

Abstract
Aptamers consist of short oligonucleotides that bind specific targets. They provide advantages over antibodies, including robustness, low cost, and reusability. Their chemical structure allows the insertion of reporter molecules and surface-binding agents in specific locations, which have been recently exploited for the development of aptamer-based biosensors and direct detection strategies. Mainstream use of these devices, however, still requires significant improvements in optimization for consistency and reproducibility. DNA aptamers are more stable than their RNA counterparts for biomedical applications but have the disadvantage of lacking the wide array of computational tools for RNA structural prediction. Here, we present the first approach to predict from sequence the three-dimensional structures of single stranded (ss) DNA required for aptamer applications, focusing explicitly on ssDNA hairpins. The approach consists of a pipeline that integrates sequentially building ssDNA secondary structure from sequence, constructing equivalent 3D ssRNA models, transforming the 3D ssRNA models into ssDNA 3D structures, and refining the resulting ssDNA 3D structures. Through this pipeline, our approach faithfully predicts the representative structures available in the Nucleic Acid Database and Protein Data Bank databases. Our results, thus, open up a much-needed avenue for integrating DNA in the computational analysis and design of aptamer-based biosensors.
Introduction
Cellular-level protein production is traditionally determined using several bioanalytical approaches, which rely on antibody or enzyme recognition. These include flow cytometry coupled with intracellular cytokine staining, enzyme-linked immunospot (ELISPOT), enzyme linked immunosorbent assay (ELISA), and polymerase chain reaction (PCR)1, 2. ELISA and PCR are robust technologies for detecting either cytokines or cytokine mRNA in blood; however, they cannot be used to identify specific populations of cytokine producing cells2. In contrast, flow cytometry and ELISPOT report the frequency of cytokine positive cells but not the cytokine concentration1. While these well-established methods can be sensitive and robust, they employ complex detection procedures, involving expensive reagents and multiple time consuming washing steps. In addition, these traditional strategies provide no information about the temporal dynamics of cytokine production, which is vital information in understanding the body’s immune response3. In order to fill this gap, aptamer-based affinity sensing strategies are emerging as viable alternatives to antibody-based immunoassays4.
Aptamer-based biorecognition elements are short nucleic acid molecules and thus are more robust and simple than antibody-based probes. Aptamers bind specifically diverse targets including ions, organic dyes, amino acids, nucleotides, RNA, biological cofactors, other small organic molecules, oligosaccharides, peptides, toxins, enzymes, growth factors, transcription factors, antibodies, viral proteins and/or components, cells, and bacteria5. The selection of aptamers is termed Systematic Evolution of Ligands by Exponential Enrichment (SELEX) and involves the discovery of full-length aptamers from large pools of randomized single-stranded DNA or RNA (1014 to 1015 variants). The selection process is highly iterative and involves exposing the oligonucleotides to a target that is either coupled to a matrix or surface. The unbound molecules are then washed away and the bound molecules are recovered and amplified. This process results in highly robust and specific aptamers that are 25–40 nucleotides long6.
The relatively simple chemical structure of aptamers allows the insertion of electrochemical or fluorescent reporter molecules7 as well as surface-binding agents8 in specific locations on the oligonucleotide9. During probe-target binding, the conformation change of the aptamer may be exploited to generate an analytical signal10. A number of aptamer-based biosensors have been successfully used to measure cell secretion of proteins11, 12; however, several opportunities for improvement remain before commercialization is feasible13. These include enhancing the sensitivity and improving the manufacturability and repeatability of aptamer-based sensors. Both of these challenges are not easily overcome without a better understanding of the molecular level interactions of the aptamer-biosensor surface (for improving manufacturability, reproducibility, and sensitivity) as well as of the aptamer-protein complex (for improving specificity and sensitivity).
Despite the growing widespread use in biotechnology and the substantial number of biomedical applications of DNA aptamers, including its clinical use as therapeutic agents for a number of human diseases14–16 and its increasing use in drug delivery17, the considerable array of computational tools available for single stranded RNA structure prediction (e.g. see refs 18–21 for recent reviews)18–21 are lacking for its DNA counterpart. Until now, the 3D computational tools available for DNA have been restricted to model mainly double-stranded DNA structures22, lacking the ability to analyze single-stranded DNA hairpins and other more complex structures. The ability to faithfully predict the 3D structure of single stranded DNA and RNA from sequence has the potential to revolutionize the way aptamers are selected and allow for crucial applications not only into aptamer design but also for biosensor set-up design and the molecular level understanding of structure and dynamics of single stranded oligonucleotide systems23.
Here, we present the first approach to predict the three-dimensional structures of single stranded DNA required for aptamer applications that extends current sequence-based computational efforts for RNA18–21. Our approach faithfully predicts the representative resolved structures available in the Nucleic Acid Database (NDB) and Protein Data Bank (PDB) databases from a pipeline that integrates 2D and 3D structural tools, including Mfold, Assemble 2, Chimera, VMD, and Molecular Dynamics (MD) simulations. Explicitly, we build ssDNA secondary structure from sequence, construct equivalent 3D ssRNA models, transform the 3D ssRNA models into ssDNA 3D structures, and finally refine the resulting ssDNA 3D structures through energy minimization. To thoroughly evaluate our approach, we considered all hairpin-like ssDNA molecules with experimentally solved 3D structures selected through an exhaustive search for ssDNA molecules and aptamers in the PDB database. Our results indicate that this approach works exceptionally well for the hairpin-like structural motif of ssDNA, the focus of the current work. To test the robustness of the results, we performed additional atomistic MD simulations for a sub-set of representative ssDNA molecules and aptamers. The atomistic details available in MD simulations with explicit solvent have been fundamental at uncovering the molecular level mechanisms of key experimental observations and deepen our understanding of the interactions and properties of biological complexes in their natural environment24–28. Partly because of the lack of solved 3D structures, very few MD simulation studies have focused on aptamers29, 30. Our results show that MD simulations can, indeed, be used to further improve the structural predictions and that the predictions are representative of those obtained from the dynamics of the systems under conditions that mimic their targeted environment.
Methods
Workflow for three-dimensional structure generation from sequence
The workflow to construct the 3D structures of the ssDNA molecules from the nucleotide sequence consists of four main steps (Fig. 1), comprising building the DNA secondary structure using Mfold in step 1, constructing refined equivalent 3D RNA models using Assemble2 and Chimera in step 2, translating the 3D RNA models into DNA models using VMD in step 3, and refining the final 3D DNA structure through minimization using VMD in step 4, as described below. Typically, this process takes about one hour to complete. Illustrative examples of the structures obtained at the end of each of the four steps of the workflow are shown in Supplementary Figure 1.
Figure 1.
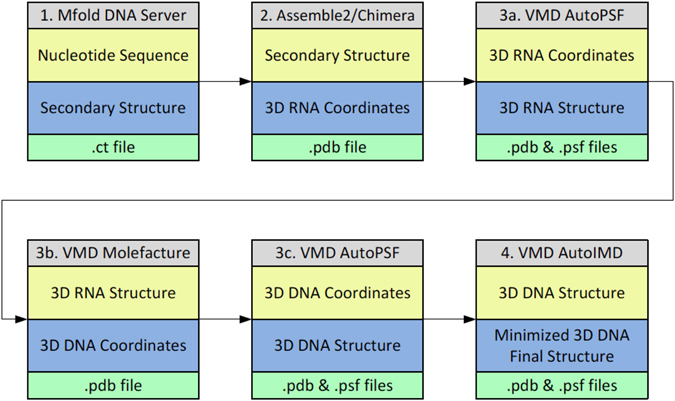
Workflow used to construct the ssDNA 3D structures from the sequence. The approach consists of four main steps, involving building the ssDNA secondary structure from the sequence using Mfold (step 1), constructing refined equivalent 3D ssRNA models using Assemble2/Chimera (step 2), translating the 3D ssRNA models into ssDNA models using VMD (step 3), and refining the 3D ssDNA structures using VMD (step 4).
