fusion HD cloning kit (Clontech). The cassette was then PCR amplified from the resulting plasmid with ∼45-bp homology regions (primers 5′-ybeX PBAD-lnt integration and ybeX PBAD-lnt integration-3′) and integrated directly upstream from lnt by Red-mediated recombination. Integrants were selected on l-arabinose and kanamycin and verified by PCR. The arabinose-dependent growth phenotype was confirmed in the resulting E. coli strain KA325, into which the lpp::Chlr allele was transduced from E. coli strain TXM327, using P1vir, to create KA349.
To create the plasmid bearing a C-terminally Strep-tagged Lpp(K58A) allele, DNA fragments comprising the Kan or Spt resistance gene, the repA gene from pCL25, and lpp(K58A) were first PCR amplified and assembled using the In-Fusion HD cloning kit. With the resulting plasmid as the template, Strep-tag was added by inverse PCR [primers 5′-lpp(K58A)-Strep and lpp(K58A)-Strep-3′].
Unmarked internal gene deletions of E. faecalis WMC_RS08810 and B. cereus BC1526 were generated using the pKFC plasmid (46). The E. faecalis deletion strain was back complemented by cloning WMC_RS08810 into shuttle expression vector pMSP3535, a gift from Gary Dunny (Addgene plasmid number 46886), under the control of a nisin-inducible promoter (47). The B. cereus deletion strain was back complemented by cloning BC1526 into shuttle expression vector pSPNprM-hp, a gift from Dieter Jahn (Addgene plasmid number 48120), under the control of a xylose-inducible promoter (48). All plasmids used in this study are listed in Table 1.
Construction of E. faecalis and B. cereus genomic fragment libraries and complementation of the lnt-inducible E. coli strain KA349.
E. faecalis and B. cereus genomic DNA was digested with NEBNext dsDNA Fragmentase (NEB) according to the manufacturer's instructions for 0.5 to 2 min at 37°C. After size fractionation by agarose gel electrophoresis, DNA fragments ranging from 3 to 5 kb were isolated. Fragments were end repaired with components from the pWEB cosmid cloning kit (Epicentre) and ligated using T4 ligase into pUC19 that had been linearized with SmaI and dephosphorylated with calf intestinal phosphatase (NEB). The resulting libraries were transformed into the conditionally lethal arabinose-regulated lnt mutant E. coli strain KA349 by electroporation. Transformants were selected on LB agar containing 100 μg/ml carbenicillin, 10 μg/ml vancomycin, 50 μg/ml palmitate, and 0.2% (wt/vol) glucose at 37°C. As a positive control, portions of the electroporated cells were also plated on the same medium with l-arabinose in place of glucose. Based on the numbers of colonies observed on the positive-control plates, the average insert size of 4 kb, and the known sizes of the E. faecalis and B. cereus genomes (2.9 and 5.4 Mb, respectively), approximately 61-fold total coverage of the E. faecalis genome and 51-fold total coverage of the B. cereus genome were screened. Plasmids were isolated from colonies growing in the absence of l-arabinose and screened by restriction digest to confirm insert size, and the junction sites were sequenced to identify the DNA insert.
Growth analysis.
Starter cultures were diluted into fresh LB medium containing 50 μg/ml carbenicillin with or without l-arabinose to an optical density at 600 nm (OD600) of 0.001. When the prediluted cultures reached an OD600 of 0.5, cultures were rediluted into fresh medium to an OD600 of 0.01. The OD600 was recorded every 30 min.
Genetic linkage analysis.
To construct a passively marked linked antibiotic resistance marker for cotransduction analysis, the full Kanr-TT-araC-PBAD-lnt cassette from KA325 was PCR amplified (primers 5′-gutA::PBAD-lnt and gutA::PBAD-lnt-3′) and integrated into the glucitol locus by the Red recombinase system. Then, the chitin metabolism gene chiQ and wild-type lnt gene were replaced with apramycin and spectinomycin resistance genes, respectively, while the culture was maintained on l-arabinose, to create E. coli strain TXM541. Donor lysates of TXM541 were prepared using P1vir and transduced into experimental recipient strains using standard protocols (49). Candidate transductants were first selected on LB agar with apramycin and then patched onto LB agar with spectinomycin to score for cotransduction of lnt::Sptr.
Separation of inner and outer membranes by discontinuous sucrose gradient centrifugation.
