

Abstract
In 2006, mutations in the granulin gene were identified in patients with familial Frontotemporal Lobar Degeneration. Granulin transcript haploinsufficiency has been proposed as a disease mechanism that leads to the loss of functional progranulin protein. Granulin mutations were initially found in tau-negative patients, though recent findings indicate that these mutations are associated with other neurodegenerative disorders with tau pathology, including Alzheimer’s disease and corticobasal degeneration. Moreover, a reduction in progranulin in tau transgenic mice is associated with increasing tau accumulation. To investigate the influence of a decline in progranulin protein on other forms of neurodegenerative-related protein accumulation, human granulin mutation cases were investigated by histochemical and biochemical analyses. Results showed a neuronal and glial tau accumulation in granulin mutation cases. Tau staining revealed neuronal pretangle forms and glial tau in both astrocytes and oligodendrocytes. Furthermore, phosphorylated α-synuclein-positive structures were also found in oligodendrocytes and the neuropil. Immunoblot analysis of fresh frozen brain tissues revealed that tau was present in the sarkosyl-insoluble fraction, and composed of three- and four-repeat tau isoforms, resembling Alzheimer’s disease. Our data suggest that progranulin reduction might be the cause of multiple proteinopathies due to the accelerating accumulation of abnormal proteins including TDP-43 proteinopathy, tauopathy and α-synucleinopathy.
Introduction
Progranulin (PGRN) is a growth factor encoded by a single gene on chromosome 17q21, extremely close to the MAPT (tau) gene. PGRN is a 593 amino acid, cysteine-rich protein with a signal peptide and 7.5 highly conserved tandem granulin repeats of a 12-cysteinyl motif. It is involved in the regulation of multiple processes, including neuronal inflammation1, wound healing2, 3, cell growth4, 5, tumorigenesis6 and chemoattraction of microglia7. Granulin (GRN) null mutations were identified in familial frontotemporal dementia (FTD) linked to chromosome 17q21 with tau-negative, ubiquitin-positive inclusions8, 9. Many mutations including those due to a frame shift by insertion/deletion or substitution of a nucleotide have been reported, and been shown to generate premature termination codons. GRN transcript haploinsufficiency has been proposed as the disease mechanism that leads to the loss of functional PGRN protein. A mutation in the signal peptide may cause mislocalization of PGRN in a protein secretion pathway or induce loss of PGRN function by impairment of its transport10, 11. Thus, these mutations are strongly involved in FTD pathogenesis.
Interestingly, loss-of-function GRN mutations have been identified in patients clinically diagnosed with Alzheimer’s disease (AD)12–19. For example, p.Gly35Arg (c.103G > A)19, and a single base pair deletion (c. 154delA) were found in AD, and the latter was shown to cause a frame shift (p.Thr52HisfsX2) creating a premature stop codon20. The rs5848 (3′ UTR + 78C > T) variant was also found in AD21 and associated with an increased risk of this disease22. In addition, GRN mutations were found in the accelerating accumulation of abnormal proteins in corticobasal syndrome10, 23–26. Furthermore, tau pathology, in addition to TAR-DNA binding protein of 43 kDa (TDP-43) pathology, was found in most members of two families harboring a GRN mutation27. These findings suggest that a decline in, or dysfunction of, PGRN may cause tau abnormalities, leading to the formation of tau pathology by activation of cyclin-dependent kinases in a P301L tau/GRN +/− mouse model28. To explore these issues, we performed immunohistochemical staining and biochemical analyses on human familial GRN mutation cases and examined whether GRN reduction accelerates the accumulation of neurodegenerative-related proteins other than TDP-43.
In this study, using a novel, highly sensitive immunohistochemical method employing free-floating sections, we noted massive phosphorylated-tau-positive staining in some familial GRN mutation cases. Notably, in these same cases, we also observed significant phosphorylated α-synuclein positive staining. Additionally, detergent-insoluble tau and α-synuclein proteins were detected by immunoblot analysis. Similar tau pathology was not seen in other GRN mutation cases when employing standard immunohistochemistry based on paraffin-embedded sections. Our results suggest that at least some cases with GRN mutations may show a hitherto unrecognized accelerated pathological accumulation of tau and α-synuclein.
Materials and Methods
Ethics Statement
All patients, or in some cases in which the patient had died, next of kin, provided written consent for autopsy and postmortem analyses for research purposes. Written informed consent was obtained from all patients. This study was approved by the Ethics Committee of the Tokyo Metropolitan Institute of Medical Science (permission No. 15-1 and 15-5(1)), the Banner Sun Health Research Institute and University of Manchester. The study was performed in accordance with the ethical standards laid down in the 1964 declaration of Helsinki and its later amendments.
Cases
The brain tissues used in Study A from four patients with GRN and three controls were from the Banner Sun Health Research Institute (Sun City, AZ), Brain and Body Donation Program29, 30. The additional nine GRN mutation cases in Study B were from the Manchester Brain Bank (UK). Ten control cases in Study B were registered in the autopsy archives of Dementia Research Project, Tokyo Metropolitan Institute of Medical Science. Case details are presented in Table 1. Seven different GRN mutations were recorded. Briefly, Case 1 had a c.1252C > T mutation resulting in p.Arg418X. Cases 2, 4, 9 and 12 had a c.1477C > T mutation resulting in p.Arg493X. A point mutation in a translation initiation codon (c.1A > C) predicted reduced mRNA levels in case 3. Three patients (cases 8, 14 and 16) shared c.1355delG mutation resulting in p.V452WfsX38. Case 10 had a c.1402C > T mutation resulting in p.Q468X and case 13 had a c.90_91insCTGC mutation resulting in p.C31LfsX34. Two patients (cases 11 and 15) shared c.388_391delCAGT mutation resulting in p.Q130SfsX124.
Table 1.
Description of the GRN mutation and control cases used in the initial study (Study A, No. 1–7) and the additional cases used in the second study (Study B, No. 8–26).
