

 An unprecedented intermolecular atom transfer thiosulfonylation reaction of alkenes was achieved by combining Au catalysis and visible-light photoredox catalysis. A SCF3 group and other functionalized thio groups together with sulfonyl group were regioselectively introduced into alkenes.
An unprecedented intermolecular atom transfer thiosulfonylation reaction of alkenes was achieved by combining Au catalysis and visible-light photoredox catalysis. A SCF3 group and other functionalized thio groups together with sulfonyl group were regioselectively introduced into alkenes.
Abstract
Regioselective difunctionalization of alkenes has attracted significant attention from synthetic chemists and has the advantage of introducing diverse functional groups into vicinal carbons of common alkene moieties in a single operation. Herein, we report an unprecedented intermolecular atom transfer thiosulfonylation reaction of alkenes by combining gold catalysis and visible-light photoredox catalysis. A trifluoromethylthio group (SCF3) and other functionalized thio groups together with a sulfonyl group were regioselectively introduced into alkenes in the most atom economical manner. A detailed mechanism study indicated that a synergistic combination of gold catalysis and photoredox catalysis is crucial for this reaction.
Introduction
Regioselective difunctionalization of alkenes has the advantage of introducing diverse functional groups into vicinal carbons of common alkene moieties in a single operation and has attracted extensive attention from synthetic chemists.1 Of the many catalytic processes that have been developed, a majority requires stoichiometric amounts of an external strong oxidant, such as PhI(OAc)2, Selectfluor (eqn (1)),2 and are typically initiated by a transition metal-catalyzed intramolecular addition. Intermolecular difunctionalization of alkenes is more challenging because of the regiochemical issue (eqn (2)). Recently, Stephenson developed an elegant visible light-mediated atom transfer radical addition reaction converting haloalkanes and α-halocarbonyl compounds into alkenes.3 These neutral redox reactions are very attractive because they are atom-economical and require no additional oxidants.
 |
1 |
 |
2 |
The trifluoromethylthio (SCF3) group is a key structural unit in many pharmaceutical and agrochemical products such as tiflorex, toltrazuril, and vaniliprole.4 It is well known that SCF3 groups in a molecule induce even higher lipophilicity than trifluoromethyl substituents, and consequently, the incorporation of SCF3 group into pharmaceuticals could greatly improve their ability to cross lipid membranes.5 Because of this, the introduction of a SCF3 group into small molecules has attracted significant attention in synthetic chemistry.6,7 Current methods for the construction of C–SCF3 bonds involve electrophilic trifluoromethylthio reagents6 or the nucleophilic AgSCF3 reagent.7 Sulfonyl groups are similar to carboxyl or phosphate groups in terms of molecular size and charge distribution, and the sulfonyl group has been introduced into bioactive molecules to improve their activity.8 A sulfonyl group has two receptors for hydrogen bonds and this can enhance the binding affinities of drug molecules with target proteins. Sulfones can be easily transformed into other functional groups, such as alkenes, via Julia olefination.9 We investigated that whether both SCF3 and sulfonyl groups could be simultaneously introduced into organic compounds in a single step. Moreover, this transformation has not been described to date. This report describes a dual gold and photoredox catalytic approach to the intermolecular atom transfer thiosulfonylation of alkenes.
A combination of visible light-mediated photoredox catalysis10 and transition-metal-catalysis is possible to bring two distinctive catalytic systems together and achieve unprecedented new reactions.11 Recently, the use of gold(i) complexes in photoredox catalysis has gained considerable attention.12,13 This photoredox catalytic cycle triggers the conversion of Au(i) into Au(iii) under mild conditions, a conversion which previously had only been achieved with stoichiometric quantities of strong oxidants.14 In 2013, Glorius reported a first dual gold and photoredox-catalyzed reaction, which achieved intramolecular oxy- and amino-arylation of alkenes with aryldiazonium salts (Scheme 1A).12b Toste et al. took advantage of the visible light-mediated Au(i)/Au(iii) cycle to produce arylative ring expansion reactions and carbon–phosphorus cross-coupling reactions (Scheme 1B).12d,e This visible light-mediated single electron oxidative reaction has been utilized to access gold(iii) complexes from gold(i) species.12k Recently, Hashmi reported an aryldiazonium salts mediated Au(i) to Au(iii) transformation upon irradiation with blue LED in the absence of a photosensitizer.12q In all these reactions,12 the same aryldiazonium salts were used. The development of new gold/photoredox catalysis mode is highly desirable. To achieve the proposed trifluoromethyl-thiosulfonylation reaction in the most atom-economical manner, a difunctionalization reagent, such as PhSO2SCF3 (2a), is required. This reagent can be easily prepared from PhSO2Na and AgSCF3 (for details, see the ESI†). We envisioned that the reaction of PhSO2SCF3 with a cationic gold catalyst in the presence of a photocatalyst would generate an LAuSCF3 species and a benzenesulfonyl radical, which can then add to the alkene forming a new alkyl radical.3a This radical may oxidize LAuSCF3 into a Au(ii) intermediate, which is further oxidized to a Au(iii) derivative by the RuIII catalyst. Subsequent reductive elimination forms the target difunctionalized product, regenerating the Au(i) catalyst (Scheme 1C). This reaction provided a new approach to introduce trifluoromethylthio (SCF3) group at the α position of styrenes.
Scheme 1. Dual gold and photoredox catalytic reactions.