Step 1: Build ssDNA secondary structure from sequence
The first step of our approach is the prediction of secondary structures from DNA sequences. The input consists of the DNA sequences, which for this study were selected after searching the PDB database with a focus on the hairpin-like common fold of ssDNA, as detailed in the results section. Starting with the nucleotide sequence, the secondary structures of the ssDNA molecules were predicted using the mfold web server (http://mfold.rna.albany.edu/?=mfold)31, based on free energy minimization techniques. In mfold, all possible secondary structures are approximated based on Watson-Crick base pairing and the most thermodynamically stable structures are selected32, 33. The initial sequences were selected as linear at a temperature of 37 °C and ionic concentration of 1 M of Na+, 0 M of Mg2+, computing only fold configurations within 5% from the minimum free energy, and considering a maximum number of 50 folds with no limit to the maximum distance between paired bases. In addition to the predicted secondary structure of the ssDNA this step provides the minimum free energy of the fold.
Step 2: Construct equivalent 3D ssRNA models and refine structures
The second step is to use the predicted secondary structures as a starting point to generate the 3D structures of the equivalent ssRNA models using Assemble2/Chimera. The 3D structures were modeled and visualized using Assemble234 and Chimera35 following a manual process of individually selecting the 2D helical and non-helical elements of the ssDNA molecule and translating the residues into equivalent 3D RNA models. The 3D RNA models were then refined using 100 iterations to remove geometric deficiencies and optimize the structural parameters such as bond length and angles, planarity of certain groups, non-bonded contacts, and restricted torsion angles. The refinement was achieved by the geometrical least squares method using the Konnert–Hendrickson algorithm36 as implemented in the Assemble2 program.
Step 3: Transform the 3D ssRNA models into ssDNA 3D structures
In the third step, the refined ssRNA 3D structures were imported into VMD37, where hydrogen atoms were added using the AutoPSF VMD plugin (step 3a), and the ssRNAs modified into ssDNA 3D structures by: (i) identifying each uracil residue and replacing the H5 atom with a methyl group using the Molefacture VMD plugin (step 3b) and (ii) replacing the ribose sugar backbone with deoxyribose using the AutoPSF VMD plugin and manually renaming the modified uracil residues to thymine in the pdb file (step 3c). The output of this step consists of both pdb and psf files that contain the atomic coordinates (.pdb) and atom type, charge, mass and bonding information (.psf).
Step 4: Refine final ssDNA 3D structures
The fourth step of our approach consists of further refinement of the ssDNA 3D structures obtained in step 3. It uses the AutoIMD VMD plugin to refine the structures through energy minimization using 10,000 iterations. The output of this step consists of the final coordinate (.pdb) file in addition to the psf file obtained in the previous step. We will use these two files, in addition to files containing the topology of the molecules and the force-field parameters, for conducting further analysis through molecular dynamics simulations as detailed below.
Molecular Dynamics Simulations
Description of the systems and initial structure set-up
We carried out molecular dynamics simulation studies for 5 ssDNA sequences from the pool of 24 structures selected after an exhaustive search from the PDB database, as detailed in the results section, using as initial configurations our predicted 3D structure and the corresponding experimentally resolved structure from the PDB database. For the sake of simplicity, throughout this paper, the five original ssDNA resolved through conventional experimental methods will be referred to as “original” and those derived here using computational modeling methods will be referred to as “predicted”. They are labeled using their corresponding PDB IDs. The predicted and original structures for the single stranded DNA hairpins corresponding to the PDB ID entries 1BJH, 1LA8, 2M8Y, 2VAH, and 2L5K were each solvated in a water box to closer represent the biological as well as the biosensor environment. In the case of the original systems, first we used the AutoPSF VMD plugin to add the hydrogen atoms to the ssDNAs, missing from the NMR solution structures. Using the Solvate plugin in VMD37, a layer of water of about 25 Å in each direction from the atom with the largest coordinate in that direction was created to fully immerse the ssDNA molecules. In addition, each system was neutralized by replacing a predetermined number of water molecules with sodium ions. Table 1 contains the resulting dimensions of the simulation cells as well as the type and number of ions used to neutralize each system.
Table 1.
Dimensions of the simulation box, number of atoms of the ssDNA molecules, ion type and number of ions in the system, and total number of atoms for each of the 10 different molecular dynamics simulations.
| Structure | L X (Å) | L Y (Å) | L Z (Å) | ssDNA atoms | Ion Type | # of Ions | Total Atoms |
|---|---|---|---|---|---|---|---|
| 1BJH original | 70 | 74 | 71 | 349 | Na+ | 10 | 32780 |
| 1BJH predicted | 75 | 73 | 70 | 349 | Na+ | 10 | 33947 |
| 1LA8 original | 72 | 79 | 77 | 411 | Na+ | 12 | 39720 |
| 1LA8 predicted | 72 | 72 | 78 | 411 | Na+ | 12 | 36867 |
| 2M8Y original | 74 | 77 | 78 | 474 | Na+ | 14 | 39899 |
| 2M8Y predicted | 74 | 71 | 81 | 474 | Na+ | 14 | 38411 |
| 2VAH original | 72 | 74 | 82 | 569 | Na+ | 17 | 38974 |
| 2VAH predicted | 74 | 74 | 87 | 569 | Na+ | 17 | 42901 |
| 2L5K original | 75 | 75 | 94 | 728 | Na+ | 22 | 47412 |
| 2L5K predicted | 75 | 75 | 94 | 728 | Na+ | 22 | 46119 |
Molecular Dynamics Simulation Details
We carried out the ten different simulations using the atomistic molecular dynamics (MD) simulation code NAMD38. The details of each system are summarized in Table 1. All the MD simulations were carried out using the NAMD2.9 software package with the recent version of the all-atom CHARMM force field for the nucleic acids39 (CHARMM27) and the rigid TIP3P model for water40, which is consistent with the nucleic acids force field. Following standard procedures, the solvated ssDNA systems were first minimized for 100,000 steps using the conjugate gradient energy minimization method as implemented in NAMD. This was followed by a NVT equilibration run at a temperature of 300 K and a NPT production run of a total of 10 ns at a temperature of 300 K and a pressure of 1 atm. To maintain these conditions, we used the Langevin dynamics method with a friction constant of 1 and the Nose-Hoover Langevin piston method41. The simulations were carried out using a time step of 2 fs. In all the simulations, we used three-dimensional periodic boundary conditions and the minimum image convention42 to calculate the short-range Lennard-Jones interactions using a spherical cutoff distance of 12 Å with a switch distance of 10 Å. The long-range electrostatic interactions were calculated by using the particle-mesh Ewald (PME) method43.
Results and Discussion
Selection of ssDNA Candidates
We performed an exhaustive search for ssDNA molecules and aptamers with experimentally solved 3D structures in the Protein Data Bank database (http://www.pdb.org). The selection method consisted of two independent searches using the following keywords: “DNA hairpin” and “aptamer DNA”. This resulted in a total of 772 entries. The search results were then filtered by ‘Polymer Type’; only including entries that were classified under “DNA”. This resulted in reducing the pool of potential candidates to 97 entries as detailed in Table 2. Subsequently, 20 entries containing ligands were excluded from the list of candidates. The remaining 77 structures were then individually reviewed and 53 additional structures that did not correspond to the hairpin-like structural motif were eliminated. The exclusion criteria in this last step eliminated the following types of structures: duplex, triplex, three-way junction, dimer, dimer G-quadruplex, quadruplex duplex hybrid, complexed with non-protein ligand, Z-DNA, and non-nucleotide sequence modifications. This last step resulted in the exclusion from the list of all the DNA sequences with a G-score greater than zero as calculated using the QGRS Mapper software program44. QGRS Mapper uses a scoring system to predict the presence of quadruplex forming G-rich sequences in nucleotide sequences and non-zero scores indicate the possibility of G-quadruplex formation with higher scores representing better G-quadruplex forming candidates. The remaining pool of 24 candidates (Table 2), with 21 different sequences, was selected as our initial set of 21 DNA sequences for 3D structural prediction.
Table 2.