Starter cultures of E. coli strains KA528 and KA532 were inoculated at a dilution of 1:1,000 into 1 liter of LB with l-arabinose and 25 μg/ml spectinomycin or 50 μg/ml each of carbenicillin and kanamycin, respectively. An additional starter culture of strain KA528 was washed three times, resuspended in fresh LB, and inoculated at a dilution of 1:2,000 into 1 liter of LB with 25-μg/ml spectinomycin in order to deplete Lnt protein. The cultures were grown to an OD600 of 1.0 to 1.2, harvested by centrifugation, washed with 25 ml phosphate-buffered saline (PBS), and stored frozen. Cell pellets were processed as previously described (50, 51) and were resuspended in 25 ml of 50 mM Tris-HCl, pH 7.8, containing 1 mM EDTA and 0.1 mg/ml each of DNase and RNase. Cell suspensions were passed through a French pressure cell four times at 14,000 lb/in2, followed by the addition of phenylmethylsulfonyl fluoride (PMSF) protease inhibitor to 1 mM and MgCl2 to 2 mM. Unbroken cells were removed by centrifugation (3,200 × g for 10 min at 4°C) before collecting membranes by ultracentrifugation (110,000 × g for 90 min at 4°C). The supernatant was discarded, and the membrane pellet was resuspended in 10 mM HEPES, pH 7.4 (adjusted with NaOH), and collected by centrifugation as before. The resulting total membrane fractions were homogenized in 3 ml of HEPES buffer with the aid of a small-bore 26-gauge needle and layered onto a discontinuous sucrose gradient containing 5 ml of 2.02 M, 18 ml of 1.44 M, and 12 ml of 0.77 M sucrose in HEPES buffer. The tubes were centrifuged at 80,000 × g in a swinging bucket rotor for 19 h at 4°C. One-milliliter fractions were collected by piercing the bottom of the centrifuge tubes with a syringe needle. The amount of protein in each fraction was measured by using Bradford reagent. The fractions containing proteins corresponding to the OM and IM were pooled, and their total volumes normalized with HEPES buffer. The membrane fractions were stored frozen at −20°C.
NADH oxidase assay.
The incubation mixtures for the NADH oxidase assay contained 50 mM Tris-HCl, pH 7.5, 0.12 mM NADH, 0.2 mM dithiothreitol, and 3 μl of the pooled and normalized membrane fraction in a volume of 1.0 ml. The rate of decrease in absorbance at 340 nm was assayed at room temperature as has been described previously (52).
Immunoblotting for Lpp(K58A)–Strep-tag, BamB, BamC, and LptE from membrane fractions.
Normalized membrane fractions were separated by SDS-PAGE using a 16.5% Tris-Tricine gel (53), transferred to a polyvinylidene difluoride (PVDF) membrane (0.2 μM), and incubated with a 1:5,000 dilution of Precision Protein StrepTactin-horseradish peroxidase (HRP) conjugate (Bio-Rad) according to the manufacturer's instructions. Then, Lpp(K58A)–Strep-tag levels were detected by enhanced chemiluminescence (SuperSignal West Pico). To detect BamB and BamC, the membrane fractions were separated by SDS-PAGE using a 10% Tris-glycine gel, and a 12% Tris-glycine gel was used for LptE. After transfer to PVDF membranes, samples were incubated with a 1:5,000 dilution of polyclonal antibodies against BamB, BamC, and LptE overnight at 4°C. The bands were visualized using HRP-conjugated secondary antibody (1:5,000) and detected by chemiluminescence.
Lipoprotein N-acylation whole-cell competition assay.
Strains expressing lnt and/or the firmicute candidate N-acyl transferase genes, along with a selection-compatible Lpp(K58A)–Strep-tag reporter low-copy-number plasmid construct (pKA522 or pKA524), were grown to mid-log phase (OD600 of 0.5 to 0.6), pelleted by centrifugation (5,000 × g for 2 min at room temperature), and washed once with PBS. The supernatant was discarded, and the cell pellet stored frozen. To obtain Lnt-depleted samples, strain KA528 was inoculated at a 1:6,000 dilution into LB with 0.2% glucose and grown until cell division stopped, as monitored by the OD600. Aliquots of OD600-normalized whole cells were boiled in SDS-PAGE sample loading buffer and separated by SDS-PAGE using a 16.5% Tris-Tricine gel. Immunoblots for Lpp(K58A)–Strep-tag were performed as described above.