| Case No. | Gender | Age at death | Clinical diagnosis | Pathological diagnosis | Mutation (cDNA) | Mutation (Protein) | APOE | Tau haplotype | Brain weight (g) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 54 | AD/PiD | FTLD-TDP Type A/HS | c.1252C > T | p.R418X | n.d. | H1/H2 | 980 |
| 2 | F | 56 | FTD/AD | FTLD-TDP Type A/LBD | c.1477C > T | p.R493X | n.d. | n.d. | 940 |
| 3 | F | 72 | AD | FTLD-TDP Type A/AD | c.1A > C | p.0 | n.d. | H1/H1 | 1120 |
| 4 | M | 55 | AD | FTLD-TDP Type A/HS/LBD | c.1477C > T | p.R493X | n.d. | H1/H1 | 800 |
| 5 | M | 73 | Myeloid leukemia | Age changes only | None | None | n.d. | n.d. | 1240 |
| 6 | M | 76 | Multiple myeloma | n.d. | n.d. | 1375 | |||
| 7 | M | 79 | Prostate cancer | n.d. | n.d. | 1428 | |||
| 8 | F | 71 | FTD | FTLD-TDP Type A | c.1355delG | p.V452WfsX38 | E3/E3 | H2/H2 | 955 |
| 9 | F | 61 | FTD | c.1477C > T | p.R493X | n.d. | n.d. | 900 | |
| 10 | F | 66 | FTD | c.1402C > T | p.Q468X | E3/E3 | H1/H1 | 1100 | |
| 11 | F | 71 | PNFA | c.388_391delCAGT | p.Q130Sfs124 | E3/E3 | H1/H1 | 863 | |
| 12 | M | 66 | FTD | c.1477C > T | p.R493X | E3/E3 | H2/H2 | 1495 | |
| 13 | M | 73 | Prog Anomia | c.90_91insCTGC | p.C31LfsX34 | E3/E3 | n.d. | 1250 | |
| 14 | M | 71 | CBD/PAX | c.1355delG | p.V452WfsX38 | E3/E4 | H2/H2 | 925 | |
| 15 | M | 73 | PiD | c.388_391delCAGT | p.Q130Sfs124 | E3/E3 | n.d. | 980 | |
| 16 | M | 72 | PNFA | c.1355delG | p.V452WfsX38 | E3/E4 | H1/H2 | 870 | |
| 17 | M | 66 | Narcolepsy | Age changes only | None | None | n.d. | n.d. | 1221 |
| 18 | F | 63 | Spinal muscular atrophy | n.d. | n.d. | 1221 | |||
| 19 | M | 51 | Sch | n.d. | n.d. | 1332 | |||
| 20 | M | 50 | Candidal meningitis | n.d. | n.d. | 1300 | |||
| 21 | F | 53 | Atypical psychosis | n.d. | n.d. | ? | |||
| 22 | M | 63 | Sch/Parkinson’s syndrome | n.d. | n.d. | ? | |||
| 23 | M | 51 | Sch/Aspiration pneumonia | n.d. | n.d. | 1494 | |||
| 24 | M | 60 | Sch/Aspiration pneumonia | n.d. | n.d. | 1435 | |||
| 25 | M | 63 | Sch/Pseudomonas aeruginosa pneumonia | n.d. | n.d. | 1430 | |||
| 26 | M | 57 | Sch/Multiple myeloma | n.d. | n.d. | 1149 |
n.d.: not determined, Gender: F, female; M, Male.
AD, Alzheimer’s disease; CBD, corticobasal degeneration; FTD, frontotemporal dementia; HS, hippocampal sclerosis; LBD, Lewy body disease; PAX, apraxia;
PiD, Pick’s disease; PNFA, progressive non-fluent aphasia; Prog Anomia, progressive anomia; Sch, schizophrenia.
Histochemical analysis
Study A: For immunohistochemistry, sections fixed in 4% paraformaldehyde and preserved in 20% sucrose were cut serially on a freezing microtome at 40 μm thickness, collected in maintenance solution, and immunostained as free-floating sections (cases 1–7). Sections were incubated with 1% H2O2 for 30 min to eliminate endogenous peroxidase activity and were pretreated by autoclaving for 10 min in 10 mM sodium citrate buffer, pH6.0, at 121 °C. Sections were incubated for 24 hours with AT8 (1:1,000, Innogenetics, Ghent, Belgium), anti-TDP-43-pS409/410 antibody (1:1,000, Dr. Hasegawa), anti-phosphorylated α-synuclein antibody (1175, 1:1,000, Dr. Akiyama), E50 (for amyloid β, 1:10,000, Dr. Akiyama), RD3 (1:1,000, Merck Millipore), anti-4R tau (1:1,000, Dr. Hasegawa) or anti-FUS (1:1000, Sigma-Aldrich, St. Louis, MO, USA).
Study B: As we could not obtain sections in cases 8–26 that had been fixed and preserved under the same conditions as cases 1–7, we used formalin-fixed, paraffin-embedded sections instead. Therefore, sections from cases 8–26 were cut at 10 μm thickness, deparaffinized, incubated with 1% H2O2 for 30 min to eliminate endogenous peroxidase activity in the tissue, then pretreated for 10 min in 10 mM sodium citrate buffer, pH6.0 at 110 °C. Sections were then treated with formic acid for 10 min (for α-synuclein staining) or 30 min (for tau staining). For tau immunostaining, sections were incubated in 10 μg/ml of trypsin (Sigma-Aldrich) at 37 °C for 10 min. They were also incubated with AT8 and anti-phosphorylated α-synuclein antibody (1175), overnight, as in Study A. Antibody labeling was performed by incubation with goat anti-rabbit IgG (1:1,000, Vector Laboratories, Burlingame, CA, USA) or horse anti-mouse IgG (1:1,000, Vector Laboratories) for 3 hours. The antibody labeling was visualized by incubation with avidin-biotinylated horseradish peroxidase complex (ABC Elite, Vector Laboratories, 1:1,000) for 3 hours, followed by incubation with a solution containing 0.01% 3,3′-diaminobenzidine, 1% nickel ammonium sulfate, 0.05 M imidazole and 0.00015% H2O2 in 0.05 M Tris-HCl buffer, pH 7.6. Counter nuclear staining was performed with Kernechtrot stain solution (Merck, Darmstadt, Germany) or hematoxylin (Muto Pure Chemicals, Tokyo, Japan). The sections were then rinsed with distilled water, mounted on glass slides, treated with xylene, and coverslipped with Entellan (Merck). Tissue sections (cases 1–4) were also stained using a modified Gallyas-Braak method. Photographs were taken with a BX53 microscope (Olympus, Tokyo, Japan).
For fluorescent immunohistochemistry, free floating sections were incubated for 10 min with 0.1% Sudan black/70% ethanol solution and then incubated for 24 hours with AT8 (1:500), anti-TDP-43-pS409/410 antibody (1:500), anti-phosphorylated α-synuclein antibody (1175, 1:500) or pSyn#64 (1:500, Wako, Osaka, Japan). Antibody labeling was visualized by incubation with Alexa 488- or 568-labeled anti-rabbit IgG (1:100, Invitrogen, Carlsbad, CA, USA) or Alexa 488- or 568-labeled anti-mouse IgG (1:100, Invitrogen) for 2 hours. The sections were coverslipped with ProLong Gold with 4′,6-diamidino-2-phenylindole (Invitrogen). Photographs were taken with a BZ-8000 (Keyence, Osaka, Japan).
