
Fig. 1.
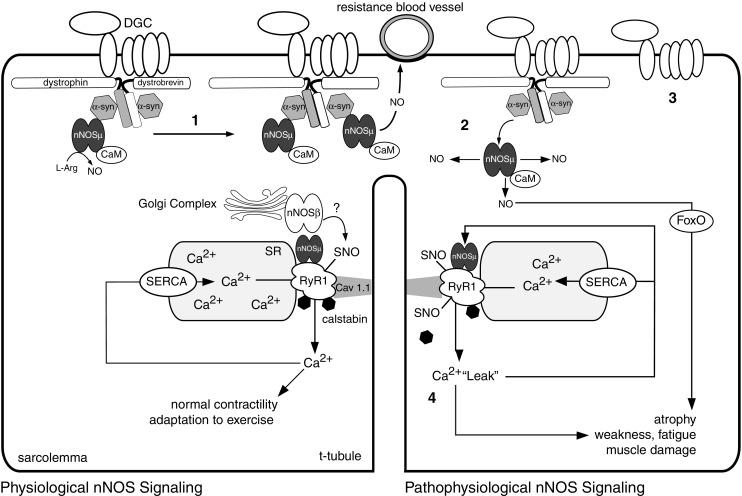
Model for the activity-dependent control of Ca2+/calmodulin (CaM) regulated neuronal nitric oxide synthase (nNOS) signaling and function. nNOSμ localizes to the dystrophin glycoprotein complex by binding a-syntrophin (a-syn), where it opposes blood vessel constriction during exercise. The spliced nNOS isoforms nNOSμ and nNOSβ (?) may regulate muscle fatigue and exercise performance by promoting normal ryanodine receptor 1 (RyR1) activity. SNO represents S-nitrosylation of regulatory cysteine thiols. RyR1 releases Ca2+ into the cytosol during contraction, while a sarcoplasmic reticulum Ca2+-ATPase (SERCA) pumps Ca2+ from the cytosol back into the sarcoplasmic reticulum (SR). 1 nNOSμ signaling is increased by endurance type exercise. 2 Prolonged inactivity leads to dissociation of nNOSμ from the sarcolemma inducing atrophy through a forkhead transcription factor (FoxO) mediated pathway. 3 In Duchenne muscular dystrophy, loss of dystrophin destabilizes the dystrophin glycoprotein complex (DGC), and impairs the vasoregulatory function of nNOSμ, leading to RyR1 destabilization, pathogenic nNOSμ-mediated RyR1 hypernitrosylation and Ca2+ leak. 4 RyR1 Ca2+ leak can lead to muscle damage, weakness, and fatigue. RyR1 Ca2+ leak can further inappropriately activate nNOSμ, leading to further RyR1 nitrosylation and progressively more damaging Ca2+ leakage in a severely detrimental feed-forward loop