Protein Data Bank database search results for the selection of ssDNA candidates.
| PMID | PDB ID | Chain Length | Sequence | QGRS | Exclude | Exclusion Criteria |
|---|---|---|---|---|---|---|
| 8069626 | 134D | 31 | TCCTCCTTTTTTAGGAGGATTTTTTGGTGGT | 15 | Yes | Triplex |
| 8069626 | 135D | 31 | TCCTCCTTTTTTAGGAGGATTTTTTGGTGGT | 15 | Yes | Triplex |
| 8069626 | 136D | 31 | TCCTCCTTTTTTAGGAGGATTTTTTGGTGGT | 15 | Yes | Triplex |
| 7613864 | 184D | 7 | GCATGCT | 0 | Yes | Dimer Quadruplex |
| 9737926 | 1A8N | 12 | GGGCTTTTGGGC | 0 | Yes | Dimer Quadruplex |
| 9737927 | 1A8W | 12 | GGGCTTTTGGGC | 0 | Yes | Dimer Quadruplex |
| 9092659 | 1AC7 | 16 | ATCCTAGTTATAGGAT | 0 | No | N/A |
| 9398169 | 1AO9 | 13 | GAGAGAXTCTCTC | 0 | Yes | Complexed with non-protein ligand |
| 7664126 | 1AU6 | 8 | CATGCATG | 0 | Yes | Complexed with non-protein ligand |
| 9384529 | 1AW4 | 27 | ACCTGGGGGAGTATTGCGGAGGAAGGT | 0 | Yes | Complexed with non-protein ligand |
| 9000625 | 1BJH | 11 | GTACAAAGTAC | 0 | No | N/A |
| 9367776 | 1C11 | 11 | TCCCGTTTCCA | 0 | Yes | Dimer Quadruplex |
| 12371856 | 1CS7 | 13 | GUTTTGXCAAAAC | 0 | Yes | Complexed with non-protein ligand |
| 2299669 | 1D16 | 16 | CGCGCGTTTTCGCGCG | 0 | Yes | Z-DNA |
| 10653638 | 1DB6 | 22 | CGACCAACGTGTCGCCTGGTCG | 0 | Yes | Complexed with non-protein ligand |
| 10756190 | 1DGO | 18 | AGGATCCTUTTGGATCCT | 0 | No | N/A |
| N/A | 1ECU | 19 | GCGCGAAACTGTTTCGCGC | 0 | No | N/A |
| 10924101 | 1EN1 | 18 | GTCCCTGTTCGGGCGCCA | 0 | No | N/A |
| 11090280 | 1EZN | 36 | CGTGCACCCGCTTGCGGCGACTTGTCGTTGTGCACG | 0 | Yes | Three way junction |
| 11790144 | 1FV8 | 11 | TATCATCGATA | 0 | Yes | Non-nucleotide modified residues |
| 11352724 | 1G5L | 6 | CCAAAG | 0 | Yes | Complexed with non-protein ligand |
| 11352724 | 1GJ2 | 6 | CCAAAG | 0 | Yes | Complexed with non-protein ligand |
| 11952790 | 1IDX | 18 | AGGATCCTTUTGGATCCT | 0 | No | N/A |
| 11952790 | 1II1 | 18 | AGGATCCUTTTGGATCCT | 0 | No | N/A |
| 11843626 | 1JU0 | 23 | CTTGCTGAAGCGCGCACGGCAAG | 0 | Yes | Dimer |
| 11843626 | 1JUA | 23 | CTTGCTGAAGCGCGCACGGCAAG | 0 | Yes | Dimer |
| 11991355 | 1JVE | 27 | CCTAATTATAACGAAGTTATAATTAGG | 0 | No | N/A |
| 12449414 | 1KR8 | 7 | GCGAAGC | 0 | No | N/A |
| 11895443 | 1L0R | 14 | ACGAAGTGCGAAGC | 0 | Yes | Complexed with non-protein ligand |
| 11849039 | 1LA8 | 13 | CGCGGTGTCCGCG | 0 | No | N/A |
| 11849039 | 1LAE | 13 | CGCGGTPTCCGCG | 0 | Yes | Non-nucleotide modified residues |
| 11849038 | 1LAI | 13 | CGCGGTGTCCGCG | 0 | Yes | Duplex |
| 11849038 | 1LAQ | 13 | CGCGGTPTCCGCG | 0 | Yes | Duplex |
| 11849038 | 1LAS | 13 | CGCGGTPTCCGCG | 0 | Yes | Duplex |
| 12560479 | 1MF5 | 7 | GCATGCT | 0 | Yes | Dimer Quadruplex |
| 12564921 | 1MP7 | 10 | GCCAGAGAGC | 0 | Yes | Complexed with non-protein ligand |
| 12755609 | 1NGO | 27 | CTCTTTTTGTAAGAAATACAAGGAGAG | 0 | No | N/A |
| 12755609 | 1NGU | 27 | CTCTCCTTGTATTTCTTACAAAAAGAG | 0 | No | N/A |
| 12758081 | 1P0U | 13 | GCATCGACGATGC | 0 | No | N/A |
| 8525381 | 1PNN | 24 | GAAGAAGAG | 0 | Yes | Triplex |
| 12449414 | 1PQT | 7 | GCGAAGC | 0 | No | N/A |
| 12952463 | 1PUY | 13 | GTTTTGXCAAAAC | 0 | Yes | Complexed with non-protein ligand |
| 10481034 | 1QE7 | 22 | CTAGAGGATCCTTTUGGATCCT | 0 | No | N/A |
| N/A | 1QYK | 7 | GCATGCT | 0 | Yes | Dimer Quadruplex |
| N/A | 1QYL | 7 | GCATGCT | 0 | Yes | Dimer Quadruplex |
| 15199171 | 1SNJ | 36 | CGTGCAGCGGCTTGCCGGCACTTGTGCTTCTGCACG | 0 | Yes | Three way junction |
| 14684897 | 1UE2 | 9 | GCGAAAGCT | 0 | Yes | Duplex |
| 14684897 | 1UE3 | 8 | GCGAAAGC | 0 | Yes | Duplex |
| 8901550 | 1XUE | 17 | GTGGAATGCAATGGAAC | 0 | No | N/A |
| 7583654 | 1ZHU | 10 | CAATGCAATG | 0 | No | N/A |
| 8548453 | 229D | 17 | CCAGACUGAAGAUCUGG | 0 | Yes | Non-nucleotide modified residues |
| 9818148 | 2ARG | 30 | TGACCAGGGCAAACGGTAGGTGAGTGGTCA | 18 | Yes | Complexed with non-protein ligand |
| 16620121 | 2AVH | 11 | GGGGTTTGGGG | 0 | Yes | G-Quadruplex |
| N/A | 2F1Q | 42 | GCACTGCATCCTTGGACGCTTGCGCCACTTGTGGTGCAGTGC | 0 | Yes | Four way junction |
| 16866556 | 2GKU | 24 | TTGGGTTAGGGTTAGGGTTAGGGA | 42 | Yes | G-Quadruplex |
| N/A | 2K67 | 17 | TTAATTTNNNAAATTAA | 0 | Yes | Non-nucleotide modified residues |
| N/A | 2K68 | 17 | TTAATTTNNNAAATTAA | 0 | Yes | Non-nucleotide modified residues |
| N/A | 2K69 | 17 | TTAATTTNNNAAATTAA | 0 | Yes | Non-nucleotide modified residues |
| 19374420 | 2K71 | 8 | GCGAAAGC | 0 | No | N/A |
| 19321501 | 2K8Z | 8 | TCGTTGCT | 0 | Yes | Dimer |
| 19070621 | 2KAZ | 13 | GGGACGTAGTGGG | 0 | Yes | Dimer Quadruplex |
| 21410196 | 2L13 | 13 | TATTATXATAATA | 0 | Yes | Non-nucleotide modified residues |
| 22129448 | 2L5K | 23 | CAGTTGATCCTTTGGATACCCTG | 0 | No | N/A |
| 22507054 | 2LO5 | 12 | GGCCGCAGTGCC | 0 | No | N/A |
| 22507054 | 2LO8 | 10 | GCCGCAGTGC | 0 | No | N/A |
| 22507054 | 2LOA | 10 | GCCGCAGTGC | 0 | Yes | Complexed with non-protein ligand |
| 22798499 | 2LSC | 12 | CGCGAAUUCGCG | 0 | Yes | Complexed with non-protein ligand |
| 23794476 | 2M8Y | 15 | CGCGAAGCATTCGCG | 0 | No | N/A |
| 23794476 | 2M8Z | 27 | GGTTGGCGCGAAGCATTCGCGGGTTGG | 9 | Yes | Quadruplex Duplex Hybrid |
| 23794476 | 2M90 | 32 | GCGCGAAGCATTCGCGGGGAGGTGGGGAAGGG | 21 | Yes | Quadruplex Duplex Hybrid |
| 23794476 | 2M91 | 30 | GGGAAGGGCGCGAAGCATTCGCGAGGTAGG | 7 | Yes | Quadruplex Duplex Hybrid |
| 23794476 | 2M92 | 34 | AGGGTGGGTGCTGGGGCGCGAAGCATTCGCGAGG | 17 | Yes | Quadruplex Duplex Hybrid |
| 23794476 | 2M93 | 32 | TTGGGTGGGCGCGAAGCATTCGCGGGGTGGGT | 29 | Yes | Quadruplex Duplex Hybrid |
| 8658168 | 2NEO | 19 | CCCGATGCXGCAATTCGGG | 0 | Yes | Complexed with non-protein ligand |
| 17362008 | 2O3M | 22 | AGGGAGGGCGCTGGGAGGAGGG | 39 | Yes | G-Quadruplex |