Sequential fractionation of brain extracts
Frozen brain samples (middle frontal gyrus, approximately, 0.2 g) were homogenized in 10 volumes of A68 buffer (10 mM Tris-HCl, pH 7.5, 0.8 M NaCl, 1 mM ethylene glycol bis-N, N, N′, N′-tetraacetic acid, 10% sucrose) containing 1% sarkosyl followed by incubation for 30 min at 37 °C. Each brain homogenate was centrifuged at 15,000 rpm for 10 min at 4 °C, and the supernatant was collected. The supernatant was then centrifuged at 136,000× g for 20 min at 4 °C. The sarkosyl-insoluble pellet was sonicated in 60 μl of SDS-PAGE sample buffer containing 4 M urea.
Histopathological assessments
Age-related plaque scores were determined using the Braak staging31. For purpose of this protocol, the letter corresponds to the following assessment: 0 = No Aβ deposits, A = initial Aβ deposits can be found in basal portions of the isocortex, B = Aβ deposits can be shown in virtually all isocortical association areas, C = Aβ deposits can be seen in all areas of the isocortex, including the sensory and motor core fields. For the evaluation of neurofibrillary changes (tau deposition), Braak staging was applied31. In this protocol, the number corresponds to the following assessment of the area of tau deposition: Stage I = transentorhinal cortex, Stage II = entorhinal cortex, Stage III = hippocampus-subiculum, Stage IV = temporal cortex, Stage V = parietal cortex, and Stage VI = occipital cortex. The degree of accumulation of tau and α-synuclein was also evaluated qualitatively and a score ranging from – (negative) to +++ (severe) was assigned.
Immunoblotting analyses
For immunoblotting, brain extracts from the GRN mutation cases were boiled for 5 minutes with SDS-PAGE sample buffer (60 mM Tris-HCl, pH 6.8, containing 2% SDS, 10% glycerol, 0.025% bromophenol blue and 5% mercaptoethanol) and loaded onto a 5–20% acrylamide minigel. Loaded samples were electrophoresed for 75 minutes at 200 V with molecular weight markers (Bio-Rad, Hercules, CA, USA). Electrophoresed proteins were transferred onto a polyvinylidene difluoride membrane (Merck Millipore) and subjected to 200 mA for 60 minutes. The printed membranes were blocked with 3% gelatin for 15 min and then incubated in a primary antibody solution, T46, (1:1,000, Innogenetics), RD3 (1:1,000, Merck Millipore), anti-4R tau (1:1,000, Dr. Hasegawa) for overnight at room temperature. Following incubation with the secondary anti-mouse or anti-rabbit antibody (1:50,000, Bio-Rad), immunoreactivity was detected by the chemiluminescence method using a Super Signal West Dura Extended Duration Substrate (Thermo Fisher Scientific, West Palm Beach, FL, USA) and was visualized with a LAS-4000 mini (GE Healthcare UK Ltd., Buckinghamshire, UK). For α-synuclein immunoblot, the printed membranes were incubated in a primary antibody solution, anti-phosphorylated α-synuclein antibody (1175, 1:1,000) or pSyn#64 (1:1,000) for overnight at room temperature. Following incubation with the biotinylated-secondary anti-rabbit or anti-mouse antibody (1:500, Vector Laboratories), followed by Vectastain Elite ABC kit (Vector Laboratories). Immunoreactivity was detected by 3,3′-diaminobenzidine with nickel chloride.
Results
GRN mutation cases used in the present study
The age, gender, clinical and pathological diagnoses and genetic information on the familial GRN mutation cases and control cases used in this study is summarized in Table 1. None of the GRN mutation cases examined in this study had MAPT mutation, and the MAPT haplotype was determined to be H1/H1 in cases 3, 4, 10 and 11, H1/H2 in cases 1 and 16, H2/H2 in cases 8, 12 and 14 (Table 1). The MAPT haplotype of cases 2, 9, 13 and 15 were unknown.
TDP-43 accumulation in GRN mutation cases
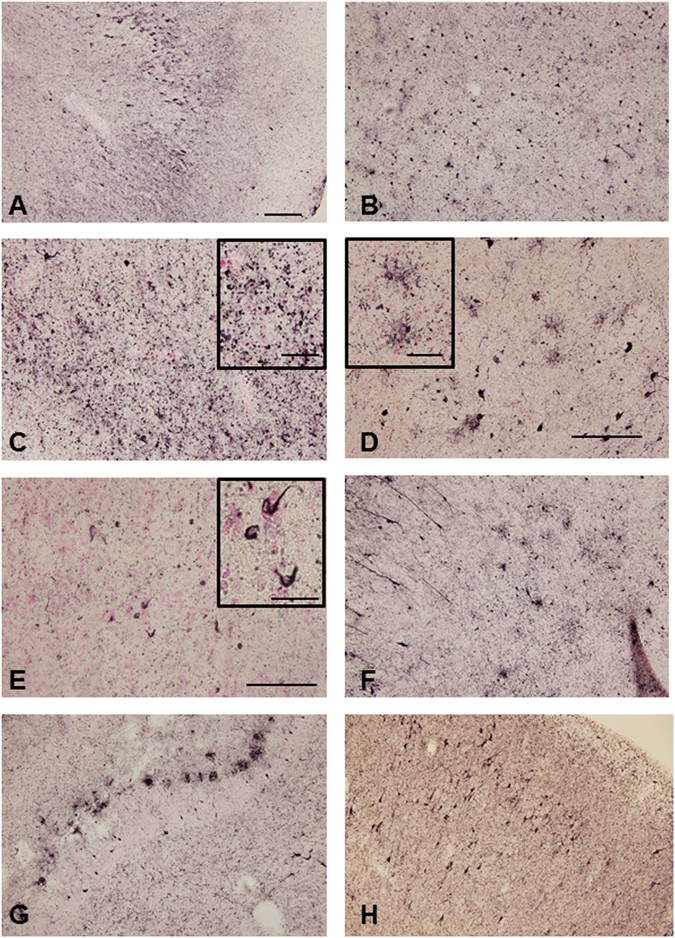
Phosphorylated TDP-43 inclusions were visualized by anti-TDP-43-pS409/410 antibody. Neuronal cytoplasmic inclusions (NCIs) and dystrophic neurites (DNs) were observed in the cerebral cortices of GRN mutation cases 1–4 (Fig. 1A–D) and cases 8–16 (data not shown). All thirteen GRN mutation cases were categorized as “Type A” according to the classification system of Mackenzie et al.32, consistent with that type of TDP-43 pathology previously reported in GRN mutation cases (Tables 2 and 3). All control cases (5–7 and 17–26) were negative for phosphorylated TDP-43 (Tables 2 and 3).
Figure 1.
Immunohistochemical staining of the temporal lobe of GRN mutation cases with antibody to phosphorylated TDP-43. Numerous neuronal cytoplasmic inclusions (arrows) and dystrophic neurites (arrowheads) were stained with anti-TDP-43-pS409/410 antibody in cases 1 (A), 2 (B), 3 (C) and 4 (D). The sections were counterstained with hematoxylin. The scale bar in (A) applies to all photomicrographs (100 μm).