LPS gel and silver stain.
The LPS profiles of the normalized membrane fractions were analyzed by SDS-PAGE (16.5% Tris-Tricine gel) and visualized by silver staining (54).
Purification of Lpp(K58A)–Strep-tag from E. coli.
One-liter cultures of E. coli expressing Lnt only (KA548) or the E. faecalis candidate N-acyltransferase WMC_RS08810 (KA532) along with Lpp(K58A)–Strep-tag were grown to late exponential phase (OD600 of 1.5) and harvested by centrifugation (3,200 × g for 10 min at 4°C). Cells were resuspended in 25 ml of 50 mM Tris-HCl, pH 7.8, containing 1 mM EDTA and 0.1 mg/ml each of DNase and RNase and broken by French press as described above. The membranes were collected by ultracentrifugation (110,000 × g for 90 min at 4°C) and stored at −20°C. The membranes were resuspended in 10 mM HEPES, pH 7.4, with 1 mM EDTA to a concentration of 1 mg/ml of protein. Membrane-bound proteins were solubilized by supplementing with Triton X-100 (2%, vol/vol) and incubating samples at room temperature for 30 min with gentle shaking (55, 56). At the end of the incubation period, samples were clarified by centrifugation (20,000 × g for 20 min at room temperature). The supernatants were diluted with 3 volumes of HEPES buffer containing 0.5 mM n-dodecyl-β-d-maltopyranoside (DDM) and passed over StrepTactin-Sepharose resin (IBA Biosciences). Protein was eluted as directed by the manufacturer, except that all wash and elution solutions were supplemented with 0.5 mM DDM. Proteins were concentrated to 1 mg/ml using a Pierce protein concentrator (9,000 molecular-weight cutoff [MWCO]), aliquoted, and frozen at −80°C.
Purification of native lipoproteins by Triton X-114 phase partitioning.
E. faecalis and B. cereus cultures were grown to late exponential phase (OD600 of 1.5) in 15 ml of TSB, washed once with TBSE solution (20 mM Tris-HCl, pH 8.0, 130 mM NaCl, and 5 mM EDTA), and frozen until use. Native bacterial lipoproteins were enriched using the Triton X-114 phase partitioning method with some modifications (12). Briefly, the harvested bacterial cells were resuspended in 800 μl TSBE with 1 mM PMSF and 0.5 mg/ml lysozyme at 37°C for 30 min. Cells were disrupted by bead beating using a MagNA lyser (7000 setting, using an equal volume of 0.1-mm zirconia silica beads in 5 20-s cycles with 2-min chilling period on ice in between cycles). Unbroken cells and beads were removed by low-speed centrifugation (3,000 × g for 5 min at 4°C). The beads were washed with a second aliquot of TBSE, and the supernatants were pooled and then supplemented with Triton X-114 to a final concentration of 2% (vol/vol). After incubation at 4°C for 1 h, the mixture was incubated at 37°C for 10 min to induce phase separation. Samples were centrifuged (10,000 × g for 10 min at room temperature), and the upper aqueous phase was removed and replaced with the same volume of TBSE solution. This procedure was repeated twice more, with subsequent incubations shortened to 10 min. The final Triton X-114 phase was chilled on ice and brought to 500 μl with ice-cold TSBE, and the single-phase solution immediately centrifuged (16,000 × g for 2 min at 4°C) to remove insoluble integral membrane proteins. The lipoprotein-enriched fraction was obtained from the supernatant by precipitation with 3 volumes of acetone and overnight incubation at −20°C. Pellets were washed twice with acetone, air dried, and resuspended in 10 mM Tris-HCl, pH 8.0, or directly in SDS-PAGE sample buffer.
Electroblotting, tryptic digestion, and lipopeptide extraction from nitrocellulose membranes.
