

Abstract
Phospholipase C-related, but catalytically inactive protein (PRIP) was previously identified as a novel inositol 1,4,5-trisphosphate-binding protein with a domain organization similar to that of phospholipase C-δ but lacking phospholipase activity. We recently showed that PRIP gene knock-out (KO) in mice increases bone formation and concomitantly decreases bone resorption, resulting in increased bone mineral density and trabecular bone volume. However, the role of PRIP in osteoclastogenesis has not yet been fully elucidated. Here, we investigated the effects of PRIP on bone remodeling by investigating dynamic tooth movement in mice fitted with orthodontic devices. Morphological analysis indicated that the extent of tooth movement was smaller in the PRIP-KO mice than in wild-type mice. Histological analysis revealed fewer osteoclasts on the bone-resorption side in maxillary bones of PRIP-KO mice, and osteoclast formation assays and flow cytometry indicated lower osteoclast differentiation in bone marrow cells isolated from these mice. The expression of genes implicated in bone resorption was lower in differentiated PRIP-KO cells, and genes involved in osteoclast differentiation, such as the transcription factor NFATc1, exhibited lower expression in immature PRIP-KO cells initiated by M-CSF. Moreover, calcineurin expression and activity were also lower in the PRIP-KO cells. The PRIP-KO cells also displayed fewer M-CSF-induced changes in intracellular Ca2+ and exhibited reduced nuclear localization of NFATc1. Up-regulation of intracellular Ca2+ restored osteoclastogenesis of the PRIP-KO cells. These results indicate that PRIP deficiency impairs osteoclast differentiation, particularly at the early stages, and that PRIP stimulates osteoclast differentiation through calcium-calcineurin-NFATc1 signaling via regulating intracellular Ca2+.
Keywords: calcineurin, calcium, differentiation, NFAT transcription factor, osteoclast, M-CSF, PRIP, RANK/RANKL
Introduction
Osteoclasts are derived from a monocyte/macrophage lineage and fuse to form large multinucleated cells that reconstitute actin cytoskeleton to attach to the bone surface for bone resorption (1). Osteoclastogenesis is under the regulation of osteoblast-lineage cells, which express two essential factors for osteoclastogenesis: macrophage colony-stimulating factor (M-CSF)3 and receptor activator of nuclear factor κB ligand (RANKL) (2, 3). M-CSF is considered to be critical for the survival and proliferation of pre-osteoclast cells. It also stimulates expression of RANK, the receptor for RANKL, in pre-osteoclasts to induce RANKL-RANK signaling (4, 5). The signaling activates downstream pathways, including mitogen-activated protein kinase (MAPK) and c-Jun N-terminal kinase (JNK). Transcription factors such as nuclear factor-activated T cell c1 (NFATc1), c-Fos, and nuclear factor-κB (NF-κB) are also activated in the process of osteoclast differentiation (6). In particular, NFATc1 is a master regulator of osteoclastogenesis because it regulates a number of osteoclast-specific genes, such as cathepsin K, calcitonin receptor, and tartrate-resistant acid phosphatase (TRAP) (7). Another transcription factor, PU.1, is also critical for osteoclastogenesis in the earliest stages of the process, as it regulates expression of the M-CSF receptor c-Fms, thereby mediating M-CSF activity during osteoclast differentiation (8). Mice deficient in PU.1 lack not only osteoclasts but also macrophages, resulting in osteopetrosis (9), an osteocondensation disease caused by increased bone formation or failure of resorption. Mice without RANKL and/or a mutation in M-CSF also develop osteopetrosis. Aberrations in RANKL, M-CSF, or their downstream factors thus result in disruption of osteoclasts, an imbalance between bone resorption and formation and ultimately bone diseases such as osteoporosis, rheumatoid arthritis, and Paget's disease (10, 11). However, the molecular and cellular mechanisms underlying these and earlier events in the process of osteoclastogenesis are unknown.
Phospholipase C-related, but catalytically inactive protein (comprising PRIP-1 and -2), was first isolated as a novel d-myo-inositol 1,4,5-trisphosphate (Ins(1,4,5)P3)-binding protein (12, 13). PRIP has a domain organization similar to phospholipase C-δ1, consisting of a pleckstrin homology (PH) domain, EF-hand motifs, catalytic X and Y, and C2 domains, but does not function as an enzyme (14, 15). We have developed PRIP-1 and -2 double knock out (KO) mice to explore the biological roles of PRIP in terms of Ins(1,4,5)P3/Ca2+ signaling (16, 17), γ-aminobutyric acid type A (GABAA) receptor signaling (18–21), protein phosphatases (22, 23), and SNARE proteins (24, 25).
In a previous study we found that homozygous KO male and female pairs have decreased numbers of litters and smaller litter sizes (26). We then determined that the cause is on the female side and is related to increased gonadotropin (luteinizing and follicle-stimulating hormones) secretion and decreased serum sex steroid hormones (estrogen and progesterone) (26). We, therefore, analyzed the bone properties of KO mice, given that the imbalance in their hormones might cause osteoporosis. Contrary to our expectation, we found increased bone mineral density and trabecular bone volume in these mice, indicating that PRIP deficiency triggers bone mass increase as a result of increased bone formation and/or decreased bone resorption, independent of the reproductive hormone imbalance (26). We further determined that PRIP deficiency stimulates bone morphogenetic protein signaling, resulting in increased bone mass (27, 28). However, the role of PRIP in osteoclastogenesis is not yet fully clarified.
To examine this further, here we use PRIP-KO mice equipped with an orthodontic device to investigate the involvement of PRIP in bone remodeling during dynamic orthodontic tooth movement. Orthodontic tooth movement triggers alveolar bone resorption by osteoclasts on the pressure side and new bone formation by osteoblasts on the tension side (29). We also performed cell biological analyses using osteoclast precursors from KO mice, demonstrating the involvement of PRIP in the regulation of osteoclast differentiation.
Results
Morphological analysis of tooth movement
Orthodontic tooth movement was executed between the upper incisor and the first molar in the right maxilla of wild-type (WT) and KO mice (Fig. 1A). Morphological analysis of their maxillary bones using micro-computed tomography (μCT) showed less tooth movement in KO mice than in WT individuals in both the transverse and the sagittal planes (Fig. 1B). To evaluate orthodontic tooth movement in more detail, we measured the distance between the upper right first and second molars in the μCT images. This revealed that the distance was significantly shorter in KO mice than in WT mice for both males (WT, 110.2 ± 23.62 μm; KO, 52.3 ± 9.9 μm) and females (WT, 113.6 ± 42.9 μm; KO, 50.3 ± 28.2 μm; Fig. 1C). These results clearly indicate that PRIP deficiency affects tooth movement induced by orthodontic force in both male and female mice.
Figure 1.

Experimental tooth movement by orthodontic force. A, illustration of the method by which the upper first molar tooth was moved by means of orthodontic force. The arrow indicates the direction of orthodontic force. B, after the experimental tooth movement, the maxillary bones of WT and KO mice were analyzed using μCT. The panels show typical images. The arrows (black and white) indicate the spaces caused by tooth movement. R, right side with mechanical loading; L, left side without mechanical loading. C, the movement distance between the first molar and the second molar was measured from digital μCT images. WT males, n = 6; KO males, n = 5; WT females, n = 5; KO females. n = 4. *, p < 0.05; **, p < 0.01.
Histological analysis of maxillary bone
Histological analysis around the target teeth was performed using sagittal sections of maxillary bone stained for TRAP. Histological images indicated that the distributions and the number of osteoclasts on the pressure side (M) in the alveolar bone were obviously different between WT and KO individuals. TRAP staining was less intense, and the bone resorption area was apparently decreased in KO maxillae (Fig. 2, A and B). The number of TRAP-positive osteoclasts was decreased in KO mice compared with WT mice (Fig. 2B). Bone resorption lacunae on the pressure side after tooth movement were smaller in KO mice than in WT mice (Fig. 2B). These results indicate that the number of osteoclasts is responsible for decreased bone resorption in addition to the possibility of functional failure.
Figure 2.