Table 2.
Summary of immunohistochemical analyses of the initial study (Study A, No. 1–7).
| Case No. | Case | Age | TDP-43 | Aβ | Tau | Tau (neuronal/glial) | α-syn | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Hip-Ent | Amygdala | Temporal | Frontal | Hip-Ent | Amygdala | Temporal | ||||||
| 1 | GRN mutation | 54 | FTLD-TDP (Type A) | A | IV | +++/+ | ++/+ | +/+ | +/+ | ++ | + | + |
| 2 | 56 | 0 | IV | +++/++ | +++/++ | +++/++ | ++/++ | + | N.A. | + | ||
| 3 | 72 | C | V | +++/+ | ++/+ | +++/++ | +++/- | N.A. | + | + | ||
| 4 | 55 | A | IV | +++/+ | +/+ | +/+ | +/+ | +++ | + | ++ | ||
| 5 | Control | 73 | Negative | B | IV | ++/− | ++/+ | ++/+ | N.A. | + | + | − |
| 6 | 76 | A | I | +/− | +/− | +/− | N.A. | + | + | − | ||
| 7 | 79 | A | I | ++/− | +/+ | +/+ | N.A. | + | + | − | ||
+mild, ++moderate, +++severe, −negative, N.A.: not available.
Table 3.
Summary of immunohistochemical analyses of the second study (Study B, No. 8–26).
| Case No. | Case | Age | TDP-43 | Aβ | Tau | α-syn |
|---|---|---|---|---|---|---|
| 8 | GRN mutation | 71 | FTLD-TDP (Type A) | A | I | Negative |
| 9 | 61 | 0 | IV | |||
| 10 | 66 | 0 | II | |||
| 11 | 71 | 0 | II | |||
| 12 | 66 | 0 | I | |||
| 13 | 73 | 0 | II | |||
| 14 | 71 | B | I | |||
| 15 | 73 | 0 | 0 | |||
| 16 | 72 | A | II | |||
| 17 | Control | 66 | Negative | 0 | II | |
| 18 | 63 | 0 | 0 | |||
| 19 | 51 | 0 | I | |||
| 20 | 50 | A | 0 | |||
| 21 | 53 | 0 | I | |||
| 22 | 63 | 0 | I | |||
| 23 | 51 | 0 | 0 | |||
| 24 | 60 | A | I | |||
| 25 | 63 | 0 | 0 | |||
| 26 | 57 | 0 | 0 |
Tau accumulation in GRN mutation cases
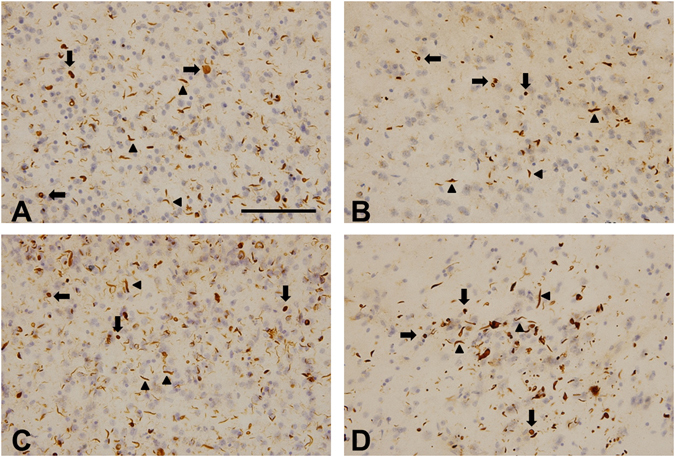
We observed a considerable number of tau-positive neurons, astrocytes and oligodendrocytes in all 4 Study A GRN mutation cases by either AT8 immunostaining (Fig. 2, and Table 2) or Gallyas silver staining (data not shown). In particular, case 2 showed massive AT8-positive structures in the entorhinal cortex, hippocampus (Fig. 2A), amygdala (Fig. 2B), temporal cortex (Fig. 2C), insula. In the temporal lobe, the majority of tau-positive neuronal cytoplasmic staining appeared as pretangle-like forms (Fig. 2A,F,G,H). In the neuropil, fine tau–positive granules were abundant (Fig. 2C). The size of most of these granules appeared smaller than the tau-positive grains observed in argyrophilic grain disease (AGD) brains, and they were negative for Gallyas-silver staining (data not shown).
Figure 2.
Photomicrograph of phosphorylated tau immunohistochemistry of the temporal lobe of the GRN mutation cases. Massive AT8-positive structures were observed in the hippocampus (A), amygdala (B), inferior temporal cortex (C) in case 2. AT8-positive astrocytes were observed in the temporal cortex (D) and AT8-positive oligodendrocytes in the white matter of temporal lobe (E) in case 2. AT8-positive deposition was also detected in amygdala in case 1 (F), hippocampus in case 3 (G) and entorhinal cortex in case 4 (H). The sections were counterstained with Kernechtrot stain solution. The scale bar in A applies to (B,C,D,F,G and H) (200 μm), in E (100 μm), respectively. The scale bars in the inserts are 50 μm (C and D) and 25 μm (E), respectively.
Furthermore, tau-positive astrocytic structures, resembling “bush-like” astrocytes previously reported in AGD33, were found in the cortex in all cases in Study A. Their morphology was significantly different from the tufted-astrocytes in progressive non-fluent aphasia (PSP) patients, the astrocytic plaques in corticobasal degeneration (CBD) or the ramified astrocytes in Pick’s disease (Fig. 2D). In the white matter, tau-positive oligodendroglial coiled bodies were observed (Fig. 2E).
Gallyas silver staining also revealed structures similar to those stained with AT8, including neurofibrillary tangles (NFTs), threads, and astrocytic and oligodendrocytic structures (data not shown). In the hippocampal region, many NFTs were found in cases 2 and 4 by both AT8 immunostaining and Gallyas silver staining (data not shown). The control cases of Study A showed mild to moderate AT8-positive structures and less glial tau deposition compared to GRN mutation cases (Table 2). Case 1, 2, 4 and 5 exhibited tau deposition that corresponded to Braak stage IV, Case 3 corresponded to Braak stage V and Case 6 and 7 corresponded to Braak stage I, respectively (Table 2). The degree of accumulation of tau was evaluated qualitatively and a score ranging from – (negative) to +++ (severe) was assigned (Supplementary Figure 1 and Table 2).
In Study B, eight of nine GRN mutation cases exhibited some AT8 immunoreactivity (Supplementary Figure 2 and Table 3), but the levels of phosphorylated tau deposition were up to Braak stage II except for Case 9 (Table 3), dissimilar to that seen in GRN mutation cases of Study A. No tau deposition or only Braak stage I-II were observed in the control cases of study B (Table 3). The tau pathology in the GRN mutation cases (Study A) was also detected by 3-repeat (3R)-tau (RD3) and 4-repeat (4R)-tau (anti-4R) specific antibodies indicating that both 3R and 4R tau accumulation was present (Data not shown).