To prepare lipopeptides for MS analysis, the strategy of Serebryakova et al. was utilized, with minor modifications (57). After SDS-PAGE separation (described above for immunoblotting), purified Lpp(K58A)–Strep-tag and enriched lipoproteins from E. faecalis and B. cereus were transferred onto BioTract NT nitrocellulose transfer membrane (PALL Life Sciences) and stained with Ponceau S (0.2% in 5% acetic acid). The stained protein bands were excised, thoroughly destained with water, and diced. The nitrocellulose pieces were washed twice with 50 mM NH4HCO3, pH 7.8, and digested overnight at 37°C in 20 μl of a 20-μg/ml solution of trypsin gold (Promega) in 50 mM NH4HCO3, pH 7.8. Hydrophilic peptides were sequentially extracted stepwise with 50 μl of aqueous 0.5% trifluoroacetic acid, 10% acetonitrile, and 20% acetonitrile, using a 10-min room temperature incubation for each solution. A final 10-min extraction with 15 μl of a 10-mg/ml α-cyano-4-hydroxycinnamic acid (CHCA; Sigma) matrix solution dissolved in chloroform-methanol (2:1, vol/vol) was performed to elute tightly bound hydrophobic peptides, including the bulk fraction of acylated N-terminal lipoprotein-derived tryptic peptides.
MALDI-TOF MS and MS-MS.
One microliter of the extracted lipopeptide in CHCA was deposited onto a steel target plate. An optional second microliter was layered onto the same spot when needed. Where indicated above, aqueous NaHCO3 was added to the tryptic peptide extract (final concentration 1 mM) to enrich sodiated adduct formation. MALDI-TOF MS was conducted using an Ultraflextreme (Bruker Daltonics) MALDI-TOF mass spectrometer in positive reflectron mode. The MS-MS spectra were acquired using a MALDI-TOF/TOF (tandem TOF) instrument (Ultraflextreme; Bruker Daltonics) in Lift mode.
Supplementary Material
ACKNOWLEDGMENTS
We thank Tatiana Laremore (Penn State Proteomics and Mass Spectrometry Core Facility, University Park, PA) for expert technical assistance with MALDI-MS-MS, Gloria Komazin (The Pennsylvania State University) for help with cloning and strain construction, Sarah Ades (The Pennsylvania State University) for P1vir, Gary Dunny for pMSP3535 (Addgene plasmid no. 46886), Dieter Jahn for pSPNprM-hp (Addgene plasmid no. 48120), and Daniel Kahne (Harvard University) for anti-BamB, anti-BamC, and anti-LptE antibodies.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00099-17.
REFERENCES
- 1.Babu MM, Priya ML, Selvan AT, Madera M, Gough J, Aravind L, Sankaran K. 2006. A database of bacterial lipoproteins (DOLOP) with functional assignments to predicted lipoproteins. J Bacteriol 188:2761–2773. doi: 10.1128/JB.188.8.2761-2773.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakayama H, Kurokawa K, Lee BL. 2012. Lipoproteins in bacteria: structures and biosynthetic pathways. FEBS J 279:4247–4268. doi: 10.1111/febs.12041. [DOI] [PubMed] [Google Scholar]
- 3.Buddelmeijer N. 2015. The molecular mechanism of bacterial lipoprotein modification—how, when and why? FEMS Microbiol Rev 39:246–261. doi: 10.1093/femsre/fuu006. [DOI] [PubMed] [Google Scholar]
- 4.Narita S, Tokuda H. 2016. Bacterial lipoproteins; biogenesis, sorting and quality control. Biochim Biophys Acta 2016:S1388-1981(16)30323-7. doi: 10.1016/j.bbalip.2016.11.009. [DOI] [PubMed] [Google Scholar]
- 5.Nguyen MT, Götz F. 2016. Lipoproteins of Gram-positive bacteria: key players in the immune response and virulence. Microbiol Mol Biol Rev 80:891–903. doi: 10.1128/MMBR.00028-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Juncker AS, Willenbrock H, Von Heijne G, Brunak S, Nielsen H, Krogh A. 2003. Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci 12:1652–1662. doi: 10.1110/ps.0303703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bagos PG, Tsirigos KD, Liakopoulos TD, Hamodrakas SJ. 2008. Prediction of lipoprotein signal peptides in Gram-positive bacteria with a hidden Markov model. J Proteome Res 7:5082–5093. doi: 10.1021/pr800162c. [DOI] [PubMed] [Google Scholar]