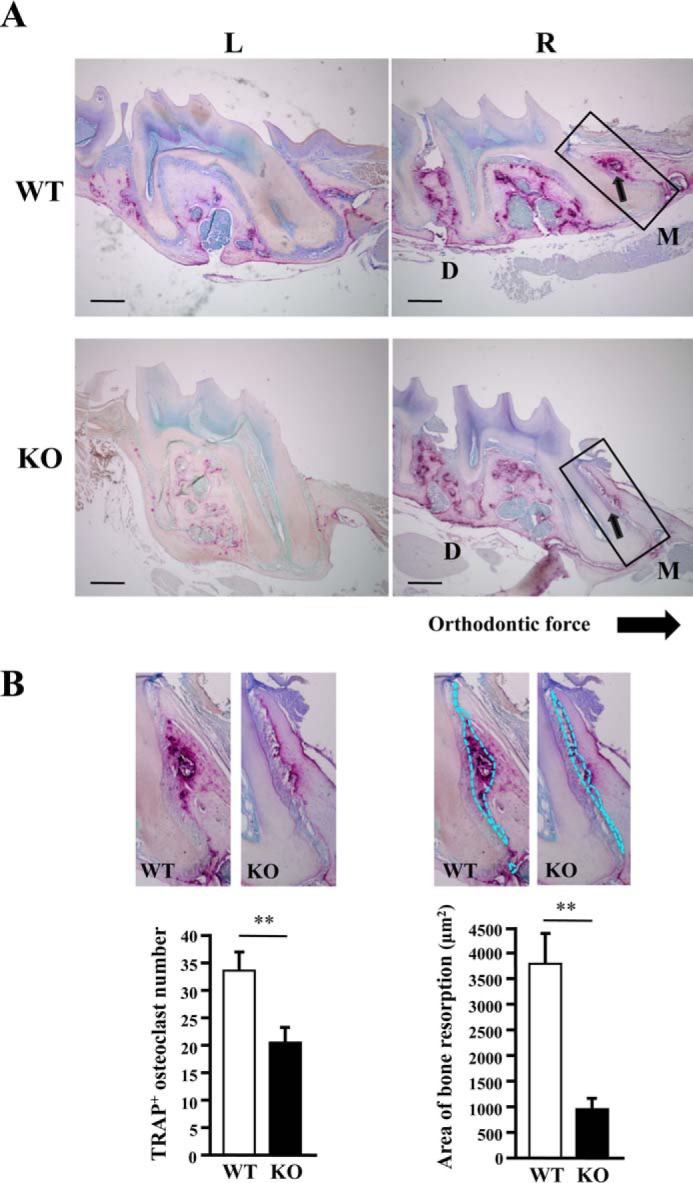
Histological analysis of maxillary bones. A, sagittal sections of maxillary bone around the first molar, analyzed in Fig. 1, were stained for TRAP. Arrows indicate TRAP-positive osteoclasts and bone resorption lacunae. R, right side, with mechanical loading; L, left side, without mechanical loading; M, mesial/pressure side; D, distal/tension side. Areas outlined with rectangles are enlarged in B. B, the left panel shows the enlarged images of TRAP-positive (TRAP+) cells on the pressure side in sagittal sections of maxillary bone and a graph of the number of TRAP+ cells in the peripheral region of lacunae (0.5 mm2). The right panel shows the same images with bone resorption lacunae outlined with dotted lines and a graph of the area of the lacunae on the pressure side. Data are shown as the mean ± S.D. of the results from several sections of five male mice each. Data from female mice were similar (not shown). **, p < 0.01. Bar = 200 μm.
Osteoclastogenesis in the co-culture system of bone marrow cells with pre-osteoblasts
Bone marrow cells derived from mice of both genotypes were co-cultured with pre-osteoblasts from calvaria from newborn WT or KO mice in a medium containing 1α,25-dihydroxycholecalciferol (1α,25(OH)2D3) for 7 days. They were then subjected to TRAP staining for multinucleated osteoclast-like cells. Of the four combinations resulting from combining bone marrow cells from each genotype with pre-osteoblasts from each genotype, those containing KO bone marrow cells showed the weakest TRAP staining (Fig. 3). This indicates that the impairment of osteoclastogenesis is attributed to pre-osteoclastic cells present in KO bone marrow. Furthermore, there were fewer TRAP-positive osteoclasts whatever the number of nuclei in KO bone marrow, suggesting that osteoclastogenesis is impaired before the formation of multinuclear osteoclasts. Few differences were found between male and female.
Figure 3.

Osteoclast differentiation in a coculture of bone marrow cells and pre-osteoblasts. Bone marrow cells (BM) derived from WT or KO femora in male (A) and female (B) mice, respectively, were cocultured with pre-osteoblasts (POB) from WT or KO calvaria. To identify osteoclastogenesis, cells were stained for TRAP. Those positive for TRAP (TRAP+) and containing three or more nuclei were identified as osteoclasts. The graphs show the number of osteoclasts with the indicated number of nuclei. Bone marrow cells were separately obtained from six mice of each genotype and independently cocultured. Data shown are the mean ± S.D. obtained by triplicate assays of the six cocultures. **, p < 0.01. Bar = 100 μm.
RANK and CD115 expression during osteoclast differentiation
We next examined induction of osteoclast formation by M-CSF and RANKL. Bone marrow cells from WT and KO mice were first cultured with M-CSF for 3 days, then with M-CSF and RANKL for 4 days, and finally stained for TRAP. Again, there were fewer TRAP-positive cells in KO cells, irrespective of the number of nuclei (Fig. 4A). During osteoclast differentiation, cells express receptors for M-CSF (CD115/c-Fms) and RANKL (RANK), and double-positive (c-Fms+/RANK+) cells differentiate into osteoclasts. We, therefore, stimulated cells from the femurs of WT and KO mice with M-CSF and RANKL and then scanned them for c-Fms+/RANK+ cells using flow cytometry. In cells derived from both male and female KO mice, the double-positive population was one-third that from WT mice (Fig. 4B), confirming the impairment of osteoclast differentiation in KO cells. Again, few differences were found between male and female. Therefore, subsequent experiments were performed with male mice only.
Figure 4.
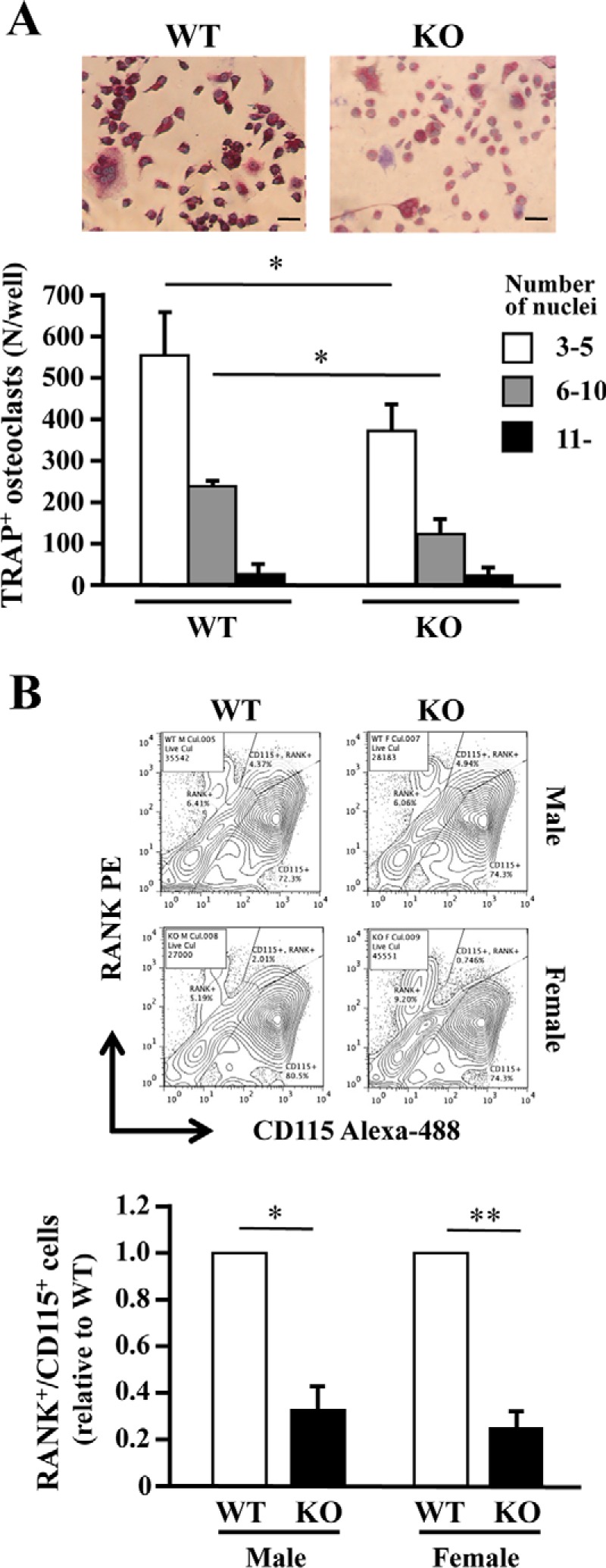
PRIP-deficient bone marrow cell cultures with M-CSF and RANKL. A, bone marrow cells from the femurs of both genotypes were cultured in 96-well plates with M-CSF for 3 days and then with M-CSF and RANKL for 4 days. The cells were stained for TRAP. TRAP-positive cells (TRAP+) containing more than three nuclei were regarded as osteoclasts, and the number of the cells is shown in the graph. Images and results shown are for males only. B, bone marrow cells were cultured as described in A in low-cell-binding plates. Cells stained with anti-RANK-phycoerythrin (PE) and anti-CD115-Alexa 488 antibodies were analyzed using flow cytometry. The panels are typical examples of the results, and the graph summarizes the analysis. Similar results were obtained for samples from three mice of each genotype cultured in triplicate. *, p < 0.05; **, p < 0.01. Bar = 20 μm.
Expression of genes involved in osteoclast differentiation and function
We then examined the expression of the marker genes for osteoclast differentiation and function using real-time polymerase chain reaction (PCR) analysis. As with Fig. 4, bone marrow cells cultured with M-CSF for 3 days and then with M-CSF and RANKL for 4 days (Fig. 5A) was examined, and it was revealed that the expression of most of the marker genes implicated in osteoclast differentiation or function, with the exception of Spp1 (encoding osteopontin) and Ror2, was significantly lower in KO cells. The down-regulation of Tnfrs11a (encoding RANK) supported the flow cytometry results (Fig. 4).
Figure 5.

Gene expression of osteoclast-related factors in bone marrow cell culture. Cells derived from both genotypes at 6–8 weeks of age were cultured, and those total RNAs were analyzed using quantitative real-time PCR. Bone marrow cells extracted from the femurs were cultured for 1 day, and non-adherent cells were collected on the next day (C). Of which some of those cells were differentiated to osteoclast by cultured with M-CSF for 3 days (B) then with M-CSF and RANKL for 4 days (A). Gene expression was normalized to that of GAPDH. Data shown are the mean ± S.D. of samples from five mice cultured in triplicate. *, p < 0.05; **, p < 0.01.