α-synuclein accumulation in GRN mutation cases
Immunohistochemistry using an antibody to phosphorylated α-synuclein, revealed small round or dot-like structures and short thread-like structures in the temporal lobe (Fig. 3A–F), and oligodendroglial coiled body-like structures in the temporal white matter (data not shown) in cases 1–4 of Study A. The degree of accumulation of α-synuclein was evaluated qualitatively and a score ranging from – (negative) to +++ (severe) was assigned. In particular, case 4 exhibited atypical α-synuclein deposition in the temporal cortex (Fig. 3A,B and Table 2). Phosphorylated α-synuclein positive structures were not found in Study B using paraffin-embedded sections of GRN mutation cases (cases 8–16, Table 3) and control cases (cases 17–26, Table 3).
Figure 3.
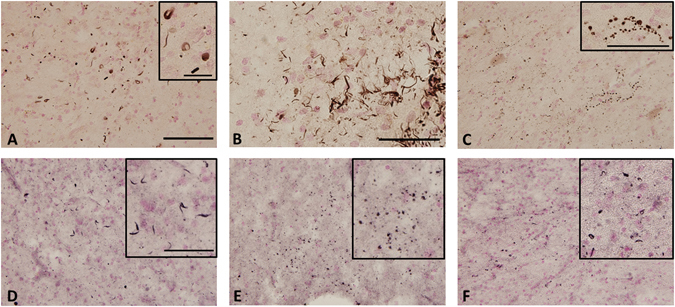
Immunohistochemical staining of phosphorylated α-synuclein in the temporal lobe of the GRN mutation cases. Phosphorylated α-synuclein -positive structures were observed in the inferior temporal cortex (A), superior temporal cortex (B) and tuberomammillary nucleus (C) in case 4, and in the temporal cortex in case 1 (D), case 2 (E) and case 3 (F). Sections were counterstained with Kernechtrot stain solution. The scale bar in (A) applies to (C–F) (200 μm). The scale bars in (B), in insert (A,C and D–F) are 50 μm.
Amyloid β deposition in GRN mutation cases
Aβ deposition was found in the temporal lobe in three of four GRN mutation cases in Study A (Fig. 4 and Table 2). In the cases 1 and 4, Aβ pathology was present mostly as diffuse plaques, corresponding to Braak stage A (Fig. 4A and D). In case 2, there was no Aβ pathology (Fig. 4B) but case 3 corresponded with Braak stage C (Fig. 4C). Of the control cases in Study A, three were similar to Braak stage A, but one (case 5) corresponded to Braak stage B. In Study B, Aβ accumulation in almost all cases corresponded to Braak stage 0, the others showing Braak stage A (Table 3).
Figure 4.
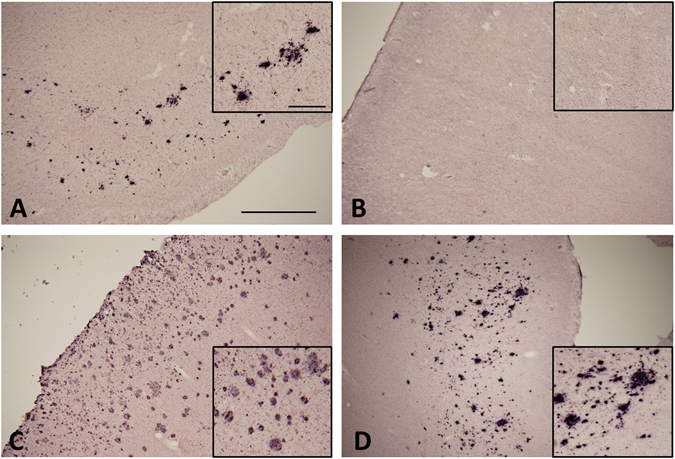
Immunohistochemical staining of amyloid β in the temporal lobe of GRN mutation cases. Amyloid β (Aβ) was observed in cases 1 (A), 3 (C) and 4 (D), but not in case 2 (B). Most Aβ-positive structures consisted of diffuse plaques. The scale bar in (A) applies to all photomicrographs (1.0 mm) and the scale bar in insert A applies to insert (B–D) (250 μm).
Immunoblot analyses
Biochemical features of accumulated tau in cases of GRN mutation (Fig. 5, Case 3: lane 1, Case 4: lane 2) were compared with those of other tauopathies including CBD (lane 3), PSP (lane 4) and AD (lane 5) by immunoblot analysis of the sarkosyl-insoluble fraction using C-terminal tau antibody (T46) (Fig. 5). The major tau band pattern in GRN mutation cases was triplets of 68, 64 and 60 kDa, similar to that in AD, but different from that in CBD and PSP (Fig. 5). GRN mutation cases were also detected by 3R-tau (RD3) and 4R-tau (anti-4R) specific antibodies indicating both 3R and 4R tau accumulation (Supplementary Figure 3). GRN mutation cases were also studied with anti-phosphorylated α-synuclein antibodies (1175 and pSyn#64) for cases 1–4. Very faint bands of phosphorylated α-synuclein were observed at 16 kDa. (Supplementary Figure 4).
Figure 5.
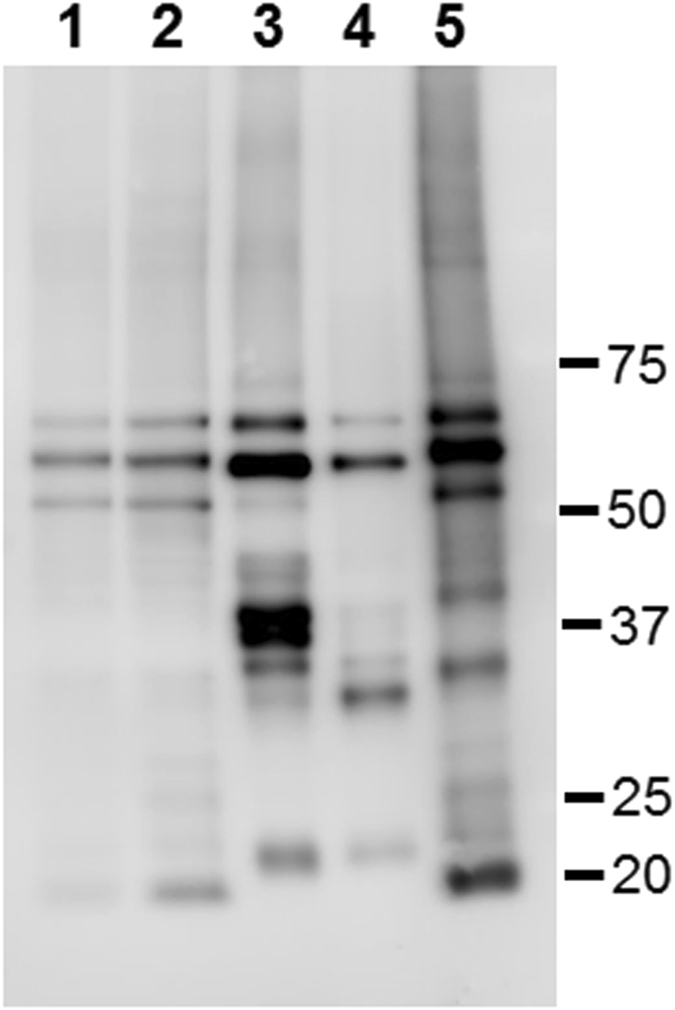
Comparison of the banding patterns of sarkosyl-insoluble tau on immunoblotting between cases with a GRN mutation and those with other tauopathies. Immunoblotting analysis was visualized using the T46 antibody for detecting tau in the sarkosyl-insoluble fraction from cases 3 (lane 1) and 4 (lane 2) with GRN mutations, and a case each of CBD (lane 3), PSP (lane 4) and AD (lane 5). Molecular weight markers are shown on the right (kDa).