- 8.Sankaran K, Wu HC. 1994. Lipid modification of bacterial prolipoprotein. Transfer of diacylglyceryl moiety from phosphatidylglycerol. J Biol Chem 269:19701–19706. [PubMed] [Google Scholar]
- 9.Hussain M, Ichihara S, Mizushima S. 1982. Mechanism of signal peptide cleavage in the biosynthesis of the major lipoprotein of the Escherichia coli outer membrane. J Biol Chem 257:5177–5182. [PubMed] [Google Scholar]
- 10.Gupta SD, Wu HC. 1991. Identification and subcellular localization of apolipoprotein N-acyltransferase in Escherichia coli. FEMS Microbiol Lett 62:37–41. doi: 10.1111/j.1574-6968.1991.tb04413.x. [DOI] [PubMed] [Google Scholar]
- 11.Tschumi A, Nai C, Auchli Y, Hunziker P, Gehrig P, Keller P, Grau T, Sander P. 2009. Identification of apolipoprotein N-acyltransferase (Lnt) in mycobacteria. J Biol Chem 284:27146–27156. doi: 10.1074/jbc.M109.022715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kurokawa K, Ryu K-H, Ichikawa R, Masuda A, Kim M-S, Lee H, Chae J-H, Shimizu T, Saitoh T, Kuwano K, Akira S, Dohmae N, Nakayama H, Lee BL. 2012. Novel bacterial lipoprotein structures conserved in low-GC content Gram-positive bacteria are recognized by Toll-like receptor 2. J Biol Chem 287:13170–13181. doi: 10.1074/jbc.M111.292235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reffuveille F, Serror P, Chevalier S, Budin-Verneuil A, Ladjouzi R, Bernay B, Auffray Y, Rincé A. 2012. The prolipoprotein diacylglyceryl transferase (Lgt) of Enterococcus faecalis contributes to virulence. Microbiology 158:816–825. doi: 10.1099/mic.0.055319-0. [DOI] [PubMed] [Google Scholar]
- 14.Robichon C, Vidal-Ingigliardi D, Pugsley AP. 2005. Depletion of apolipoprotein N-acyltransferase causes mislocalization of outer membrane lipoproteins in Escherichia coli. J Biol Chem 280:974–983. doi: 10.1074/jbc.M411059200. [DOI] [PubMed] [Google Scholar]
- 15.Vidal-Ingigliardi D, Lewenza S, Buddelmeijer N. 2007. Identification of essential residues in apolipoprotein N-acyl transferase, a member of the CN hydrolase family. J Bacteriol 189:4456–4464. doi: 10.1128/JB.00099-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fukuda A, Matsuyama S-I, Hara T, Nakayama J, Nagasawa H, Tokuda H. 2002. Aminoacylation of the N-terminal cysteine is essential for Lol-dependent release of lipoproteins from membranes but does not depend on lipoprotein sorting signals. J Biol Chem 277:43512–43518. doi: 10.1074/jbc.M206816200. [DOI] [PubMed] [Google Scholar]
- 17.Yakushi T, Masuda K, Narita S, Matsuyama S, Tokuda H. 2000. A new ABC transporter mediating the detachment of lipid-modified proteins from membranes. Nat Cell Biol 2:212–218. doi: 10.1038/35008635. [DOI] [PubMed] [Google Scholar]
- 18.Matsuyama SI, Yokota N, Tokuda H. 1997. A novel outer membrane lipoprotein, LolB (HemM), involved in the LolA (p20)-dependent localization of lipoproteins to the outer membrane of Escherichia coli. EMBO J 16:6947–6955. doi: 10.1093/emboj/16.23.6947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao XJ, Wu HC. 1992. Nucleotide sequence of the Staphylococcus aureus signal peptidase II (lsp) gene. FEBS Lett 299:80–84. doi: 10.1016/0014-5793(92)80105-P. [DOI] [PubMed] [Google Scholar]
- 20.Qi H-Y, Sankaran K, Gan K, Wu HC. 1995. Structure-function relationship of bacterial prolipoprotein diacylglyceryl transferase: functionally significant conserved regions. J Bacteriol 177:6820–6824. doi: 10.1128/jb.177.23.6820-6824.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saito HE, Harp JR, Fozo EM. 2014. Incorporation of exogenous fatty acids protects Enterococcus faecalis from membrane-damaging agents. Appl Environ Microbiol 80:6527–6538. doi: 10.1128/AEM.02044-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yakushi T, Tajima T, Matsuyama S, Tokuda H. 1997. Lethality of the covalent linkage between mislocalized major outer membrane lipoprotein and the peptidoglycan of Escherichia coli. J Bacteriol 179:2857–2862. doi: 10.1128/jb.179.9.2857-2862.