| 17388570 | 2OEY | 25 | CCATCGTCTACCTTTGGTAGGATGG | 0 | Yes | Complexed with non-protein ligand |
| 9020982 | 2PIK | 23 | CACTCCTGGTTTTTCCAGGAGTG | 0 | Yes | Complexed with non-protein ligand |
| 18515837 | 2VAH | 18 | AGGATCCTUTTGGATCCT | 0 | No | N/A |
| 18515837 | 2VAI | 18 | AGGATCCTUTTGGATCCT | 0 | No | N/A |
| 22287624 | 3QXR | 22 | AGGGAGGGCGCUGGGAGGAGGG | 39 | Yes | G-Quadruplex |
| N/A | 3T86 | 7 | GCATGCT | 0 | Yes | Complexed with non-protein ligand |
| 22409313 | 4DKZ | 12 | CGCGAAXXCGCG | 0 | Yes | Non-nucleotide modified residues |
| 8107090 | 148D | 15 | GGTTGGTGTGGTTGG | 20 | Yes | G-Quadruplex |
| 9384529 | 1AW4 | 27 | ACCTGGGGGAGTATTGCGGAGGAAGGT | 14 | Yes | Complexed with non-protein ligand |
| 9799703 | 1BUB | 15 | GGTTGGTGTGGTTGG | 20 | Yes | G-Quadruplex |
| 10756199 | 1C32 | 15 | GGTTGGTGTGGTTGG | 20 | Yes | G-Quadruplex |
| 10756199 | 1C34 | 15 | GGTTGGTGTGGTTGG | 20 | Yes | G-Quadruplex |
| 10756199 | 1C35 | 15 | GGTTGGTGTGGTTGG | 20 | Yes | G-Quadruplex |
| 10756199 | 1C38 | 15 | GGTTGGTGTGGTTGG | 20 | Yes | G-Quadruplex |
| 10653638 | 1DB6 | 22 | CGACCAACGTGTCGCCTGGTCG | 0 | Yes | Complexed with non-protein ligand |
| 8757801 | 1QDF | 15 | GGTTGGTGTGGTTGG | 20 | Yes | G-Quadruplex |
| 8757801 | 1QDH | 15 | GGTTGGTGTGGTTGG | 20 | Yes | G-Quadruplex |
| 15214802 | 1RDE | 15 | GGTTGGTGTGGTTGG | 20 | Yes | G-Quadruplex |
| 15637158 | 1Y8D | 16 | GGGGTGGGAGGAGGGT | 21 | Yes | Dimer Quadruplex |
| 9818148 | 2ARG | 30 | TGACCAGGGCAAACGGTAGGTGAGTGGTCA | 18 | Yes | Complexed with non-protein ligand |
| 17145716 | 2IDN | 15 | GGTTGGTGTGGTTGG | 20 | Yes | G-Quadruplex |
| 23935071 | 2M53 | 25 | TGTGGGGGTGGACGGGCCGGGTAGA | 21 | Yes | G-Quadruplex |
The details of the selection criteria are discussed in the text (see results section). For each experimentally resolved structure from the PDB database, denoted by its PDB ID, the table provides the corresponding publication denoted by its PubMed identifier (PMID), the sequence as provided in the PDB database, the chain length, the G-score denoted by QGRS obtained using the QGRS Mapper, and, if the DNA candidate was excluded from our list of potential candidates, the exclusion criteria.
The 24 ssDNA molecules had 21 unique sequences ranging in length from 7 to 27 nucleotides, all with structures solved by NMR spectroscopy, and comprising a wide variety of systems of biological and biomedical interest, ranging from models of biologically relevant sequences, such as those to which HIV-1 nucleocapsid protein binds during reverse transcription45 or those with uracil (a constituent of RNA) bases46, to DNA aptamers, such as those that bind Mucin 1, a cell-surface glycoprotein overexpressed in a number of cancers47.
In addition, five representative ssDNA hairpins (Table 2; entries in bold typesetting), with PDB IDs 1BJH48, 1LA849, 2M8Y50, 2VAH46, and 2L5K47, were selected from the 24 candidates for additional analysis through atomistic molecular dynamics simulations. The length of these molecules ranges from 11 (1BJH) to 24 (2L5K) nucleotides.
Three-dimensional Structure Prediction from Sequence
The approach followed, as indicated in the workflow (Fig. 1) and detailed in the methods section, successfully captures the 3D structures predicted from the 21 different sequences of the 24 ssDNA hairpins identified from the PDB database. In order to test the accuracy of the 3D prediction method, each predicted ssDNA structure was aligned with the corresponding ssDNA structure downloaded from the PDB database. To measure the degree of similarity, the corresponding Root Mean Square Deviation (RMSD) of the sugar-phosphate backbone between each pair of structures was calculated. Figure 2 shows an overlay of each of the 21 predicted 3D structures (ssDNA colored red) and the corresponding 24 NMR structures downloaded from the PDB database (ssDNA colored blue), together with calculated values of the RMSDs. Our results indicate that our approach is able to faithfully predict the 3D structure of a wide variety of ssDNA hairpins, with sequences ranging from 7 to 27 nucleotides. In all cases, the RMSD values obtained ranged from 1.9 Å (PDB ID: 1PQT) for the shortest sequences to 8.59 Å (1NGU) for the longest cases, and were typically around 3.5 Å, with an average value of 4 Å. Interestingly, our approach was also able to accurately predict the 3D structure for ssDNA sequences containing uracil bases, namely 1DGO, 1IDX, 1II1, 1QE7, 2VAH, and 2VAI. Three particular predicted structures, namely 2LO8, 1EN1, and 1NGU, differed more than those corresponding to typical values we obtained for other ssDNAs with similar sequence lengths. Figure 3 shows the RMSD values obtained for the 24 predicted structures with respect to the experimental ones as a function of chain length of the DNA. For instance, the RMSD obtained for 2LO8 with a chain length of 10 nucleotides is 5.53 Å, while the corresponding value for 1ZHU is 3.05 Å. Interestingly, the sequence 2LO8 is only 10nt, and is part of 2LO5. However, its RMSD is bigger than the latter because the two additional bases in 2LO5 make a CG base-pairing and increase the stability of the loop. In the case of 1EN1, the higher RMSD value (compared for instance with the value of 3.01 obtained for 1II1 with also 18 nucleotides) mostly originates from the unstructured tail at the 3’ end of the hairpin, which was predicted as more structured. In this case, additional molecular dynamics simulations, as the ones we present in the next section for five of the ssDNA sequences, are likely to improve the prediction of the model by relaxing the positions of the nucleotides at the tail.
Figure 2.