Other down-regulated genes included those implicated in sealing zone formation (Itgb3 and Itga5 encoding Integrin β3 and αV respectively), chloride conductance (Clcn7 encoding chloride channel, voltage-sensitive 7), pH regulation (Tcirg1 encoding T-cell, immune regulator 1 ATPase, H+ transporting, lysosomal V0 subunit A3) (30), degradation of collagen and other proteins (CtsK and Mmp9 encoding cathepsin K and matrix metalloproteinase 9, a type IV collagenase, respectively), and adhesion and cellular fusion of osteoclasts (Icam1, Fermt3, and Dc-stamp encoding intercellular adhesion molecule-1, Kindlin3, and dendritic cell-specific transmembrane protein, respectively) (31, 32), all of which are important for bone resorption. Expression of Traf6 (tumor necrosis factor receptor-associated factor 6) was also lower in KO cells, which plays an essential role, together with RANK, in osteoclast cytoskeletal organization (33). The lower expression of Acp5 (encoding TRAP) confirmed the results of the TRAP assays (Figs. 3 and 4). PU.1 (proviral integration 1, encoded by Spi1) and NFATc1 (nuclear factor of activated T cells, cytoplasmic 1, encoded by Nfatc1) were also down-regulated, both of which are essential transcription factors that regulate the expression of osteoclast differentiation factors such as c-Fms, RANK, and integrin β3 in pre-osteoclasts. These results suggest that the decreased expression of genes required in the early stages of osteoclast differentiation, such as the transcription factors PU.1 and NFATc1, influence the expression of other marker genes required for osteoclast differentiation and function.
Then we observed the gene expression of some of those factors at earlier stages because most of those genes exhibited lower expression in KO cells than WT at the late stages of osteoclast differentiation induced by M-CSF and RANKL (Fig. 5A). As well as in the late stage, decreased expression of Csf1r, Nfatc1, Spi1, Tln1, Itga5, Itgb3, and Itgb5 were observed in KO cells induced by M-CSF for 3 days (Fig. 5B). The down-regulation of Csf1r (encoding c-Fms) supported the flow cytometry results. Tln1 (encoding Talin) is an adaptor protein linking β-integrins to the actin cytoskeleton, which is critical for osteoclast function (34), and Itgb5-encoding integrin β5 is expressed in immature osteoclast precursors instead of Itgb3 (35). However, in bone marrow cells or non-adherent cells collected from that bone marrow flushed from femur was cultured for 24 h, basal low expression of related genes was observed, and there was no significant difference of the expression of Csf1r, Nfatc1, and Spi1 in between WT and KO cells (Fig. 5C).
These results suggest that PRIP deficiency impairs the process of osteoclast differentiation at the early stage initiated by M-CSF. We also observed osteoclast differentiation using bone marrow cells from both genotypes cultured with M-CSF. Adherent cells, which we identified as osteoclast precursors, were much less abundant in the KO cultures for both 1.5 and 3 days (Fig. 6). This phenotype is probably the result from decreased expression of at least adhesion molecules such as αV and β5 or β3 integrins.
Figure 6.
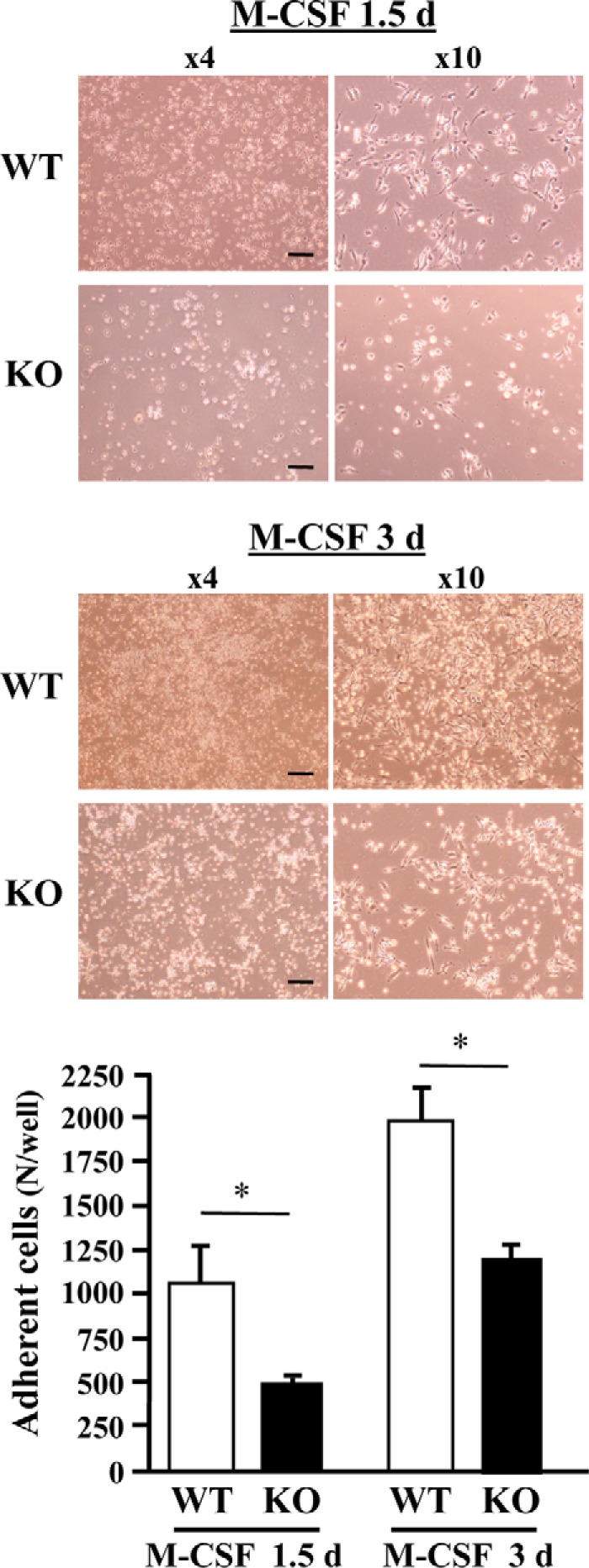
Cell adhesion in the early stages of osteoclast differentiation. Bone marrow cells from the femurs of male mice of both genotypes were cultured with M-CSF for 24 h. Nonadherent cells were harvested and cultured with M-CSF for a further 1.5 or 3 days. The upper panels are images of cell cultures. Bright cells are nonadherent. Adherent cells were identified as osteoclast precursors. The graph shows the number of adherent cells. Bar = 200 μm. Results shown are the mean ± S.D. of samples from three mice cultured in triplicate. *, p < 0.05.
Cell signaling stimulated by M-CSF during osteoclast differentiation
We then investigated the effects of PRIP deficiency in M-CSF-induced signaling during osteoclast differentiation. M-CSF is an upstream activator of ERK signals for the survival of osteoclast precursors and, therefore, is essential for the differentiation of pre-osteoclasts into osteoclasts (36). The PI3K/Akt signaling pathway also regulates the survival and differentiation of osteoclasts (37). Based on this consideration, we stimulated cultured bone marrow cells from the femurs of both genotype mice with 10 ng/ml M-CSF for 0, 10, or 30 min. Cell lysates were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by Western blotting for activation of ERK and Akt assessed by their phosphorylation levels, but there were no differences in the molecular expression or phosphorylation levels between the genotypes (data not shown). However, expression of calcineurin in KO cells was decreased (Fig. 7A). We then measured cellular phosphatase activity by calcineurin in cultured bone marrow cells; the activities were significantly lower in KO mice than in WT mice (Fig. 7B). We also examined intracellular Ca2+ changes in response to M-CSF and found that M-CSF stimulation gradually increased intracellular Ca2+ concentration for up to 6 h, but significantly less increase was observed in KO cells (Fig. 7C). These results suggest that PRIP deficiency triggers less increase of intracellular Ca2+ level and less expression of calcineurin, resulting in the down-regulation of the activity leading to lesser osteoclast differentiation.
Figure 7.
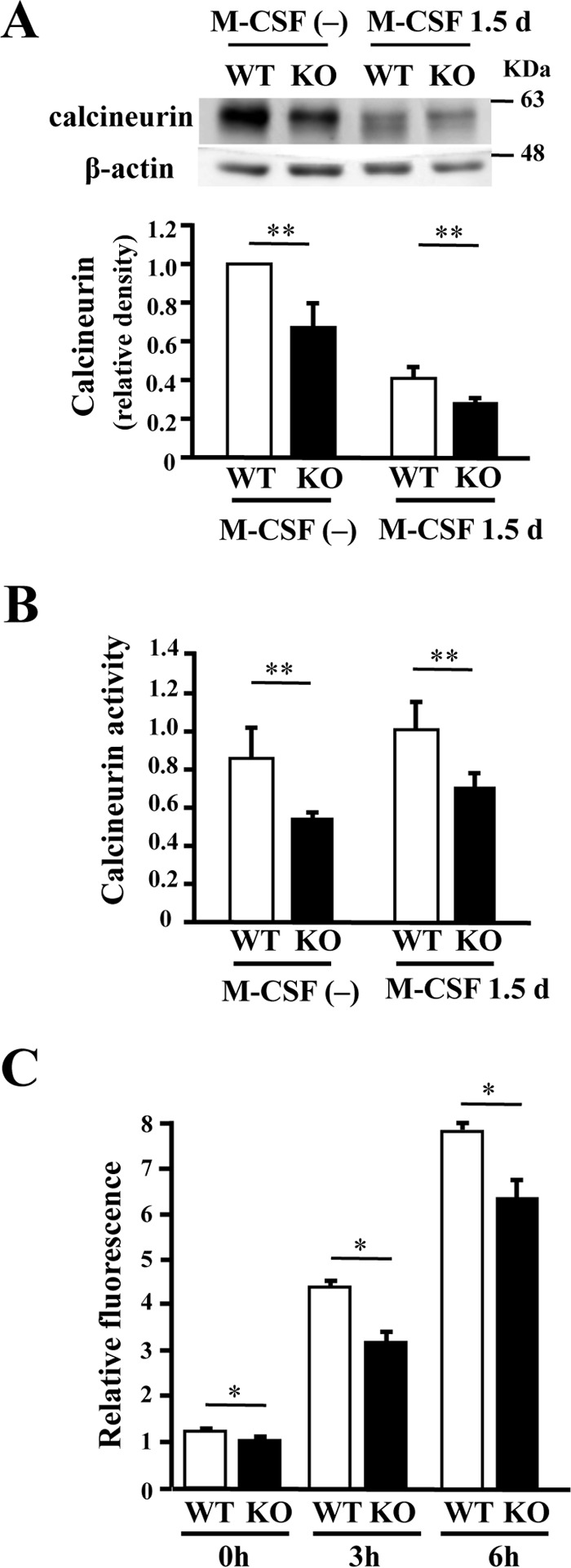
Expression and activity of calcineurin in bone marrow cells. A, cultured bone marrow cells from mice femurs of both genotypes were stimulated with M-CSF for 1.5 days. Cell lysates were examined for expression of calcineurin by Western blotting. Typical blots are shown. Density of calcineurin was normalized by the total amount of β-actin. B, bone marrow cells cultured in the same way were lysed to examine for calcineurin activity, summarized in the graph. Results shown are the mean ± S.D. of samples from six cultures done in triplicate. C, intracellular Ca2+ changes induced by M-CSF in pre-osteoclasts. Pre-osteoclasts from the femurs of male mice of both genotypes were cultured with M-CSF for 24 h. After incorporation of Fluo-4 AM, cells were stimulated with 20 ng/ml M-CSF. The graph showed relative fluorescence intensity at 0, 3, and 6 h after stimulation with M-CSF. *, p < 0.05; **, p < 0.01.