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo MS, Updegrove TB, Gogol EB, Shabalina SA, Gross CA, Storz G. 2014. MicL, a new σE-dependent sRNA, combats envelope stress by repressing synthesis of Lpp, the major outer membrane lipoprotein. Genes Dev 28:1620–1634. doi: 10.1101/gad.243485.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hirota Y, Suzuki H, Nishimura Y, Yasuda S. 1977. On the process of cellular division in Escherichia coli: a mutant of E. coli lacking a murein-lipoprotein. Proc Natl Acad Sci U S A 74:1417–1420. doi: 10.1073/pnas.74.4.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chimalakonda G, Ruiz N, Chng S-S, Garner RA, Kahne D, Silhavy TJ. 2011. Lipoprotein LptE is required for the assembly of LptD by the beta-barrel assembly machine in the outer membrane of Escherichia coli. Proc Natl Acad Sci U S A 108:2492–2497. doi: 10.1073/pnas.1019089108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nikaido H, Varra M. 1985. Molecular basis of bacterial outer membrane permeability. Microbiol Mol Biol Rev 49:1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu TT. 1966. A model for three-point analysis of random general transduction. Genetics 54:405–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Braun V, Rotering H, Ohms J-P, Haganmaier H. 1976. Conformational studies on murein-lipoprotein from the outer membrane of Escherichia coli. Eur J Biochem 70:601–610. doi: 10.1111/j.1432-1033.1976.tb11051.x. [DOI] [PubMed] [Google Scholar]
- 29.Al-Saad KA, Zabrouskov V, Siems WF, Knowles NR, Hannan RM, Hill HH. 2003. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry of lipids: ionization and prompt fragmentation patterns. Rapid Commun Mass Spectrom 17:87–96. doi: 10.1002/rcm.858. [DOI] [PubMed] [Google Scholar]
- 30.Brülle JK, Tschumi A, Sander P. 2013. Lipoproteins of slow-growing Mycobacteria carry three fatty acids and are N-acylated by apolipoprotein N-acyltransferase BCG_2070c. BMC Microbiol 13:223. doi: 10.1186/1471-2180-13-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mohiman N, Argentini M, Batt SM, Cornu D, Masi M, Eggeling L, Besra G, Bayan N. 2012. The ppm operon is essential for acylation and glycosylation of lipoproteins in Corynebacterium glutamicum. PLoS One 7:e46225. doi: 10.1371/journal.pone.0046225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Widdick DA, Hicks MG, Thompson BJ, Tschumi A, Chandra G, Sutcliffe IC, Brülle JK, Sander P, Palmer T, Hutchings MI. 2011. Dissecting the complete lipoprotein biogenesis pathway in Streptomyces scabies. Mol Microbiol 80:1395–1412. doi: 10.1111/j.1365-2958.2011.07656.x. [DOI] [PubMed] [Google Scholar]
- 33.Ruiz N, Falcone B, Kahne D, Silhavy TJ. 2005. Chemical conditionality: a genetic strategy to probe organelle assembly. Cell 121:307–317. doi: 10.1016/j.cell.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 34.Ruiz N, Wu T, Kahne D, Silhavy TJ. 2006. Probing the barrier function of the outer membrane with chemical conditionality. ACS Chem Biol 1:385–395. doi: 10.1021/cb600128v. [DOI] [PubMed] [Google Scholar]
- 35.Narita S, Tokuda H. 2011. Overexpression of LolCDE allows deletion of the Escherichia coli gene encoding apolipoprotein N-acyltransferase. J Bacteriol 193:4832–4840. doi: 10.1128/JB.05013-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gélis-Jeanvoine S, Lory S, Oberto J, Buddelmeijer N. 2015. Residues located on membrane-embedded flexible loops are essential for the second step of the apolipoprotein N-acyltransferase reaction. Mol Microbiol 95:692–705. doi: 10.1111/mmi.12897. [DOI] [PubMed] [Google Scholar]
- 37.Omasits U, Ahrens CH, Müller S, Wollscheid B. 2014. Protter: Interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics 30:884–886. doi: 10.1093/bioinformatics/btt607. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Y-M, Rock CO. 2008. Thematic review series: Glycerolipids. Acyltransferases in bacterial glycerophospholipid synthesis. J Lipid Res 49:1867–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yao J, Rock CO. 2013. Phosphatidic acid synthesis in bacteria. Biochim Biophys Acta 1831:495–502. doi: 10.1016/j.bbalip.2012.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rock C, Goelzg SE, Grant A. 1981. Phospholipid synthesis in Escherichia coli. Characteristics of fatty acid transfer from acyl-acyl carrier protein to sn-glycerol-3-phosphate. J Biol Chem 258:462–464. [PubMed] [Google Scholar]