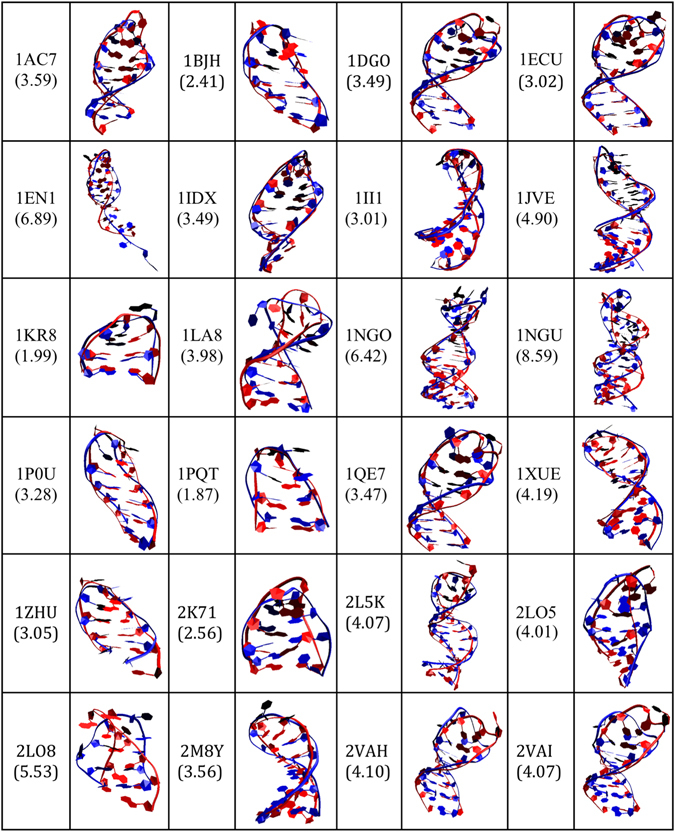
Overlay of the 3D predicted structures (ssDNA colored red) and the corresponding experimental structures downloaded from the PDB database (ssDNA colored blue) for the 24 ssDNA hairpin structures. Each structure is labeled by its corresponding PDB ID and the calculated RMSD values (in Angstroms) are shown in parenthesis.
Figure 3.
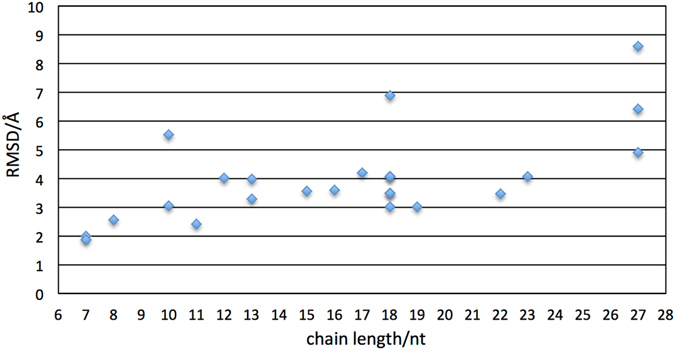
Values of the RMSD for the 24 ssDNA predicted structures with respect to the experimental ones as a function of the nucleotide chain length.
The results obtained for the 3D predictions of the five ssDNA hairpins selected from the pool of 24 candidates for additional analysis through atomistic molecular dynamics simulations, namely 1BJH, 1LA8, 2M8Y, 2VAH, and 2L5K, were in good agreement with the experimental data. Specifically, we obtained RMSD values of ranging from ~2.4 Å for 1BJH with 11 nucleotides to ~4.1 Å for 2VAH and 2L5K with 18 and 23 nucleotides, respectively. In these five cases, the secondary structure prediction obtained in step 1 of our approach using Mfold as described in the methods section resulted in the following: 1BJH is characterized by a minimum free energy of 0.04 kcal/mol and consists of a 4-base pair stacked stem and a 3-nucleotide hairpin loop; 1LA8 is characterized by a minimum free energy of −4.9 kcal/mol and consists of a 5-base pair stacked stem and a 3-nucleotide hairpin loop; 2M8Y is characterized by a minimum free energy of −6.3 kcal/mol and consists of a 6-base pair helical stacked stem followed by a 3-nucleotide hairpin loop; 2VAH is characterized by a minimum free energy of −6.2 kcal/mol and consists of a 7-base pair helical stacked stem and a 4-nucleotide hairpin loop; and 2L5K is characterized by a minimum free energy of 0.05 kcal/mol and consists of a 3-base pair stacked stem attached to a 4-base pair stacked stem by two strings of 3-base single stranded nucleotides, followed by a 3-nucleotide hairpin loop. As in the other cases, the predicted secondary structures of these five ssDNA molecules are in agreement with the resolved structures available through the Protein Data Bank database.
Refinement of 3D Predictions and Dynamics through Molecular Dynamics Simulations
We have used fully atomistic molecular dynamics (MD) simulations to further improve our structural predictions for the 1BJH, 1LA8, 2M8Y, 2VAH, and 2L5K cases and study the dynamics of the systems. For these cases, the 3D structural predictions obtained from sequence (Fig. 2) were solvated in water and used as initial configurations of the molecular dynamics simulations, as detailed in the methods section. Figure 4 shows the initial configuration of the MD simulation corresponding to the 1LA8 ssDNA structure, solvated in water containing 12 sodium ions. In all five cases, the DNA structures evolve as a function of time during the 10-ns length of the simulations. In Fig. 5, we display three snapshots for each of the five different ssDNAs obtained from the simulations at different time intervals, which correspond to 1 ns, 5 ns, and 10 ns. Our results indicate that, in all cases, the hairpin structure of the ssDNAs remains stable during the 10 ns of the simulations. The overlays of Fig. 5 correspond to the ssDNA structures at the different time intervals (ssDNA colored red) with respect to the corresponding structure downloaded from the PDB database (ssDNA colored blue). The corresponding RMSD values calculated at different times along the 10-ns simulation, corresponding to 1 ns, 2 ns, 3 ns, 4 ns, 5 ns, 6 ns, 7 ns, 8 ns, and 9 ns, are summarized in Table 3. These values are typical and in the same range as values used to demonstrate similarity of proteins51, 52. The quantitative RMSD calculation and the qualitative visual comparison of the pairs of structures show that the approach successfully predicts the three-dimensional structure of ssDNA hairpins.
Figure 4.

Initial configuration of the MD simulation of the predicted 3D ssDNA structure for the sequence CGCGGTGTCCGCG, corresponding to 1LA8. Only the molecules within the central simulation cell are shown. The ssDNA molecule was solvated in water and the system charge neutralized with 12 sodium ions. The atoms of the water molecules are represented as white (hydrogen) and red (oxygen) lines and the sodium ions are displayed as yellow spheres with radii corresponding to its atomic van der Waals radius. The ssDNA molecule is shown in blue with the backbone and bases represented as new ribbons and the additional components represented as lines. The unit cell dimensions are L x = 72 Å, L y = 72 Å, and L z = 78 Å.
Figure 5.