Nuclear localization of NFATc1 during osteoclast differentiation
The results so far indicate that PRIP deficiency might cause the impairment of nuclear translocation of NFATc1, a master regulator of osteoclast differentiation that function to promote the transcription of multiple genes that are important for osteoclast differentiation. We then performed immunofluorescent staining for NFATc1 using bone marrow cells from two genotypes. Fluorescent intensity was apparently lower in KO cells, probably because of the lower amount, and the nuclear signal was hardly observed in KO cells after stimulation with M-CSF plus RANKL. On the other hand, WT cells clearly showed nuclear localization of NFATc1 (Fig. 8). A similar difference between WT and KO cells was observed in those stimulated with M-CSF alone (data not shown). The results suggest that the impairment of osteoclastogenesis in KO mice is at least partly caused by the failure of nuclear translocation of NFATc1.
Figure 8.
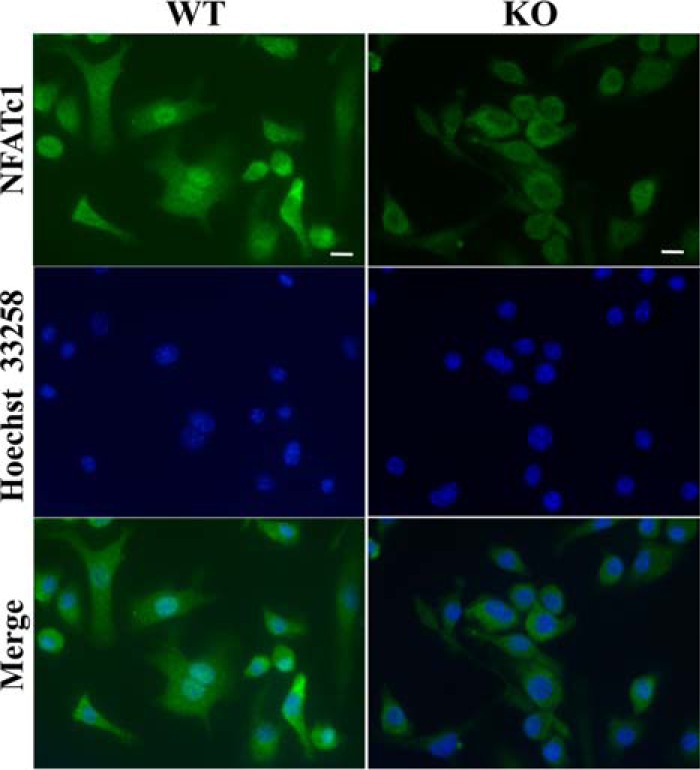
Immunofluorescent staining of NFATc1 nuclear localization in cells during osteoclast differentiation. Cultured bone marrow cells of both genotypes, with M-CSF for 3 days followed by with M-CSF plus RANKL for another 4 days, were immunostained with NFATc1 antibody. The left panels show the localization of NFATc1 (green). Nuclei were stained with Hoechst 33258 (blue; center panels). Merged images are provided in the right panels. Similar results were observed in cells stimulated with only M-CSF for 3 days. Images are of typical results were; similar results were obtained from cells of three mice of each genotype cultured in triplicate. Bar = 10 μm.
Recovery of osteoclastogenesis in KO cells by thapsigargin
A lower intracellular Ca2+ increase in KO cells might be the initial event for the impairment of osteoclastogenesis, which is followed by lower calcineurin activity and less nuclear translocation of NFATc1. We then performed experiments for osteoclastogenesis by M-CSF plus RANKL in the presence of thapsigargin (see the next section for further discussion of this drug). The addition of thapsigargin slightly promoted osteoclastogenesis compared with the control in WT cells, as assessed by TRAP staining (Fig. 9A). On the other hand, full recovery to or beyond the WT level was observed in KO cells in the presence of thapsigargin (Fig. 9A). Nuclear localization of NFATc1 in KO cells was clearly shown in the presence of thapsigargin, in clear contrast to cells in which the drug was absent (Fig. 9B). Quantitative analysis of NFATc1 localization also showed a remarkable increase of the nuclear localization in KO cells by the treatment with thapsigargin (Fig. 9C).
Figure 9.

The effect of intracellular Ca2+ change on osteoclast differentiation. A, bone marrow cells from each genotype were cultured with or without 10 nm thapsigargin (TG) in medium containing M-CSF and RANKL as described above. Cells were stained for TRAP, and those positive for TRAP (TRAP+) containing three or more nuclei were identified as osteoclasts. The graphs show the number of osteoclasts with the indicated number of nuclei. Bar = 100 μm. Results are shown as the mean ± S.D. of samples from three cultures done in triplicate. *, p < 0.05; **, p < 0.01. B, the same culture in the presence of TG was also performed for the analysis of localization of NFATc1. Panels show the results of immunofluorescent staining of NFATc1. Images are of typical results; similar results were obtained from cells of three mice of each genotype cultured in triplicate. Bar = 10 μm. C, a graph shows the result of quantification of fluorescence intensity of NFATc1 in cells (nucleus/cytosol) as assessed by the immunofluorescent staining as shown in Figs. 8 and 9B. Data are shown as the mean ± S.D. of the results from fluorescence intensity in nucleus and cytosol respectively in five or more cultured cells of three mice of each genotype. **, p < 0.01.
Discussion
We previously reported that bone mineral density and trabecular bone volume are higher in KO mice (26), and a subsequent study revealed that increased bone mass in KO mice is partly caused by up-regulation of bone morphogenetic protein signaling required for osteoblast differentiation (27). However, the implications of decreased bone resorption for the bone properties of KO mice have not yet been fully examined. This study was, therefore, undertaken to examine the role of PRIP in osteoclastic activity by performing dynamic orthodontic tooth movement in KO mice.
The use of orthodontic tooth movement devices is clinically practical and a useful model for understanding the pathophysiological mechanism of bone remodeling and, therefore, suitable for in vivo studies of dynamic mechanical loading-induced bone remodeling using various animal species (38, 39). Studies using this methodology in mice have shown the functional significance of factors related to bone metabolism, including inflammatory cytokines, chemokines, and their receptors (39–42). We adopted the force level of ∼10–15 g, which corresponds to a typical force level of 30–40 g produced in rats using the same method (43). Using this methodology, we clearly showed less tooth movement and bone resorption in KO mice, indicating the involvement of PRIP in the promotion of osteoclast activity and/or osteoclastogenesis in vivo.
In vitro osteoclast formation assays using a combination of pre-osteoclasts and osteoblastic/stromal cells from WT and KO mice showed conclusively that pre-osteoclasts from KO mice are deficient in their capacity for differentiation into osteoclasts. We also found that mRNA levels of osteoclast markers such as RANK, c-Fms, and TRAP were lower in cultured cells from KO mice based on assessment by flow cytometry and real-time PCR analysis. This is a direct indication of lower osteoclast differentiation ability. The reduction of specific interacting proteins and transcription factors such as DC-STAMP, Kindlin3, PU.1, and NFATc1 leads to the impairment of osteoclast differentiation in KO mice. PU.1, an ETS family transcription factor, regulates the expression of various genes for osteoclast differentiation (7, 44–46). Expression of PU.1 does not change or increase slightly during osteoclast differentiation (9, 48). PU.1 cooperates with NFATc1 and MITF (microphthalmia-associated factor) to regulate the expression of specific genes such as Itgb3 and Ctsk in response to M-CSF and RANKL during osteoclast differentiation (49, 7), and differentiation from myeloid progenitors to osteoclasts is dependent on PU.1 and MITF as well as M-CSF (50). Decreased expression of PU.1 and NFATc1, in addition to c-Fms, in KO mice probably leads to the down-regulation of many osteoclast-specific genes during osteoclast differentiation, and PRIP would be involved in the process of osteoclast differentiation, not on bone marrow stage or late stage but on the early stage initiated by M-CSF.