Fluorescence immunohistochemistry
Fluorescent double-staining of the temporal lobe of the GRN mutation case 4 was performed to examine whether TDP-43/tau (Fig. 6A), TDP-43/α-synuclein (Fig. 6B) or tau/α-synuclein (Fig. 6C) were co-localized in the abnormal structures. Colocalization of these proteins was very infrequent in most abnormal structures.
Figure 6.
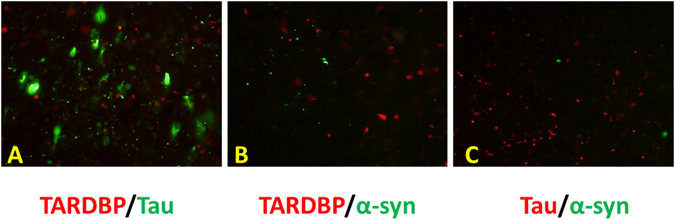
Immunofluorescent double-staining of accumulated proteins in the temporal lobe in GRN mutation case. Phosphorylated TDP-43, α-synuclein and tau immunoreactivity were obserbed in the temporal lobe of GRN mutation case 4. pTDP-43 (red)/pTau (green) (A), pTDP-43 (red)/pα-synuclein (green) (B) and pTau (red)/pα-synuclein (green) (C).
Discussion
The results of the present study show that GRN mutations causing PGRN reduction may accelerate the intracellular accumulation of not only TDP-43 but also tau and α-synuclein in the brains of familial FTD patients with GRN mutations. This suggests that GRN mutations causing PGRN reduction may be causative or represent risk factors for multiple proteinopathies (TDP-43 proteinopathy, tauopathy or α-synucleinopathy).
Immunohistochemical analyses of phosphorylated TDP-43 revealed a considerable number of neuronal cytoplasmic inclusions and dystrophic neurites in all GRN mutation cases (Fig. 1, Tables 2 and 3). In FTLD-TDP, TDP-43 pathology falls within four histological subtypes (types A-D) based on the predominant type of TDP-43-positive structures exhibited32. Type A is characterized by numerous short dystrophic neurites and crescentic or oval neuronal cytoplasmic inclusions. Cases of FTLD-TDP with a GRN mutation invariably display type A pathology34–36, and present observations were in accordance with this.
The very high sensitivity staining method that we performed for Study A (cases 1–7) revealed Case 2 to show atypical tauopathy with massive tau deposition in neuron, astrocytes and oligodendrocytes without Aβ deposition. Case 4 was atypical synucleinopathy with diffuse α-synuclein positive structures that were observed mainly in the neocortex. Case 3 exhibited massive Aβ deposition corresponding to Braak Stage C and tau deposition corresponding to Braak stage V in AD pathology. Case 1 exhibited tau deposition that corresponded to Braak stage IV. It is possible that case 3 might be an incidental complication of AD because the age was late 70 s’. The pathology in the other two cases (case 2 and 4), however, is very rarely observed in the normal aging brain at mid-50 years of age. The control cases in Study A also had levels of tau deposition that corresponded to Braak stage I to IV (Table 2), but the average age was higher than that of the GRN mutation cases. We compared abnormal tau deposition using the paraffin-embedded tissues of GRN mutation and control cases, and there were significantly differences (Study B, Table 3). Though Study B was less obvious differences than Study A. Braak et al. reported that for Braak NFT stage III-IV, the ratio was less than 10% at ages 50 s’ to 60 s’37, so that our GRN mutation cases in Study A showed tau accumulation atypical for normal aging.
It has been widely accepted for the past decade that there is no tau deposition in GRN mutation brains. However, using high-sensitivity immunohistochemical staining, we have found that hyper-accumulated tau and α-synuclein can occur in younger GRN mutation cases. Part B of the present study, using paraffin-embedded tissues, however, showed only mild tau deposition, as has been previously reported8, 9. Hence, GRN mutation may accelerate deposition of tau and α-synuclein but the level of abnormal protein deposition seen in routine paraffin-embedded sections from GRN cases might not be as strong as that seen than in free-floating sections and therefore go unrecognized. Re-analysis might be necessary in other GRN mutation cases using this high-sensitivity immunohistochemical staining method, or immunoblot analyses on frozen brain tissue, in order to gain a fuller appreciation of the level of tau pathology present in such cases.
Our previous report made mention of the fact that a GRN mutation in P301L tau transgenic mice affected phosphorylated tau deposition28. The results of the present study support our previous observations in mice. It has been reported that PGRN deficiency causes lysosomal dysfunction38. We hypothesized that lysosomal dysfunction might reduce protein degradation in brain cells allowing aggregation-prone neurodegenerative disease-related proteins to deposit more easily.
The features of tau pathology in GRN mutation cases in this study are of predominantly neuronal pretangles, abundant fine granules in the neuropil, and astrocytic and oligodendroglial pathology. It is interesting that fine tau-positive granules were reported in the striatum of a brain with a GRN c.709-2A > G mutation27. Among tauopathies, the tau pathology most similar to our cases might be found in AGD. However, the size of the fine granules in our cases seemed smaller than that of the grains in AGD and they were negative for Gallyas-silver staining (data not shown). Although the form of tau-positive astrocytes in our cases was similar to the “bush-like” astrocytes in AGD, their Gallyas-positive status in contrast to the Gallyas-negative status of the “bush-like” AGD astrocytes33. No FUS accumulation was found in any GRN mutation cases in Study A, thus there might be no or little relationship between the GRN mutation and FUS deposition (data not shown).