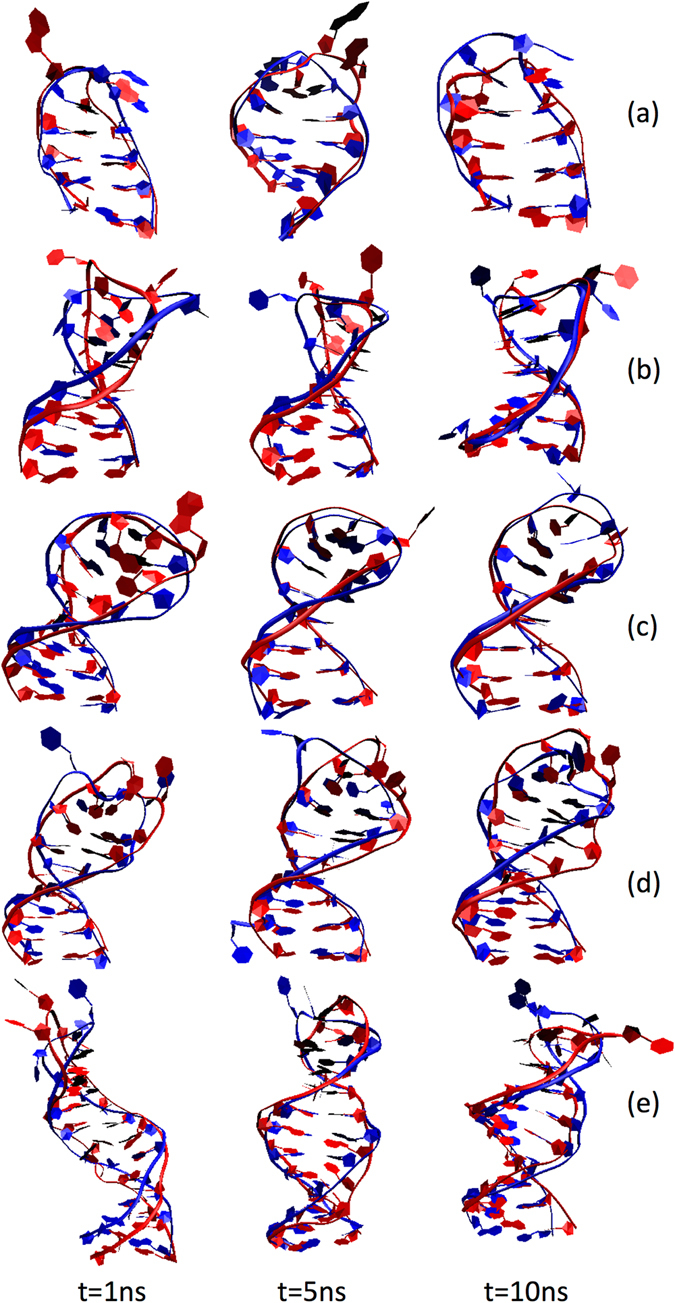
Evolution of the ssDNA molecules as a function of time. The overlays of the predicted structures taken from the MD simulations (ssDNA colored red) and the corresponding experimental structures downloaded from the PDB database (ssDNA colored blue) correspond to (a) 1BJH, (b) 1LA8, (c) 2M8Y, (d) 2VAH, and (e) 2L5K. The snapshots for the predicted ssDNA molecules correspond to (from left to right) 1 ns, 5 ns, and 10 ns. The corresponding RMSD values are summarized in Table 3. Water molecules and ions within the simulation cell are not shown.
Table 3.
RMSD values (in Angstroms) of the sugar-phosphate backbone for the predicted ssDNA structures at different time points along the MD simulations with respect to the corresponding experimental structures downloaded from the PDB database.
| 1BJH | 1LA8 | 2M8Y | 2VAH | 2L5K | |
|---|---|---|---|---|---|
| 3D-Prediction | 2.41 | 3.98 | 3.56 | 4.10 | 4.07 |
| 1 ns | 2.00 | 3.44 | 1.46 | 3.16 | 7.02 |
| 2 ns | 4.40 | 4.28 | 1.56 | 3.06 | 5.77 |
| 3 ns | 2.11 | 2.73 | 2.04 | 3.26 | 4.69 |
| 4 ns | 2.36 | 3.69 | 2.05 | 3.36 | 4.69 |
| 5 ns | 2.17 | 2.95 | 1.62 | 3.07 | 2.79 |
| 6 ns | 2.47 | 2.92 | 1.35 | 3.53 | 2.90 |
| 7 ns | 3.22 | 1.78 | 1.65 | 2.93 | 3.59 |
| 8 ns | 2.64 | 2.28 | 2.90 | 2.84 | 3.12 |
| 9 ns | 3.32 | 2.32 | 1.85 | 2.69 | 4.32 |
The corresponding values for the 3D predictions (Fig. 2) are included for completeness.
Similar MD simulations were performed using the experimentally resolved structures as initial configurations, following the approach detailed in the methods section. To explore the structural deviation of the ssDNA molecules with respect to their corresponding starting structure, the RMSD of the sugar-phosphate backbone of each structure was calculated independently over the course of the 10-ns trajectory. The RMSDs provides valuable information on the conformational flexibility of the biomolecule and provides a means to evaluate the structural deviation of the biomolecule through time. The results of the RMSD studies (Fig. 6) show that the predicted structures have similar time-dependent behavior in comparison to the original structures.
Figure 6.
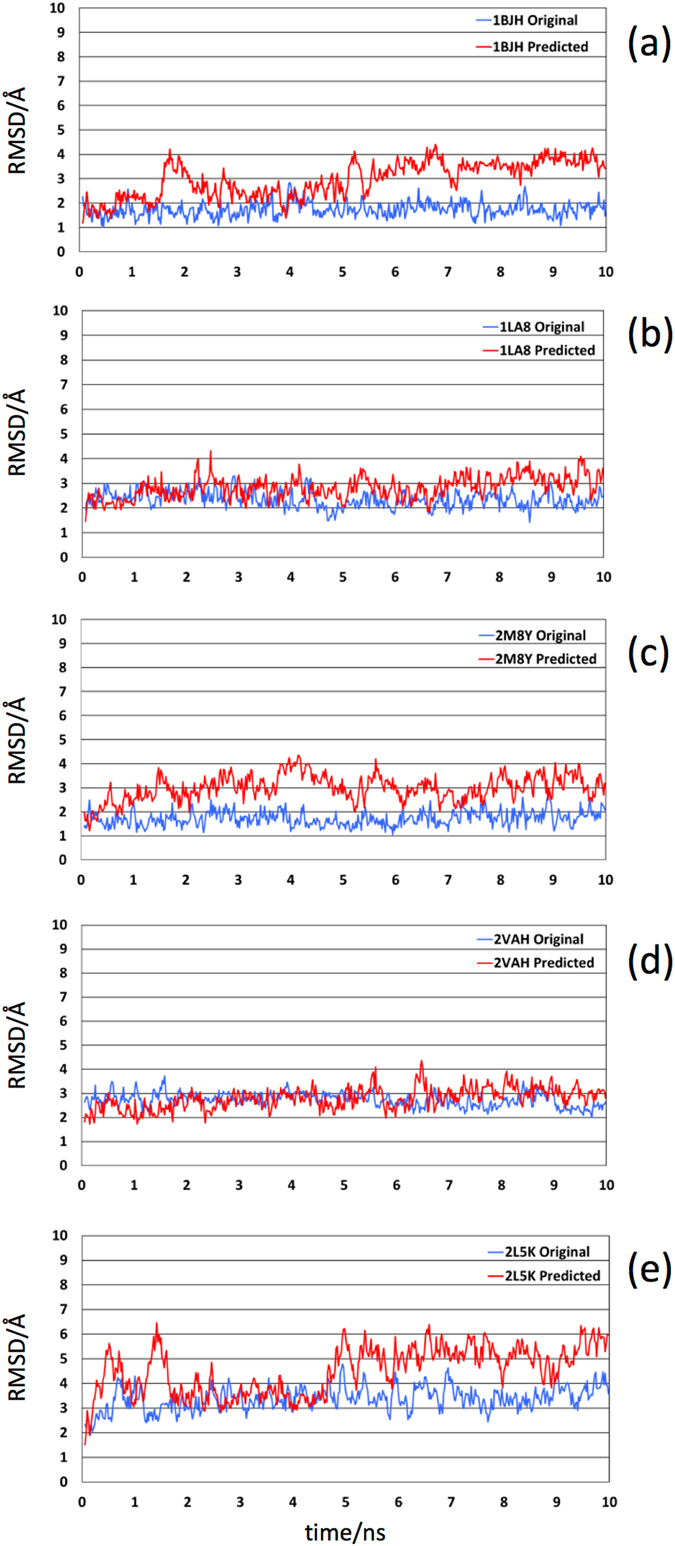
Time evolution of the RMSDs for the sugar-phosphate backbone with respect to the structures at time 0 for the predicted (red curves) and original (blue curves) structures. The panels correspond to (a) 1BJH, (b) 1LA8, (c) 2M8Y, (d) 2VAH, and (e) 2L5K.