The functional properties of osteoclasts of KO mice would also be impaired if indeed osteoclasts are fully formed. Many reports indicate that there is a robust increase in αVβ3 integrin during osteoclast formation and maturation, which is required for forming a sealing zone (actin ring) on the bone surface (35, 51, 52). Integrin β5 (encoded by Itgb5), which is also strongly expressed in pre-osteoclasts, is utilized as a αVβ5 integrin complex for the attachment of immature osteoclasts to the bone matrix (35, 53). In KO cells we found that expression of not only integrin β3 and αV, but also β5, was depressed, indicating down-regulation of αVβ5 integrin heterodimer and resulting in reduced adhesion of pre-osteoclasts and a reduced formation of the sealing zone. Lower expression of TCIRG1, Clcn7, MMP9, and cathepsin K also indicates the impairment of osteoclastic functional activities in KO cells.
In contrast, expression of Ror2 mRNA was higher in KO cells. Wnt5a-Ror2 signaling is reported to play a role in osteoclastogenesis (54). To examine the role of PRIP in Wnt5a signaling and subsequent JNK signaling in osteoclast formation, we examined Wnt5a-stimulated osteoclastogenesis and Wnt5a-induced phosphorylation of JNK, but there were no significant differences in bone marrow cells from two genotypes (data not shown). The cause and effects of the higher expression of Ror2 in KO cells are yet to be determined.
We previously reported that there was little difference in the number of TRAP-positive multinucleated cells between two genotypes, but their activities were less in KO cells, likely because of an impaired formation of actin ring (27). However, in this study, the number of the TRAP-positive osteoclastic cells was fewer in KO cells, although the reason for this discrepancy is unclear.
We then explored possible signaling molecules involved in the early stages of osteoclastogenesis but found no significant difference in the expression or phosphorylation of signaling factors implicated in M-CSF-related pathways such as ERK and PI3K/Akt. However, calcineurin expression was decreased in KO cells. Calcineurin is activated through calmodulin after increased intracellular Ca2+ concentration and activates NFATc1 by dephosphorylation, which translocates to the nucleus and thus induces osteoclast-specific gene transcription to allow differentiation of osteoclasts. Ca2+ oscillation/calcineurin-dependent activation and amplification of NFATc1 in osteoclast precursors is essential for their differentiation into osteoclasts (55, 56). KO cells exhibited decreased phosphatase activity of calcineurin and reduced intracellular Ca2+ changes, suggesting down-regulation of calcium-calcineurin-NFATc1 signaling during osteoclast differentiation in KO mice.
During osteoclast differentiation, M-CSF signaling is followed by the Akt/PI3K, Grb2 (growth factor receptor protein 2)/Sos (son of sevenless), and Src pathways, which stimulate phospholipase C-γ (PLCγ) directly or indirectly to contribute to up-regulation of the Ca2+ concentration (35). Alternatively, the SH2 domains of PLCγ2 bind to phosphorylated Tyr-721 of c-Fms to be activated in myeloid cell line (57). RANKL also contributes to up-regulation of intracellular Ca2+ concentration through the cooperation of RANK and the RANK costimulatory receptors such as ITAM (immunoreceptor tyrosine-based activation motif) signals to activate PLCγ (58). Thus, the signaling pathways for osteoclastogenesis involves Ins(1,4,5)P3-mediated Ca2+ signaling. We previously reported that PRIP binds Ins(1,4,5)P3 via the PH domain, regulating Ca2+ release from the endoplasmic reticulum (ER) (12–16). Ins(1,4,5)P3 produced by PLC just beneath the plasma membrane is hydrolyzed by the 5-phosphatase, but a certain Ins(1,4,5)P3 avoids hydrolyzation by binding to PRIP. This remaining Ins(1,4,5)P3 reaches the receptors on the ER membrane to activate Ca2+ release (59). In the absence of PRIP, the produced Ins(1,4,5)P3 was highly hydrolyzed by the phosphatase, resulting in down-regulation of Ca2+ release from the ER (16, 17, 59).
Furthermore, depletion of Ca2+ in the lumen of the ER triggers store-operated Ca2+ entry from the extracellular space via the activation of the Stim1-Orai system, resulting in an increase in the intracellular Ca2+ concentration (60). PRIP deficiency triggered down-regulation of Ins(1,4,5)P3-mediated Ca2+ release from the ER (17) and would also be expected to reduce subsequent Ca2+ entry through the Stim1-Orai system, resulting in the observed lower cytosolic Ca2+ level in bone marrow cells from KO mice. This hypothesis was examined by testing the effect of thapsigargin, a specific inhibitor of ER Ca2+-ATPase that causes the depletion of Ca2+ storage in the ER independent of the action of Ins(1,4,5)P3. As observed, the inclusion of thapsigargin in the culture medium for osteoclastogenesis recovered the process to WT levels and indeed resulted in enhanced promotion; this latter effect was probably driven by an enhanced rate of increase of cytosolic Ca2+, as Ca2+ entry in brain cortex slices of KO mice and B-cells in PRIP type 2 KO mice was found to be enhanced (61, 47). Thapsigargin slightly enhanced osteoclastogenesis even in WT cells, possibly caused by full activation of the Ca2+ entry system.
In conclusion, we found that PRIP is positively involved in osteoclast differentiation in the early phases of the process through the regulation of calcium-calcineurin-NFATc1 signaling via the regulation of intracellular Ca2+ concentrations and possibly other mechanisms. PRIP regulate the activity of calcineurin via Ca2+ signaling, and PRIP deficiency reduced the expression of calcineurin and NFATc1, but this might be caused by reduced Ca2+ signaling. PRIP could, therefore, be a new therapeutic target for the treatment of diseases related to bone remodeling. Further researches are apparently needed to get more insights into the molecular mechanisms underlying PRIP function in calcium-calcineurin-NFATc1 signaling.
Experimental procedures
Experimental animals
PRIP-1 and -2 double knock out (KO) mouse strains and the corresponding WT mice were generated as described previously (19). In brief, KO mice were bred by crossing PRIP-1 KO and PRIP-2 KO mice, each on a C57BL/6J (Charles River Laboratories Japan, Yokohama, Japan) background. The resultant PRIP KO and WT mice were then backcrossed for a further four generations onto C57BL/6J. The handling of the mice and all procedures were approved by the Animal Care Committee of Kyushu University, according to the guidelines of the Japanese Council on Animal Care.
Experimental tooth movement by orthodontic force
To move the upper right first molar with the upper incisors as anchors, a Ni-Ti closed coil spring (Tomy, Tokyo, Japan) was inserted between the upper incisors and the upper right first molar in 8-week-old mice. A ligature wire with a diameter of 0.2 mm (Tomy) was laced between the upper right first and second molar, and the closed coil spring was tied to the mesial first molar. The end of the closed coil spring was tied to the upper incisors with ligature wire. The force level was approximately 12 g. The treatment was continued for 8 days, during which the mice were provided with powdered food.
Microcomputed tomography and measurement of movement distance
After the tooth movement treatment, skulls were taken from 4–6 individuals of each genotype. The skulls were fixed with 70% ethanol and analyzed using μCT (Skyscan-1076 in vivo micro-CT scanner; Bruker, Kontich, Belgium). Tooth movement distance was evaluated using images from the μCT. The shortest distance between the upper right distal surface of the first molar and the mesial surface of the second molar was measured using Skyscan-1076 software.
Histological analysis
Extracted maxillary bones were fixed in 70% ethanol for 1 day and decalcified in a buffer containing 10% EDTA and 0.1 m Tris-HCl at pH 7.4 for 7 days. Decalcified bones were embedded in paraffin and sectioned in the sagittal plane at a thickness of 7 μm. TRAP staining was carried out using a TRAP/ALP (alkaline phosphatase) Stain Kit (Wako, Osaka, Japan) according to the manufacturer's instructions. The sections were visualized using BZ-9000 Fluorescence Microscope (KEYENCE, Osaka, Japan), and their Alveolar bone resorption lacunae on the pressure side (M) were measured with BZ-II Analyzer (KEYENCE).
Cell culture
Bone marrow cells prepared from the femurs of 6–8-week-old mice from each genotype were cultured in α-minimal essential medium (α-MEM) (Wako) with 10% fetal bovine serum (FBS), 100 units/ml penicillin, and 0.1 mg/ml streptomycin. The cells were maintained at 37 °C in a humidified 5% CO2 incubator. After 1 day, the non-adherent cells (pre-osteoclasts) were seeded in cell culture dishes (Corning, NY) or low-cell-binding dishes (Thermo Fisher Scientific, Waltham, MA) and cultured in α-MEM with 10% FBS, containing 20 ng/ml recombinant human M-CSF (Peprotech, Rocky Hill, NJ) for 3 days. The precursor cells were then cultured with 30 ng/ml M-CSF and 50 ng/ml RANKL (Wako) for 4 days. For the examination of the effect of intracellular Ca2+ increase on osteoclastogenesis, the cells were cultured with 10 nm thapsigargin (Sigma) in the medium containing M-CSF and RANKL as described above. Differentiated adherent cells were used for TRAP staining or immunofluorescence staining. Non-adherent cells were used for flow cytometric analysis. Real-time PCR was carried out using non-adherent and adherent cells as described below.
In vitro osteoclast formation
Osteoclast differentiation was examined in co-cultures of bone marrow cells with pre-osteoblasts that were isolated from the calvaria of newborn mice. Pre-osteoblasts (1 × 104 per well) and bone marrow cells (1 × 105 per well) were co-cultured in α-MEM with 10% FBS containing 1α,25(OH)2D3 (10−8 m) in a 48-well plate for 7 days. Osteoclast formation was analyzed by staining for TRAP (TRAP KIT; Sigma) and counting the number of TRAP-positive multinucleated cells.