Immunoblot analysis of our cases using C-terminal tau antibody revealed that the banding patterns of the full-length tau in the sarkosyl-insoluble fraction appeared to be essentially the same as that seen in AD (Fig. 5). The staining using three and four repeat tau specific antibodies revealed that tau in the sarkosyl-insoluble fraction consists of both forms of tau (Supplementary Figure 2). These results suggest that accumulated tau in cases of GRN mutation cases contains six tau isoforms just as in AD. However, the distribution of fine granular tau and the lack of any or only light Aβ accumulation (Fig. 4) is different from AD pathology. Tau isoforms in GRN mutation cases were biochemically different from those in CBD and PSP (Fig. 5). Cases of GRN mutation may therefore represent a different tauopathy from that of AD, CBD, PSP and AGD.
In addition to neuronal and glial tau accumulation, the present study also revealed α-synuclein-positive structures, including small round, dot-like or thread-like structures in the cortex and oligodendroglial coiled body-like structures in the white matter in the GRN mutation cases in Study A (cases 1–4, Fig. 3). Case number 4 showed particularly striking phosphorylated α-synuclein pathology. However, the nine paraffinized GRN mutation cases showed no α-synuclein-positive structures. This discrepancy might be caused by fixation or preservation methods. Leverenz and colleagues reported that α-synuclein pathology was observed in two of seven brains with a familial GRN mutation. One case showed brainstem α-synuclein pathology while the other was cortical27.
Accumulations of phosphorylated-tau, α-synuclein and TDP-43 were reported in the brains of Guam/Kii-amyotrophic lateral sclerosis (ALS)-parkinsonism-dementia complex (PDC) patients39. The triplet tau band patterns (68, 64, and 60 kDa) of immunoblot analysis in the sarkosyl-insoluble fraction of GRN mutation cases (Fig. 5) appeared to be essentially the same among cases with the GRN mutation, AD and Guam/Kii ALS-PDC. Fine tau-positive granules were also reported in the cerebral white matter of Guam-PDC cases, in which the morphology seemed to resemble that of our cases. Hazy astrocytes were observed in Guam-PDC cases, but their morphology seemed to differ from that of our cases. To date, no GRN mutations in Guam/Kii ALS-PDC cases have been reported. Very recently we reported that the GRN mutation leads lysosomal dysfunction40. We speculated that there could be common pathway(s) in lysosomal function or that these diseases have something common features of protein aggregation because of the similarities (TDP-43, tau, α-synuclein deposition and 3R/4R tau isoform aggregation).
In conclusion, when using a highly sensitive free-floating immunohistochemical technique combined with western blotting, we have shown widespread pathological tau and α-synuclein deposition in neurons and glial cells in familial GRN mutation cases that are not apparent when using standard immunohistochemical methods based on routine paraffin embedded sections. Although, the number of samples for this study was small and we recognize the limits of this study, our findings suggest that the pathologies seen in GRN mutation cases may possibly be renamed “neuronoglial multiple proteinopathies”.
Electronic supplementary material
Acknowledgements
This research was partially supported by the Japan Society for the Promotion of Science, a Grant-in-Aid for Scientific Research (C) [JSPS KAKENHI grant number JP24591738] to M. Hosokawa and by a Daiwa Securities Research Foundation grant to M. Hosokawa. We are grateful to the Banner Sun Health Research Institute Brain and Body Donation Program of Sun City, Arizona for the provision of human brain tissue. The Brain and Body Donation Program is supported by the National Institute of Neurological Disorders and Stroke (U24 NS072026 National Brain and Tissue Resource for Parkinson’s Disease and Related Disorders), the National Institute on Aging (P30 AG19610 Arizona Alzheimer’s Disease Core Center), the Arizona Department of Health Services (contract 211002, Arizona Alzheimer’s Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05-901 and 1001 to the Arizona Parkinson’s Disease Consortium) and the Michael J. Fox Foundation for Parkinson’s Research. We are also grateful to The Manchester Brain Bank, which is part of the Brains for Dementia Research programme, jointly funded by Alzheimer’s Research UK and Alzheimer’s Society. The authors would like to thank Dr. Edith G. McGeer, Dr. William Campbell and Mrs Catherine Campbell for editing of the manuscript.
Author Contributions
Study concept and design: M.Ho. and T.A. Experiment: M.Ho., H.K. and M.Ha. Providing brain samples and patients information: G.S., T.B., A.R. and D.M. Analysis and interpretation of data: M.Ho., T.B., D.M., M.Ha., H.A. and T.A. Writing the manuscript: M.Ho. and T.A.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-01587-6
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Yin F, et al. Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice. J Exp Med. 2010;207:117–128. doi: 10.1084/jem.20091568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.He Z, Ong CH, Halper J, Bateman A. Progranulin is a mediator of the wound response. Nat Med. 2003;9:225–229. doi: 10.1038/nm816. [DOI] [PubMed] [Google Scholar]
- 3.Zhu J, et al. Conversion of proepithelin to epithelins: roles of SLPI and elastase in host defense and wound repair. Cell. 2002;111:867–878. doi: 10.1016/S0092-8674(02)01141-8. [DOI] [PubMed] [Google Scholar]
- 4.Daniel R, Daniels E, He Z, Bateman A. Progranulin (acrogranin/PC cell-derived growth factor/granulin-epithelin precursor) is expressed in the placenta, epidermis, microvasculature, and brain during murine development. Dev Dyn. 2003;227:593–599. doi: 10.1002/dvdy.10341. [DOI] [PubMed] [Google Scholar]
- 5.Van Damme P, et al. Progranulin functions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J Cell Biol. 2008;181:37–41. doi: 10.1083/jcb.200712039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ong CH, Bateman A. Progranulin (granulin-epithelin precursor, PC-cell derived growth factor, acrogranin) in proliferation and tumorigenesis. Histol Histopathol. 2003;18:1275–1288. doi: 10.14670/HH-18.1275. [DOI] [PubMed] [Google Scholar]
- 7.Pickford F, et al. Progranulin is a chemoattractant for microglia and stimulates their endocytic activity. Am J Pathol. 2011;178:284–295. doi: 10.1016/j.ajpath.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baker M, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- 9.Cruts M, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–924. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- 10.Gass J, et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet. 2006;15:2988–3001. doi: 10.1093/hmg/ddl241. [DOI] [PubMed] [Google Scholar]