- 41.Jackowski S, Rock CO. 1986. Transfer of fatty acids from the 1-position of phosphatidylethanolamine to the major outer membrane lipoprotein of Escherichia coli. J Biol Chem 261:11328–11333. [PubMed] [Google Scholar]
- 42.Hillmann F, Argentini M, Buddelmeijer N. 2011. Kinetics and phospholipid specificity of apolipoprotein N-acyltransferase. J Biol Chem 286:27936–27946. doi: 10.1074/jbc.M111.243519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kurokawa K, Lee H, Roh K-B, Asanuma M, Kim YS, Nakayama H, Shiratsuchi A, Choi Y, Takeuchi O, Kang HJ, Dohmae N, Nakanishi Y, Akira S, Sekimizu K, Lee BL. 2009. The triacylated ATP binding cluster transporter substrate-binding lipoprotein of Staphylococcus aureus functions as a native ligand for Toll-like receptor 2. J Biol Chem 284:8406–8411. doi: 10.1074/jbc.M809618200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Asanuma M, Kurokawa K, Ichikawa R, Ryu KH, Chae JH, Dohmae N, Lee BL, Nakayama H. 2011. Structural evidence of α-aminoacylated lipoproteins of Staphylococcus aureus. FEBS J 278:716–728. doi: 10.1111/j.1742-4658.2010.07990.x. [DOI] [PubMed] [Google Scholar]
- 45.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kato F, Sugai M. 2011. A simple method of markerless gene deletion in Staphylococcus aureus. J Microbiol Methods 87:76–81. doi: 10.1016/j.mimet.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 47.Bryan EM, Bae T, Kleerebezem M, Dunny GM. 2000. Improved vectors for nisin-controlled expression in Gram-positive bacteria. Plasmid. 44:183–190. doi: 10.1006/plas.2000.1484. [DOI] [PubMed] [Google Scholar]
- 48.Stammen S, Müller BK, Korneli C, Biedendieck R, Gamer M, Franco-Lara E, Jahn D. 2010. High-yield intra- and extracellular protein production using Bacillus megaterium. Appl Environ Microbiol 76:4037–4046. doi: 10.1128/AEM.00431-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thomason LC, Costantino N, Court DL, Thomason LC, Costantino N, Court DL. 2007. E. coli genome manipulation by P1 transduction. Curr Protoc Mol Biol 79:1.17.1–1.17.8. doi: 10.1002/0471142727.mb0117s79. [DOI] [PubMed] [Google Scholar]
- 50.Koplow J, Goldfine H. 1974. Alterations in the outer membrane of the cell envelope of heptose-deficient mutants of Escherichia coli. J Bacteriol 117:527–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meredith TC, Aggarwal P, Mamat U, Lindner B, Woodard RW. 2006. Redefining the requisite lipopolysaccharide structure in Escherichia coli. ACS Chem Biol 1:33–42. doi: 10.1021/cb0500015. [DOI] [PubMed] [Google Scholar]
- 52.Osborn MJ, Gander JE, Parisi E, Carson J. 1972. Mechanism of assembly of the outer membrane of Salmonella typhimurium. J Biol Chem 247:3962–3972. [PubMed] [Google Scholar]
- 53.Schägger H. 2006. Tricine-SDS-PAGE. Nat Protoc 1:16–22. doi: 10.1038/nprot.2006.4. [DOI] [PubMed] [Google Scholar]
- 54.Hitchcock PJ, Brown TM. 1983. Morphological heterogeneity among Salmonella lipopolysaccharide chemotypes in silver-stained polyacrylamide gels. J Bacteriol 154:269–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schnaitman CA. 1971. Solubilization of the cytoplasmic membrane of Escherichia coli by Triton X-100. J Bacteriol 108:545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Filip C, Fletcher G, Wulff JL, Earhart CF. 1973. Solubilization of the cytoplasmic membrane of Escherichia coli by the ionic detergent sodium-lauryl sarcosinate. J Bacteriol 115:717–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Serebryakova MV, Demina IA, Galyamina MA, Kondratov IG, Ladygina VG, Govorun VM. 2011. The acylation state of surface lipoproteins of Mollicute Acholeplasma laidlawii. J Biol Chem 286:22769–22776. doi: 10.1074/jbc.M111.231316. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.