Conclusions
DNA aptamers are more stable than their RNA counterparts, which is especially relevant for biomedical applications, but lack the wide range of computational tools for structural predictions currently available for single stranded RNA. Here, we have presented the first approach to predict the prototypical 3D structures of ssDNA required for aptamer applications. So far, there has not been a computational tool available for 3D structure prediction of DNA aptamers from their sequence and very few structures have been resolved experimentally. These limitations have constituted a major bottleneck for the emergent application of aptamers in biotechnology and for clinical use in biomedical applications. Our results strongly support that it is possible to accurately determine the structure of single-stranded DNA from sequence and provide an approach that works exceptionally well for the hairpin-like structural motif.
We have shown that our approach faithfully predicts representative structures available in the Protein Data Bank and Nucleic Acids databases. Specifically, we have extensively validated the prediction capabilities of our approach against a pool of 24 ssDNA molecules with experimentally solved 3D structures selected through an exhaustive search for ssDNA molecules and aptamers in the PDB database (http://www.pdb.org), with sequences ranging from 7 to 27 nucleotides. The studied cases are representative of a wide variety of systems of biological and biomedical interest, including Mucin 1-binding aptamers and ssDNAs with uracil bases. The resulting structures can subsequently be used as inputs in computational docking methods to study the interactions with ligands53.
We have shown that atomistic MD simulations can be used to further improve the structural predictions by studying the dynamics of the systems under conditions that mimic their targeted environment. Our results indicate that our approach works exceptionally well for the most common hairpin-like structural motif of ssDNA and it is expected to work for other systems with similar interactions between bases, like bulge loops and internal loops. Our results break the ground for future work to expand the applicability of our approach to other ssDNA folds, including G-quadruplex folds, typical of G-rich sequences such as those in the thrombin-binding aptamer, as well as the effect of covalent modifications to increase the stability of aptamers. For instance, it has been shown that phosphorothioate substitutions can substantially alter RNA conformation54.
Understanding the specific interactions involved in stabilizing biomolecular complexes immobilized on solid substrates is essential for designing biosensors with improved sensitivity, specificity, and reliability. Several key studies have shown that the sensitivity of aptamer-based biosensors is related to the surface crowding of the immobilized aptamers on the biosensor surface11, 55, 56 and have suggested that steric hindrance or electrostatic repulsion of the charged aptamer molecules at higher concentrations on the surface of the biosensor have an impact on biosensor performance. However, this area is still poorly understood because of the lack of understanding of the molecular level interactions occurring at the biosensor surface. Furthermore, understanding how small sequence changes can lead to a difference in affinity can be used to make marked improvements in biosensor performance by employing rational design techniques57–60. Our approach, therefore, provides a much-needed starting point to gain better insights in the performance of aptamer-based biosensors with the aim of improving biosensor performance by rational design techniques.
Electronic supplementary material
Acknowledgements
This work was supported by the University of California, Davis (to L.S.).
Author Contributions
L.S. conceived research; I.J. and L.S. designed research; I.J. performed the steps of the workflow and computer simulations; I.J. and L.S. analyzed the data; I.J. and L.S. wrote the paper; L.S. supervised the project.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-01348-5
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Karlsson AC, et al. Comparison of the ELISPOT and cytokine flow cytometry assays for the enumeration of antigen-specific T cells. Journal of immunological methods. 2003;283:141–153. doi: 10.1016/j.jim.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 2.Cox JH, Ferrari G, Janetzki S. Measurement of cytokine release at the single cell level using the ELISPOT assay. Methods. 2006;38:274–282. doi: 10.1016/j.ymeth.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 3.Tuleuova N, et al. Development of an aptamer beacon for detection of interferon-gamma. Analytical chemistry. 2010;82:1851–1857. doi: 10.1021/ac9025237. [DOI] [PubMed] [Google Scholar]
- 4.Jayasena SD. Aptamers: an emerging class of molecules that rival antibodies in diagnostics. Clinical chemistry. 1999;45:1628–1650. [PubMed] [Google Scholar]
- 5.Liu J, Cao Z, Lu Y. Functional nucleic acid sensors. Chemical reviews. 2009;109:1948–1998. doi: 10.1021/cr030183i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 7.Nutiu R, Li Y. Aptamers with fluorescence-signaling properties. Methods. 2005;37:16–25. doi: 10.1016/j.ymeth.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 8.Balamurugan S, Obubuafo A, Soper SA, Spivak DA. Surface immobilization methods for aptamer diagnostic applications. Analytical and bioanalytical chemistry. 2008;390:1009–1021. doi: 10.1007/s00216-007-1587-2. [DOI] [PubMed] [Google Scholar]
- 9.Kim B, Jung IH, Kang M, Shim HK, Woo HY. Cationic conjugated polyelectrolytes-triggered conformational change of molecular beacon aptamer for highly sensitive and selective potassium ion detection. Journal of the American Chemical Society. 2012;134:3133–3138. doi: 10.1021/ja210360v. [DOI] [PubMed] [Google Scholar]
- 10.Juskowiak B. Nucleic acid-based fluorescent probes and their analytical potential. Analytical and bioanalytical chemistry. 2011;399:3157–3176. doi: 10.1007/s00216-010-4304-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Y, Tuleouva N, Ramanculov E, Revzin A. Aptamer-based electrochemical biosensor for interferon gamma detection. Analytical chemistry. 2010;82:8131–8136. doi: 10.1021/ac101409t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rowe AA, White RJ, Bonham AJ, Plaxco KW. Fabrication of electrochemical-DNA biosensors for the reagentless detection of nucleic acids, proteins and small molecules. Journal of visualized experiments. 2011;52:2922. doi: 10.3791/2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou W, Jimmy Huang PJ, Ding J, Liu J. Aptamer-based biosensors for biomedical diagnostics. The Analyst. 2014;139:2627–2640. doi: 10.1039/c4an00132j. [DOI] [PubMed] [Google Scholar]
- 14.Keefe AD, Pai S, Ellington A. Aptamers as therapeutics. Nature reviews in drug discovery. 2010;9:537–550. doi: 10.1038/nrd3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou J, Rossi J. Aptamers as targeted therapeutics: current potential and challenges. Nat Rev Drug Discov. 2017;16:181–202. doi: 10.1038/nrd.2016.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nimjee SM, White RR, Becker RC, Sullenger BA. Aptamers as Therapeutics. Annu Rev Pharmacol Toxicol. 2017;57:61–79. doi: 10.1146/annurev-pharmtox-010716-104558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun H, et al. Oligonucleotide aptamers: new tools for targeted cancer therapy. Molecular therapy. Nucleic acids. 2014;3:e182. doi: 10.1038/mtna.2014.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cruz JA, et al. RNA-Puzzles: a CASP-like evaluation of RNA three-dimensional structure prediction. RNA. 2012;18:610–625. doi: 10.1261/rna.031054.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laing C, Schlick T. Computational approaches to RNA structure prediction, analysis, and design. Current opinion in structural biology. 2011;21:306–318. doi: 10.1016/j.sbi.2011.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Das R, Karanicolas J, Baker D. Atomic accuracy in predicting and designing noncanonical RNA structure. Nature methods. 2010;7:291–294. doi: 10.1038/nmeth.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dufour D, Marti-Renom MA. Software for predicting the 3D structure of RNA molecules. WIREs Comput Mol Sci. 2015;5:56–61. doi: 10.1002/wcms.1198. [DOI] [Google Scholar]
- 22.Schlick, T. Molecular Modeling and Simulation: An Interdisciplinary Guide. 2nd edn, (2010).