Flow cytometry
Cultured cells in low-cell-binding plates were collected and stained with anti-RANK antibody (9A725) (phycoerythrin) (Abcam, Cambridge, UK) and Alexa Fluor 488 anti-mouse CD115 antibody (eBioscience, San Diego, CA). Conjugated isotype phosphatidylethanolamine and isotype Alexa 488 were used as controls. Stained cells were analyzed using a FACScalibur analyzer running CellQuest software (BD Biosciences).
Quantitative real-time PCR analysis
Total RNA was extracted from cultured cells using the RNeasy Micro Kit (Qiagen, Venlo, The Netherlands) according to the manufacturer's instructions. Total RNA (1 μg) was transcribed to cDNA using ReverTra Ace (Toyobo, Osaka, Japan). Real-time PCR was performed using THUNDERBIRD SYBR qPCR Mix (Toyobo) with Thermal Cycler Dice Real Time System II (TaKaRa, Shiga, Japan). Glyceraldehyde-3-phosphate dehydrogenase gene (GAPDH) expression served as an internal control. Primer sequences were as follows: Tnfrs11a, 5′-ACCTTGGACCAACTGCACC-3′ (forward) and 5′-TGAGACTGGGCAGGTAAGC-3′ (reverse); Csf1r, 5′-TGTCATCGAGCCTAGTGGC-3′ (forward) and 5′-GGGAGATTCAGGGTCCAAG-3′ (reverse); Itga5, 5′-CCGTGGACTTCTTCGAGCC-3′ (forward) and 5′-GAATCAAACTCAATGGGCTGG-3′ (reverse); Itgb3, 5′-CCACACGAGGCGTGAACTC-3′ (forward) and 5′-CTTCAGGTTACATCGGGGTG-3′ (reverse); Ror2, 5′-AGAGCCACGAAGGGTCATC-3′ (forward) and 5′-GTTGGTAGCCACACACACTGG-3′ (reverse); Tcirg1, 5′-CACAGGGTCTGCTTACAAC-3′ (forward) and 5′-GTCTACCACGAAGCGTCTC-3′ (reverse); Clcn7, 5′-GATTTTGTCTCAGCTGGAGC-3′ (forward) and 5′-GGAACTGATTCCAGAAGGAG-3′ (reverse); Mmp9, 5′-GGACCCGAAGCGGACATTG-3′ (forward) and 5′-CGTCGTCGAAATGGGCATC-3′ (reverse); CtsK, 5′-GAAGAAGACTCACCAGAAGC-3′ (forward) and 5′-GGTTATGGGCAGAGATTTGC-3′ (reverse); Icam1, 5′-GTGATGCTCAGGTATCCATC-3′ (forward) and 5′-CACAGTTCTCAAAGCACAGC-3′ (reverse); Fermt3, 5′-GACTGAGGCTTGTTACCAC-3′ (forward) and 5′-ATACCTTGCTGCATGAGGC-3′ (reverse); Dcstamp, 5′-CCGTGGGCCAGAAGTTGC-3′ (forward) and 5′-GCCAGTGCTGACTAGGATG-3′ (reverse); Traf6, 5′-CCAGTTGCACATGAGACTGT-3′ (forward) and 5′-CGGACGCAAAGCAAGGTTAA-3′ (reverse); Acp5, 5′-CACTCCCACCCTGAGATTTG-3′ (forward) and 5′-CATCGTCTGCACGGTTCTG-3′ (reverse); Spp1, 5′-TCCCTCGATGTCATCCCTG-3′ (forward) and 5′-ACTCTCCTGGCTCTCTTTGG-3′ (reverse); Tln1, 5′-CTGCCGCATGATTCGTGAG-3′ (forward) and 5′-TCGGAGCATGTAGTAGTCC-3′ (reverse); Spi1, 5′-GATTCGCCTGTACCAGTTC-3′ (forward) and 5′-CTTGGACGAGAACTGGAAG-3′ (reverse); Nfatc1, 5′-GTTGGTATTAAAATTCTGAAACC-3′ (forward) and 5′-TTGCTGCCCTTTCACTGATG-3′ (reverse); Gapdh, 5′-AACTTTGGCATTGTGGAAGG-3′ (forward) and 5′-ACACATTGGGGGTAGGAACA-3′ (reverse).
Western blot analysis
Bone marrow cells or pre-osteoclasts were lysed with a buffer containing 25 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1% Triton X-100, 5 mm EDTA, 5 μg/ml leupeptin, 5 μg/ml pepstatin A, 2 μg/ml aprotinin, and 0.1 mm 4-amidinophenyl methanesulfonyl fluoride. Fifteen micrograms of each protein sample were separated using 10% SDS-polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane (Merck Millipore, Darmstadt, Germany). After blocking, the membrane was blotted with Pan-Calcineurin A antibody (1:1000; Cell Signaling Technology, Danvers, MA) overnight at 4 °C or monoclonal anti-β-actin antibody (1:3000; Sigma) for 1 h at room temperature then blotted with horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG (GE Healthcare) for 1 h at room temperature and visualized using an ECL system (GE Healthcare). Digital images were analyzed to measure the density of each band using an ImageQuant LAS 4000 mini (GE Healthcare).
Calcineurin cellular activity assay
Cell lysates were extracted from bone marrow cells and pre-osteoclasts. Calcineurin cellular phosphatase activity was measured using a Calcineurin Cellular Activity Assay Kit (Merck Millipore) according to the manufacturer's instructions. Briefly, after the free phosphate was removed by gel filtration, cell extracts were assayed for total phosphatase activity by incubating in a calcineurin substrate RII phosphopeptide for 30 min at room temperature. The color development by a BIOMOL GREEN reagent in the kit was measured at 620 nm on the microplate spectrophotometer.
Measurement of intracellular calcium level
Intracellular calcium levels were measured using Calcium KitII-Fluo4 (DOJINDO, Kumamoto Japan), according to the manufacturer's instructions. Pre-osteoclasts of both genotypes were cultured in Amine 96-well Black/Clear Plate (BD Biosciences) with M-CSF for 24 h. After loading with Fluo-4 AM for 1 h at 37 °C, pre-osteoclasts were stimulated with 20 ng/ml M-CSF, and fluorescence was measured using VICTORTM X4 Multilabel Plate Reader (PerkinElmer Life Sciences) with excitation at 485 nm and emission at 535 nm every 2-min period for 6 h. Values of intracellular Ca2+ changes were evaluated in accordance with the relative fluorescence intensity.
Immunofluorescent staining
Bone marrow cells from femurs of male mice (5–7 weeks) of both genotypes were cultured in an 8-well slide chamber (Watson BioLab, Kobe, Japan) with 20 ng/ml M-CSF for 3 days or cultured additionally with 30 ng/ml M-CSF and 50 ng/ml RANKL (Wako) for 4 days. The cells were then fixed in 4% paraformaldehyde in PBS for 15 min and permeabilized with 0.5% Triton X-100 for 30 min. After blocking with 2.5% BSA in PBS for 1 h, slide glasses were incubated with the NFATc1 (NFAT2) antibody (1:200; Cell Signaling Technology) overnight at 4 °C, washed with PBS, and then incubated with Alexa Fluor 488 donkey anti-rabbit IgG (1:1000; Molecular Probes, Eugene, OR) for 1 h at room temperature. Nuclei were counterstained with Hoechst 33258 (Sigma), and images were obtained using a BZ-9000 microscope (KEYENCE). Fluorescence intensity of NFATc1 was measured in the nucleus and cytosol, respectively, using BZ-II Analyzer (KEYENCE), and the mean value of the ratio of the nucleus to the cytosol was determined.
Statistical analysis
Data are presented as the mean ± S.D. of at least three independent experiments. Statistical significance was assessed using Student's t test, with significance inferred at p < 0.05 (*) and p < 0.01 (**).
Author contributions
A. M. and M. M. contributed to conception, design, data acquisition, analysis, and drafting of the manuscript. Y. H. performed and analyzed the experiments shown in Fig. 4. M. H. contributed to data analysis and critically revised the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Prof. Y. Yonemitsu (Kyushu University) for supporting the experimental apparatus and Dr. M. Kotani (Kyushu University) for providing pre-osteoblasts.
This work was supported by Grants-in-aid for Scientific Research (KAKENHI nos. 24229009 (to M. H.) and 24592804 (to M. M.)) from the Japan Society for the Promotion of Science. The authors declare that they have no conflicts of interest with the contents of this article.
- M-CSF
- macrophage colony-stimulating factor
- α-MEM
- α-minimal essential medium
- Clcn7
- chloride channel, voltage-sensitive 7
- Dc-stamp
- dendritic cell-specific transmembrane protein
- ER
- endoplasmic reticulum
- Icam1
- intercellular adhesion molecule-1
- μCT
- micro-computed tomography
- MITF
- microphthalmia-associated factor
- Mmp9
- matrix metalloproteinase 9
- NFATc1
- nuclear factor- activated T cell c1
- Orai (CRACM)
- calcium release-activated calcium modulator
- PH
- pleckstrin homology
- PLCγ
- phospholipase C-γ
- PRIP (Prip)
- phospholipase C-related but catalytically inactive protein (gene)
- RANKL
- receptor activator of nuclear factor κB ligand
- Stim1
- stromal interaction molecule 1
- Tcirg1
- T-cell, immune regulator 1 ATPase, H+ transporting, lysosomal V0 subunit A3
- Traf6
- tumor necrosis factor receptor-associated factor 6
- TRAP
- tartrate-resistant acid phosphatase
- Ins(1,4,5)P3
- d-myo-inositol 1,4,5-trisphosphate
- 1α,25(OH)2D3
- 1α,25-dihydroxycholecalciferol.