- 11.Mukherjee O, et al. HDDD2 is a familial frontotemporal lobar degeneration with ubiquitin-positive, tau-negative inclusions caused by a missense mutation in the signal peptide of progranulin. Ann Neurol. 2006;60:314–322. doi: 10.1002/ana.20963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brouwers N, et al. Alzheimer and Parkinson diagnoses in progranulin null mutation carriers in an extended founder family. Arch Neurol. 2007;64:1436–1446. doi: 10.1001/archneur.64.10.1436. [DOI] [PubMed] [Google Scholar]
- 13.Le Ber I, et al. Phenotype variability in progranulin mutation carriers: a clinical, neuropsychological, imaging and genetic study. Brain. 2008;131:732–746. doi: 10.1093/brain/awn012. [DOI] [PubMed] [Google Scholar]
- 14.Kelley BJ, et al. Prominent phenotypic variability associated with mutations in Progranulin. Neurobiol Aging. 2009;30:739–751. doi: 10.1016/j.neurobiolaging.2007.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rademakers R, et al. Phenotypic variability associated with progranulin haploinsufficiency in patients with the common 1477C–>T (Arg493X) mutation: an international initiative. Lancet Neurol. 2007;6:857–868. doi: 10.1016/S1474-4422(07)70221-1. [DOI] [PubMed] [Google Scholar]
- 16.Sleegers K, et al. Serum biomarker for progranulin-associated frontotemporal lobar degeneration. Ann Neurol. 2009;65:603–609. doi: 10.1002/ana.21621. [DOI] [PubMed] [Google Scholar]
- 17.Finch N, et al. Plasma progranulin levels predict progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain. 2009;132:583–591. doi: 10.1093/brain/awn352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carecchio M, et al. Progranulin plasma levels as potential biomarker for the identification of GRN deletion carriers. A case with atypical onset as clinical amnestic Mild Cognitive Impairment converted to Alzheimer’s disease. J Neurol Sci. 2009;287:291–293. doi: 10.1016/j.jns.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 19.Cortini F, et al. Novel exon 1 progranulin gene variant in Alzheimer’s disease. Eur J Neurol. 2008;15:1111–1117. doi: 10.1111/j.1468-1331.2008.02266.x. [DOI] [PubMed] [Google Scholar]
- 20.Kelley BJ, et al. Alzheimer disease-like phenotype associated with the c.154delA mutation in progranulin. Arch Neurol. 2010;67:171–177. doi: 10.1001/archneurol.2010.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fenoglio C, et al. Rs5848 variant influences GRN mRNA levels in brain and peripheral mononuclear cells in patients with Alzheimer’s disease. J Alzheimers Dis. 2009;18:603–612. doi: 10.3233/JAD-2009-1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee MJ, Chen TF, Cheng TW, Chiu M. rs5848 variant of progranulin gene is a risk of Alzheimer's disease in the Taiwanese population, J. rs5848 variant of progranulin gene is a risk of Alzheimer’s disease in the Taiwanese population. Neurodegener Dis. 2011;8:216–220. doi: 10.1159/000322538. [DOI] [PubMed] [Google Scholar]
- 23.Masellis M, et al. Novel splicing mutation in the progranulin gene causing familial corticobasal syndrome. Brain. 2006;129:3115–3123. doi: 10.1093/brain/awl276. [DOI] [PubMed] [Google Scholar]
- 24.Spina S, et al. Corticobasal syndrome associated with the A9D Progranulin mutation. Journal of neuropathology and experimental neurology. 2007;66:892–900. doi: 10.1097/nen.0b013e3181567873. [DOI] [PubMed] [Google Scholar]
- 25.Benussi L, et al. A novel deletion in progranulin gene is associated with FTDP-17 and CBS. Neurobiol Aging. 2008;29:427–435. doi: 10.1016/j.neurobiolaging.2006.10.028. [DOI] [PubMed] [Google Scholar]
- 26.Perry DC, et al. Progranulin mutations as risk factors for Alzheimer disease. JAMA neurology. 2013;70:774–778. doi: 10.1001/2013.jamaneurol.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leverenz JB, et al. A novel progranulin mutation associated with variable clinical presentation and tau, TDP43 and alpha-synuclein pathology. Brain. 2007;130:1360–1374. doi: 10.1093/brain/awm069. [DOI] [PubMed] [Google Scholar]
- 28.Hosokawa M, et al. Progranulin reduction is associated with increased tau phosphorylation in P301L tau transgenic mice. Journal of neuropathology and experimental neurology. 2015;74:158–165. doi: 10.1097/NEN.0000000000000158. [DOI] [PubMed] [Google Scholar]
- 29.Birdsill AC, Walker DG, Lue L, Sue LI, Beach TG. Postmortem interval effect on RNA and gene expression in human brain tissue. Cell and tissue banking. 2011;12:311–318. doi: 10.1007/s10561-010-9210-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beach TG, et al. Arizona Study of Aging and Neurodegenerative Disorders and Brain and Body Donation Program. Neuropathology: official journal of the Japanese Society of Neuropathology. 2015;35:354–389. doi: 10.1111/neup.12189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta neuropathologica. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 32.Mackenzie IR, et al. A harmonized classification system for FTLD-TDP pathology. Acta neuropathologica. 2011;122:111–113. doi: 10.1007/s00401-011-0845-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Botez G, Probst A, Ipsen S, Tolnay M. Astrocytes expressing hyperphosphorylated tau protein without glial fibrillary tangles in argyrophilic grain disease. Acta neuropathologica. 1999;98:251–256. doi: 10.1007/s004010051077. [DOI] [PubMed] [Google Scholar]
- 34.Mackenzie IR, et al. The neuropathology of frontotemporal lobar degeneration caused by mutations in the progranulin gene. Brain. 2006;129:3081–3090. doi: 10.1093/brain/awl271. [DOI] [PubMed] [Google Scholar]
- 35.Cairns NJ, et al. TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol. 2007;171:227–240. doi: 10.2353/ajpath.2007.070182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Josephs KA, et al. Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions with progranulin gene (PGRN) mutations. Journal of neuropathology and experimental neurology. 2007;66:142–151. doi: 10.1097/nen.0b013e31803020cf. [DOI] [PubMed] [Google Scholar]
- 37.Braak H, Del Tredici K. Evolutional aspects of Alzheimer’s disease pathogenesis. J Alzheimers Dis. 2013;33:S155–161. doi: 10.3233/JAD-2012-129029. [DOI] [PubMed] [Google Scholar]
- 38.Tanaka Y, Chambers JK, Matsuwaki T, Yamanouchi K, Nishihara M. Possible involvement of lysosomal dysfunction in pathological changes of the brain in aged progranulin-deficient mice. Acta neuropathologica communications. 2014;2:78. doi: 10.1186/s40478-014-0078-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Forman MS, et al. Tau and alpha-synuclein pathology in amygdala of Parkinsonism-dementia complex patients of Guam. Am J Pathol. 2002;160:1725–1731. doi: 10.1016/S0002-9440(10)61119-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tanaka Y, et al. Progranulin regulates lysosomal function and biogenesis through acidification of lysosomes. Hum Mol Genet. 2017;26:ddx011–988. doi: 10.1093/hmg/ddx011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.