
Fig. 2.
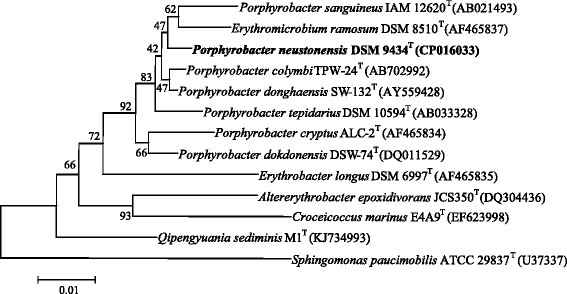
Phylogenetic tree based on 16S rRNA gene sequences was constructed by neighbor-joining algorithms. Related sequences were aligned with Clustal W [21]. Evolutionary distances were calculated according to the algorithm of the Kimura two-parameter model with bootstraps analysis set to 1000 replicates. Bar, 0.01 substitutions per nucleotide position