References
- 1. Boyle W. J., Simonet W. S., and Lacey D. L. (2003) Osteoclast differentiation and activation. Nature 423, 337–342 [DOI] [PubMed] [Google Scholar]
- 2. Takahashi N., Akatsu T., Udagawa N., Sasaki T., Yamaguchi A., Moseley J. M., Martin T. J., and Suda T. (1988) Osteoblastic cells are involved in osteoclast formation. Endocrinology 123, 2600–2602 [DOI] [PubMed] [Google Scholar]
- 3. Suda T., Takahashi N., Udagawa N., Jimi E., Gillespie M. T., and Martin T. J. (1999) Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr. Rev. 20, 345–357 [DOI] [PubMed] [Google Scholar]
- 4. Wiktor-Jedrzejczak W., Bartocci A., Ferrante A. W., Jr, Ahmed-Ansari A., Sell K. W., Pollard J. W., and Stanley E. R. (1990) Total absence of colony-stimulating factor 1 in the macrophage-deficient osteopetrotic (op/op) mouse. Proc. Nat.l Acad. Sci. U.S.A. 87, 4828–4832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yoshida H., Hayashi S., Kunisada T., Ogawa M., Nishikawa S., Okamura H., Sudo T., Shultz L. D., and Nishikawa S. (1990) The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature 345, 442–444 [DOI] [PubMed] [Google Scholar]
- 6. Takayanagi H., Kim S., Koga T., Nishina H., Isshiki M., Yoshida H., Saiura A., Isobe M., Yokochi T., Inoue J., Wagner E. F., Mak T. W., Kodama T., and Taniguchi T. (2002) Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev. Cell. 3, 889–901 [DOI] [PubMed] [Google Scholar]
- 7. Matsumoto M., Hisatake K., Nogi Y., and Tsujimoto M. (2001) Regulation of receptor activator of NF-κB ligand-induced tartrate-resistant acid phosphatase gene expression by PU.1- interacting protein/interferon regulatory factor-4: synergism with microphthalmia transcription factor. J. Biol. Chem. 276, 33086–33092 [DOI] [PubMed] [Google Scholar]
- 8. Inaba T., Gotoda T., Ishibashi S., Harada K., Ohsuga J. I., Ohashi K., Yazaki Y., and Yamada N. (1996) Transcription factor PU1 mediates induction of c-fms in vascular smooth muscle cells: a mechanism for phenotypic change to phagocytic cells. Mol. Cell. Biol. 16, 2264–2273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tondravi M. M., McKercher S. R., Anderson K., Erdmann J. M., Quiroz M., Maki R., and Teitelbaum S. L. (1997) Osteopetrosis in mice lacking haematopoietic transcription factor PU.1. Nature 386, 81–84 [DOI] [PubMed] [Google Scholar]
- 10. Teitelbaum S. L. (2000) Bone resorption by osteoclasts. Science. 289, 1504–1508 [DOI] [PubMed] [Google Scholar]
- 11. Helfrich M. H. (2003) Osteoclast diseases. Microsc. Res. Tech. 61, 514–532 [DOI] [PubMed] [Google Scholar]
- 12. Kanematsu T., Takeya H., Watanabe Y., Ozaki S., Yoshida M., Koga T., Iwanaga S., and Hirata M. (1992) Putative inositol 1,4,5-trisphosphate binding proteins in rat brain cytosol. J. Biol. Chem. 267, 6518–6525 [PubMed] [Google Scholar]
- 13. Yoshida M., Kanematsu T., Watanabe Y., Koga T., Ozaki S., Iwanaga S., and Hirata M. (1994) d-myo-Inositol 1,4,5-trisphosphate-binding proteins in rat brain membranes. J. Biochem. 115, 973–980 [DOI] [PubMed] [Google Scholar]
- 14. Kanematsu T., Misumi Y., Watanabe Y., Ozaki S., Koga T., Iwanaga S., Ikehara Y., and Hirata M. (1996) A new inositol 1,4,5-trisphosphate binding protein similar to phospholipase C-δ1. Biochem. J. 313, 319–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kanematsu T., Yoshimura K., Hidaka K., Takeuchi H., Katan M., and Hirata M. (2000) Domain organization of p130, PLC-related catalytically inactive protein, and structural basis for the lack of enzyme activity. Eur. J. Biochem. 267, 2731–2737 [DOI] [PubMed] [Google Scholar]
- 16. Takeuchi H., Oike M., Paterson H. F., Allen V., Kanematsu T., Ito Y., Erneux C., Katan M., and Hirata M. (2000) Inhibition of Ca2+ signalling by p130, a phospholipase-C-related catalytically inactive protein: critical role of the p130 pleckstrin homology domain. Biochem. J. 349, 357–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Harada K., Takeuchi H., Oike M., Matsuda M., Kanematsu T., Yagisawa H., Nakayama K. I., Maeda K., Erneux C., and Hirata M. (2005) Role of PRIP-1, a novel Ins(1,4,5)P3-binding protein, in Ins(1,4,5)P3-mediated Ca2+ signaling. J. Cell. Physiol. 202, 422–433 [DOI] [PubMed] [Google Scholar]
- 18. Terunuma M., Jang I. S., Ha S. H., Kittler J. T., Kanematsu T., Jovanovic J. N., Nakayama K. I., Akaike N., Ryu S. H., Moss S. J., and Hirata M. (2004) GABAA receptor phospho-dependent modulation is regulated by phospholipase C-related inactive protein type 1, a novel protein phosphatase 1 anchoring protein. J. Neurosci. 24, 7074–7084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kanematsu T., Yasunaga A., Mizoguchi Y., Kuratani A., Kittler J. T., Jovanovic J. N., Takenaka K., Nakayama K. I., Fukami K., Takenawa T., Moss S. J., Nabekura J., and Hirata M. (2006) Modulation of GABAA receptor phosphorylation and membrane trafficking by phospholipase C-related inactive protein/protein phosphatase 1 and 2A signaling complex underlying brain-derived neurotrophic factor-dependent regulation of GABAergic inhibition. J. Biol. Chem. 281, 22180–22189 [DOI] [PubMed] [Google Scholar]
- 20. Mizokami A., Kanematsu T., Ishibashi H., Yamaguchi T., Tanida I., Takenaka K., Nakayama K. I., Fukami K., Takenawa T., Kominami E., Moss S. J., Yamamoto T., Nabekura J., and Hirata M. (2007) Phospholipase C-related inactive protein is involved in trafficking of gamma2 subunit-containing GABAA receptors to the cell surface. J. Neurosci. 27, 1692–1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fujii M., Kanematsu T., Ishibashi H., Fukami K., Takenawa T., Nakayama K. I., Moss S. J., Nabekura J., and Hirata M. (2010) Phospholipase C-related but catalytically inactive protein is required for insulin-induced cell surface expression of gamma-aminobutyric acid type A receptors. J. Biol. Chem. 285, 4837–4846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yoshimura K., Takeuchi H., Sato O., Hidaka K., Doira N., Terunuma M., Harada K., Ogawa Y., Ito Y., Kanematsu T., and Hirata M. (2001) Interaction of p130 with, and consequent inhibition of, the catalytic subunit of protein phosphatase 1α. J. Biol. Chem. 276, 17908–17913 [DOI] [PubMed] [Google Scholar]
- 23. Sugiyama G., Takeuchi H., Kanematsu T., Gao J., Matsuda M., and Hirata M. (2013) Phospholipase C-related but catalytically inactive protein, PRIP as a scaffolding protein for phospho-regulation. Adv. Biol. Regul. 53, 331–340 [DOI] [PubMed] [Google Scholar]
- 24. Gao J., Takeuchi H., Zhang Z., Fukuda M., and Hirata M. (2012) Phospholipase C-related but catalytically inactive protein (PRIP) modulates synaptosomal-associated protein 25 (SNAP-25) phosphorylation and exocytosis. J. Biol. Chem. 287, 10565–10578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang Z., Takeuchi H., Gao J., Wang D., James D. J., Martin T. F., and Hirata M. (2013) PRIP (phospholipase C-related but catalytically inactive protein) inhibits exocytosis by direct interactions with syntaxin 1 and SNAP-25 through its C2 domain. J. Biol. Chem. 288, 7769–7780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Matsuda M., Tsutsumi K., Kanematsu T., Fukami K., Terada Y., Takenawa T., Nakayama K. I., and Hirata M. (2009) Involvement of phospholipase C-related inactive protein in the mouse reproductive system through the regulation of gonadotropin levels. Biol. Reprod. 81, 681–689 [DOI] [PubMed] [Google Scholar]
- 27. Tsutsumi K., Matsuda M., Kotani M., Mizokami A., Murakami A., Takahashi I., Terada Y., Kanematsu T., Fukami K., Takenawa T., Jimi E., and Hirata M. (2011) Involvement of PRIP, phospholipase C-related, but catalytically inactive protein, in bone formation. J. Biol. Chem. 286, 31032–31042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kotani M., Matsuda M., Murakami A., Takahashi I., Katagiri T., and Hirata M. (2015) Involvement of PRIP (Phospholipase C-related but catalytically inactive protein) in BMP-induced smad signaling in osteoblast differentiation. J. Cell. Biochem. 116, 2814–28123 [DOI] [PubMed] [Google Scholar]
- 29. Storey E. (1973) The nature of tooth movement. Am. J. Orthod. 63, 292–314 [DOI] [PubMed] [Google Scholar]
- 30. Kornak U., Kasper D., Bösl M. R., Kaiser E., Schweizer M., Schulz A., Friedrich W., Delling G., and Jentsch T. J. (2001) Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell 104, 205–215 [DOI] [PubMed] [Google Scholar]