- 23.Mortimer SA, Kidwell MA, Doudna JA. Insights into RNA structure and function from genome-wide studies. Nature reviews. Genetics. 2014;15:469–479. doi: 10.1038/nrg3681. [DOI] [PubMed] [Google Scholar]
- 24.Saiz L, Bandyopadhyay S, Klein ML. Towards an understanding of complex biological membranes from atomistic molecular dynamics simulations. Bioscience reports. 2002;22:151–173. doi: 10.1023/A:1020130420869. [DOI] [PubMed] [Google Scholar]
- 25.Saiz L. The physics of protein-DNA interaction networks in the control of gene expression. Journal of physics: Condensed matter. 2012;24:193102. doi: 10.1088/0953-8984/24/19/193102. [DOI] [PubMed] [Google Scholar]
- 26.Sinha SK, Saiz L. Determinants of protein–ligand complex formation in the thyroid hormone receptor α: a Molecular Dynamics simulation study. Computational and Theoretical Chemistry. 2014;1038:57–66. doi: 10.1016/j.comptc.2014.03.034. [DOI] [Google Scholar]
- 27.Tan H, Wei K, Bao J, Zhou X. In silico study on multidrug resistance conferred by I223R/H275Y double mutant neuraminidase. Molecular Biosystems. 2013;9:2764–2774. doi: 10.1039/c3mb70253g. [DOI] [PubMed] [Google Scholar]
- 28.Lin PH, Tsai CW, Wu JW, Ruaan RC, Chen WY. Molecular dynamics simulation of the induced-fit binding process of DNA aptamer and L-argininamide. Biotechnology journal. 2012;7:1367–1375. doi: 10.1002/biot.201200003. [DOI] [PubMed] [Google Scholar]
- 29.Shcherbinin DS, Veselovslii AV. [Investigation of interaction of thrombin-binding aptamer with thrombin and prethrombin-2 by simulation of molecular dynamics] Biofizika. 2013;58:415–424. [PubMed] [Google Scholar]
- 30.Rhinehardt KL, Srinivas G, Mohan RV. Molecular Dynamics Simulation Analysis of Anti-MUC1 Aptamer and Mucin 1 Peptide Binding. J Phys Chem B. 2015;119:6571–6583. doi: 10.1021/acs.jpcb.5b02483. [DOI] [PubMed] [Google Scholar]
- 31.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic acids research. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zuker M, Sankoff D. RNA secondary structures and their prediction. Bulletin of Mathematical Biology. 1984;46:591–621. doi: 10.1007/BF02459506. [DOI] [Google Scholar]
- 33.Zuker M. On finding all suboptimal foldings of an RNA molecule. Science. 1989;244:48–52. doi: 10.1126/science.2468181. [DOI] [PubMed] [Google Scholar]
- 34.Jossinet F, Ludwig TE, Westhof E. Assemble: an interactive graphical tool to analyze and build RNA architectures at the 2D and 3D levels. Bioinformatics. 2010;26:2057–2059. doi: 10.1093/bioinformatics/btq321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pettersen EF, et al. UCSF Chimera–a visualization system for exploratory research and analysis. Journal of computational chemistry. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 36.Hendrickson WA, Konnert JH. Diffraction analysis of motion in proteins. Biophysical journal. 1980;32:645–647. doi: 10.1016/S0006-3495(80)85007-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. Journal of molecular graphics14, 33–38, 27–38 (1996). [DOI] [PubMed]
- 38.Phillips JC, et al. Scalable molecular dynamics with NAMD. Journal of computational chemistry. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.MacKerell AD, Jr., Banavali N, Foloppe N. Development and current status of the CHARMM force field for nucleic acids. Biopolymers. 2000;56:257–265. doi: 10.1002/1097-0282(2000)56:4<257::AID-BIP10029>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 40.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. The Journal of Chemical Physics. 1983;79:926–935. doi: 10.1063/1.445869. [DOI] [Google Scholar]
- 41.Feller SE, Zhang Y, Pastor RW, Brooks BR. J. Chem. Phys. 1995;103:4693–4693. doi: 10.1063/1.470656. [DOI] [Google Scholar]
- 42.Allen, M. P. & Tildesley, D. J. Computer simulation of liquids. (Clarendon Press; Oxford University Press, 1989).
- 43.Darden T, York D, Pedersen L. Particle Mesh Ewald - an N.Log(N) Method for Ewald Sums in Large Systems. J Chem Phys. 1993;98:10089–10092. doi: 10.1063/1.464397. [DOI] [Google Scholar]
- 44.Kikin O, D’Antonio L, Bagga PS. QGRS Mapper: a web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic acids research. 2006;34:W676–682. doi: 10.1093/nar/gkl253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnson PE, et al. A mechanism for plus-strand transfer enhancement by the HIV-1 nucleocapsid protein during reverse transcription. Biochemistry. 2000;39:9084–9091. doi: 10.1021/bi000841i. [DOI] [PubMed] [Google Scholar]
- 46.Wilton DJ, Ghosh M, Chary KV, Akasaka K, Williamson MP. Structural change in a B-DNA helix with hydrostatic pressure. Nucleic acids research. 2008;36:4032–4037. doi: 10.1093/nar/gkn350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baouendi M, et al. Solution structure of a truncated anti-MUC1 DNA aptamer determined by mesoscale modeling and NMR. The FEBS journal. 2012;279:479–490. doi: 10.1111/j.1742-4658.2011.08440.x. [DOI] [PubMed] [Google Scholar]
- 48.Chou SH, Zhu L, Gao Z, Cheng JW, Reid BR. Hairpin loops consisting of single adenine residues closed by sheared A.A and G.G pairs formed by the DNA triplets AAA and GAG: solution structure of the d(GTACAAAGTAC) hairpin. Journal of molecular biology. 1996;264:981–1001. doi: 10.1006/jmbi.1996.0691. [DOI] [PubMed] [Google Scholar]
- 49.Weisenseel JP, Reddy GR, Marnett LJ, Stone MP. Structure of the 1,N(2)-propanodeoxyguanosine adduct in a three-base DNA hairpin loop derived from a palindrome in the Salmonella typhimurium hisD3052 gene. Chemical research in toxicology. 2002;15:140–152. doi: 10.1021/tx010107f. [DOI] [PubMed] [Google Scholar]
- 50.Lim KW, Phan AT. Structural basis of DNA quadruplex-duplex junction formation. Angewandte Chemie. 2013;52:8566–8569. doi: 10.1002/anie.201302995. [DOI] [PubMed] [Google Scholar]
- 51.Maiorov VN, Crippen GM. Significance of root-mean-square deviation in comparing three-dimensional structures of globular proteins. Journal of molecular biology. 1994;235:625–634. doi: 10.1006/jmbi.1994.1017. [DOI] [PubMed] [Google Scholar]
- 52.Maiorov VN, Crippen GM. Size-independent comparison of protein three-dimensional structures. Proteins. 1995;22:273–283. doi: 10.1002/prot.340220308. [DOI] [PubMed] [Google Scholar]
- 53.Dominguez C, Boelens R, Bonvin AM. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. Journal of the American Chemical Society. 2003;125:1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- 54.Smith JS, Nikonowicz EP. Phosphorothioate substitution can substantially alter RNA conformation. Biochemistry. 2000;39:5642–5652. doi: 10.1021/bi992712b. [DOI] [PubMed] [Google Scholar]
- 55.Ricci F, Lai RY, Heeger AJ, Plaxco KW, Sumner JJ. Effect of molecular crowding on the response of an electrochemical DNA sensor. Langmuir. 2007;23:6827–6834. doi: 10.1021/la700328r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.White RJ, Phares N, Lubin AA, Xiao Y, Plaxco KW. Optimization of electrochemical aptamer-based sensors via optimization of probe packing density and surface chemistry. Langmuir. 2008;24:10513–10518. doi: 10.1021/la800801v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Porchetta A, Vallee-Belisle A, Plaxco KW, Ricci F. Using distal-site mutations and allosteric inhibition to tune, extend, and narrow the useful dynamic range of aptamer-based sensors. Journal of the American Chemical Society. 2012;134:20601–20604. doi: 10.1021/ja310585e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vilar JMG, Saiz L. Reliable Prediction of Complex Phenotypes from a Modular Design in Free Energy Space: An Extensive Exploration of the lac Operon. ACS synthetic biology. 2013;2:576–586. doi: 10.1021/sb400013w. [DOI] [PubMed] [Google Scholar]
- 59.Vilar JMG, Saiz L. Multiprotein DNA looping. Physical review letters. 2006;96:238103. doi: 10.1103/PhysRevLett.96.238103. [DOI] [PubMed] [Google Scholar]
- 60.Saiz L, Vilar JMG. Ab initio thermodynamic modeling of distal multisite transcription regulation. Nucleic acids research. 2008;36:726–731. doi: 10.1093/nar/gkm1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.