- 31. Yagi M., Miyamoto T., Sawatani Y., Iwamoto K., Hosogane N., Fujita N., Morita K., Ninomiya K., Suzuki T., Miyamoto K., Oike Y., Takeya M., Toyama Y., and Suda T. (2005) DC-STAMP is essential for cell-cell fusion in osteoclasts and foreign body giant cells. J. Exp. Med. 202, 345–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schmidt S., Nakchbandi I., Ruppert R., Kawelke N., Hess M. W., Pfaller K., Jurdic P., Fässler R., and Moser M. (2011) Kindlin-3-mediated signaling from multiple integrin classes is required for osteoclast-mediated bone resorption. J. Cell Biol. 192, 883–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Armstrong A. P., Tometsko M. E., Glaccum M., Sutherland C. L., Cosman D., and Dougall W. C. (2002) A RANK/TRAF6-dependent signal transduction pathway is essential for osteoclast cytoskeletal organization and resorptive function. J. Biol. Chem. 277, 44347–44356 [DOI] [PubMed] [Google Scholar]
- 34. Zou W., Izawa T., Zhu T., Chappel J., Otero K., Monkley S. J., Critchley D. R., Petrich B. G., Morozov A., Ginsberg M. H., and Teitelbaum S. L. (2013) Talin1 and Rap1 are critical for osteoclast function. Mol. Cell. Biol. 33, 830–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Inoue M., Namba N., Chappel J., Teitelbaum S. L., and Ross F. P. (1998) Granulocyte macrophage-colony stimulating factor reciprocally regulates αV-associated integrins on murine osteoclast precursors. Mol. Endocrinol. 12, 1955–1962 [DOI] [PubMed] [Google Scholar]
- 36. Long C. L., and Humphrey M. B. (2012) Osteoimmunology: the expanding role of immunoreceptors in osteoclasts and bone remodeling. Bonekey. Rep. 1, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Verna C., Dalstra M., Lee T. C., Cattaneo P. M., and Melsen B. (2004) Microcracks in the alveolar bone following orthodontic tooth movement: a morphological and morphometric study. Eur. J. Orthod. 26, 459–467 [DOI] [PubMed] [Google Scholar]
- 38. Ren Y., Hazemeijer H., de Haan B., Qu N., and de Vos P. (2007) Cytokine profiles in crevicular fluid during orthodontic tooth movement of short and long durations. J. Periodontol. 78, 453–458 [DOI] [PubMed] [Google Scholar]
- 39. Yoshimatsu M., Shibata Y., Kitaura H., Chang X., Moriishi T., Hashimoto F., Yoshida N., and Yamaguchi A. (2006) Experimental model of tooth movement by orthodontic force in mice and its application to tumor necrosis factor receptor-deficient mice. J. Bone. Miner. Metab. 24, 20–27 [DOI] [PubMed] [Google Scholar]
- 40. Andrade I. Jr, Silva T. A., Silva G. A., Teixeira A. L., and Teixeira M. M. (2007) The role of tumor necrosis factor receptor type 1 in orthodontic tooth movement. J. Dent. Res. 86, 1089–1094 [DOI] [PubMed] [Google Scholar]
- 41. Andrade I. Jr, Taddei S. R., Garlet G. P., Garlet T. P., Teixeira A. L., Silva T. A., and Teixeira M. M. (2009) CCR5 down-regulates osteoclast function in orthodontic tooth movement. J. Dent. Res. 88, 1037–1041 [DOI] [PubMed] [Google Scholar]
- 42. Taddei S. R., Moura A. P., Andrade I. Jr, Garlet G. P., Garlet T. P., Teixeira M. M., and da Silva T. A. (2012) Experimental model of tooth movement in mice: a standardized protocol for studying bone remodeling under compression and tensile strains. J. Biomech. 45, 2729–2735 [DOI] [PubMed] [Google Scholar]
- 43. Pavlin D., Goldman E. S, Gluhak-Heinrich J., Magness M., and Zadro R. (2000) Orthodontically stressed periodontium of transgenic mouse as a model for studying mechanical response in bone: the effect on the number of osteoblasts. Clin. Orthod. Res. 3, 55–66 [PubMed] [Google Scholar]
- 44. Luchin A., Suchting S., Merson T., Rosol T. J., Hume D. A., Cassady A. I., and Ostrowski M. C. (2001) Genetic and physical interactions between Microphthalmia transcription factor and PU.1 are necessary for osteoclast gene expression and differentiation. J. Biol. Chem. 276, 36703–36710 [DOI] [PubMed] [Google Scholar]
- 45. Matsumoto M., Kogawa M., Wada S., Takayanagi H., Tsujimoto M., Katayama S., Hisatake K., and Nogi Y. (2004) Essential role of p38 mitogen-activated protein kinase in cathepsin K gene expression during osteoclastogenesis through association of NFATc1 and PU.1. J. Biol. Chem. 279, 45969–45979 [DOI] [PubMed] [Google Scholar]
- 46. Sharma S. M., Bronisz A, Hu R., Patel K., Mansky K. C., Sif S., and Ostrowski M. C. (2007) MITF and PU.1 recruit p38 MAPK and NFATc1 to target genes during osteoclast differentiation. J. Biol. Chem. 282, 15921–15929 [DOI] [PubMed] [Google Scholar]
- 47. Takenaka K., Fukami K., Otsuki M., Nakamura Y., Kataoka Y., Wada M., Tsuji K., Nishikawa S., Yoshida N., and Takenawa T. (2003) Role of phospholipase C-L2, a novel phospholipase C-like protein that lacks lipase activity, in B-cell receptor signaling. Mol. Cell. Biol. 23, 7329–7338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. So H., Rho J., Jeong D., Park R., Fisher D. E., Ostrowski M. C., Choi Y., and Kim N. (2003) (2003) Microphthalmia transcription factor and PU.1 synergistically induce the leukocyte receptor osteoclast-associated receptor gene expression. J. Biol. Chem. 278, 24209–24216 [DOI] [PubMed] [Google Scholar]
- 49. Crotti T. N., Sharma S. M., Fleming J. D., Flannery M. R., Ostrowski M. C., Goldring S. R., and McHugh K. P. (2008) (2008) PU.1 and NFATc1 mediate osteoclastic induction of the mouse β3 integrin promoter. J. Cell. Physiol. 215, 636–644 [DOI] [PubMed] [Google Scholar]
- 50. Soysa N. S., Alles N., Aoki K., and Ohya K. (2012) Osteoclast formation and differentiation: an overview. J. Med. Dent. Sci. 59, 65–74 [PubMed] [Google Scholar]
- 51. Li C. F., Ross F. P., Cao X., and Teitelbaum S. L. (1995) Estrogen enhances αVβ3 integrin: expression by avian osteoclast precursors via stabilization of β3 integrin mRNA. Mol. Endocrinol. 9, 805–813 [DOI] [PubMed] [Google Scholar]
- 52. Zhu H. J., Ross F. P., Cao X., and Teitelbaum S. L. (1996) Phorbol myristate acetate transactivates the avian β 3 integrin gene and induces αVβ3 integrin expression. J. Cell. Biochem. 61, 420–429 [DOI] [PubMed] [Google Scholar]
- 53. Shinar D. M., Schmidt A., Halperin D., Rodan G. A., and Weinreb M. (1993) Expression of αV and β3 integrin subunits in rat osteoclasts in situ. J. Bone. Miner. Res. 8, 403–414 [DOI] [PubMed] [Google Scholar]
- 54. Maeda K., Kobayashi Y., Udagawa N., Uehara S., Ishihara A., Mizoguchi T., Kikuchi Y., Takada I., Kato S., Kani S., Nishita M., Marumo K., Martin T. J., Minami Y., and Takahashi N. (2012) Wnt5a-Ror2 signaling between osteoblast-lineage cells and osteoclast precursors enhances osteoclastogenesis. Nat. Med. 18, 405–412 [DOI] [PubMed] [Google Scholar]
- 55. Hirotani H., Tuohy N. A., Woo J. T., Stern P. H., and Clipstone N. A. (2004) The calcineurin/nuclear factor of activated T cells signaling pathway regulates osteoclastogenesis in RAW264.7 cells. J. Biol. Chem. 279, 13984–13992 [DOI] [PubMed] [Google Scholar]
- 56. Asagiri M., Sato K., Usami T., Ochi S., Nishina H., Yoshida H., Morita I., Wagner E. F., Mak T. W., Serfling E., and Takayanagi H. (2005) Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J. Exp. Med. 202, 1261–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bourette R. P., Myles G. M., Choi J. L., and Rohrschneider L. R. (1997) Sequential activation of phoshatidylinositol 3-kinase and phospholipase C-γ2 by the M-CSF receptor is necessary for differentiation signaling. EMBO J. 16, 5880–5893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Negishi-Koga T., and Takayanagi H. (2009) Ca2+-NFATc1 signaling is an essential axis of osteoclast differentiation. Immunol. Rev. 231, 241–256 [DOI] [PubMed] [Google Scholar]
- 59. Kanematsu T., Takeuchi H., Terunuma M., and Hirata M. (2005) PRIP, a novel Ins(1,4,5)P3 binding protein, functional significance in Ca2+ signaling and extension to neuroscience and beyond. Mol. Cells 20, 305–314 [PubMed] [Google Scholar]
- 60. Park C. Y., Hoover P. J., Mullins F. M., Bachhawat P., Covington E. D., Raunser S., Walz T., Garcia K. C., Dolmetsch R. E., and Lewis R. S. (2009) STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to orai1. Cell 136, 876–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Toyoda H., Saito M., Sato H., Tanaka T., Ogawa T., Yatani H., Kawano T., Kanematsu T., Hirata M., and Kang Y. (2015) Enhanced desensitization followed by unusual resensitization in GABAA receptors in phospholipase C-related catalytically inactive protein-1/2 double-knockout mice. Pflugers Arch. 467, 267–284 [DOI] [PubMed] [Google Scholar]