Results and discussion
To validate this concept, styrene (1a) and PhSO2SCF3 (2a) were selected as model substrates to test the feasibility of this hypothesis. After a detailed optimization of reaction conditions, the proposed alkene trifluoromethylthio-sulfonylation product (3a) was achieved in 94% yield under the standard conditions: a mixture of IPrAuCl (10 mol%), AgSbF6 (15 mol%), Ru(bpy)3Cl2 (2.5 mol%) in DCE (1 mL) was stirred under irradiation for 1–3 h with 100 W blue LED in a N2 atmosphere (Table 1, entry 1). The dual catalytic nature of this reaction was investigated by control experiments, which confirmed that the gold catalyst, the Ru(ii) photosensitizer, and visible light irradiation are all necessary for the reaction (Table S2,† entries 2–5). In the absence of silver salt, the reaction led to a dramatic decrease in the yields (NMR yields <5%), showing that the formation of a cationic gold species is highly important (Table S2,† entry 4). Other gold catalysts, such as AuCl, Ph3PAuCl, and Gagosz catalyst (Ph3PAuNTf2), are all less effective than IPrAuCl (entries 2–5). A silver free system using IPrAuSbF6 and Ru(bpy)3(PF6)2 led to a slightly lower yield (88%, entry 6); thus, silver is not necessary in this reaction. Reactions in other solvents, such as acetonitrile and methanol, led to only traces of products and no solvent addition products, indicating that a carbocation mechanism is not involved (entries 7 and 8). Other iridium photocatalysts including, Ir[dF(CF3)ppy]2(dtbbpy)PF6 and fac-Ir(ppy)3, were also tested and no product was observed in the reaction system (entries 10–11). The attempt to lower the catalysts loading also led to a lower reaction yield (entry 12).
Table 1. Optimization of the reaction conditions a .
| ||
| Entry | Variation from the “standard” conditions | Yield b (%) |
| 1 | None | 94 (87) |
| 2 | AuCl instead of IPrAuCl | 0 |
| 3 | PPh3AuCl instead of IPrAuCl | 56 |
| 4 | IMesAuCl instead of IPrAuCl | 72 |
| 5 | PPh3AuNTf2 instead of IPrAuCl and AgSbF6 | 67 |
| 6 c | IPrAuSbF6 instead of IPrAuCl and AgSbF6 | 88 |
| 7 | CH3CN instead of DCE | 5 |
| 8 | CH3OH instead of DCE | 4 |
| 9 | Ru(phen)3Cl2 instead of Ru(bpy)3Cl2 | 67 |
| 10 | Ir[dF(CF3)ppy]2(dtbbpy)PF6 | 0 |
| 11 | fac-Ir(ppy)3 instead of Ru(bpy)3Cl2 | 0 |
| 12 | IPrAuCl (5 mol%), AgSbF6 (7.5 mol%), Ru(bpy)3Cl2 (1.2 mol%) | 64 |
aReaction conditions: a mixture of 1a (0.4 mmol), 2a (0.2 mmol), IPrAuCl (10 mol%), AgSbF6 (15 mol%), Ru(bpy)3Cl2 (2.5 mol%), in DCE (1 mL) was stirred at rt under irradiation with a 100 W blue LED at N2 atmosphere.
bDetermined by 19F NMR using (trifluoromethyl)benzene as the internal standard. The number in parentheses is the isolated yield. IPr = 1,3-bis(2,6-diisopropyl-phenyl)imidazol-2-ylidene, ppy = 2-phenylpyridine.
cRu(bpy)3(PF6)2 instead of Ru(bpy)3Cl2.
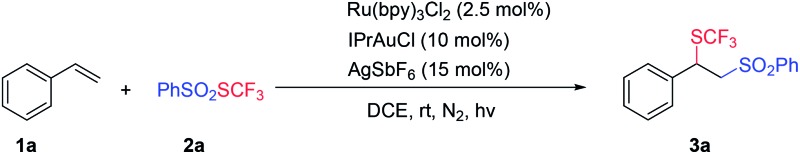
With these optimized conditions established, the scope of the dual gold and visible light-mediated alkene difunctionalization reaction was explored. As summarized in Table 2, a wide range of styrene-type alkenes were applicable and moderate to good isolated yields of the product were achieved (3a–3l). Substrates bearing different electron-withdrawing or electron-donating groups at different positions in the aromatic ring were all compatible with this reaction. A series of functional groups, such as ester, cyano, trifluoromethyl, and halogen, were all well-tolerated under the reaction conditions. In particular, the alkene 1m, containing an alkyne moiety, was also applicable to this reaction, giving the corresponding alkene difunctionalized product (3m) in moderate yield. Note that dienes, such as isoprene, were also amenable to this transformation, generating the 1,4-addition product (3n) as the major product in 55% yield, and its structure was confirmed by single crystal X-ray analysis.
Table 2. Substrate scope of alkene trifluoromethylthiosulfonylation reactions a .
|
aStandard conditions were employed. Isolated yields were reported. Diastereoselectivities were determined by GC-mass.
bFrom (E)-alkene.
cFrom (Z)-alkene.
dFrom (Z)-alkene : (E)-alkene = 7 : 3.
Internal alkenes, including acyclic and cyclic alkenes, were also suitable substrates. Most of the reactions exhibited excellent diastereoselectivities. Interestingly, trans-alkenes and cis-alkenes afforded the same major products (4o) in similar yields with similar diastereoselectivities, which is a very important feature and also an advantage of this reaction. The relative configuration of the seven-membered product 3s was unambiguously characterized by single X-ray crystallography. However, the reactions of aliphatic alkenes were unsuccessful under the same conditions.
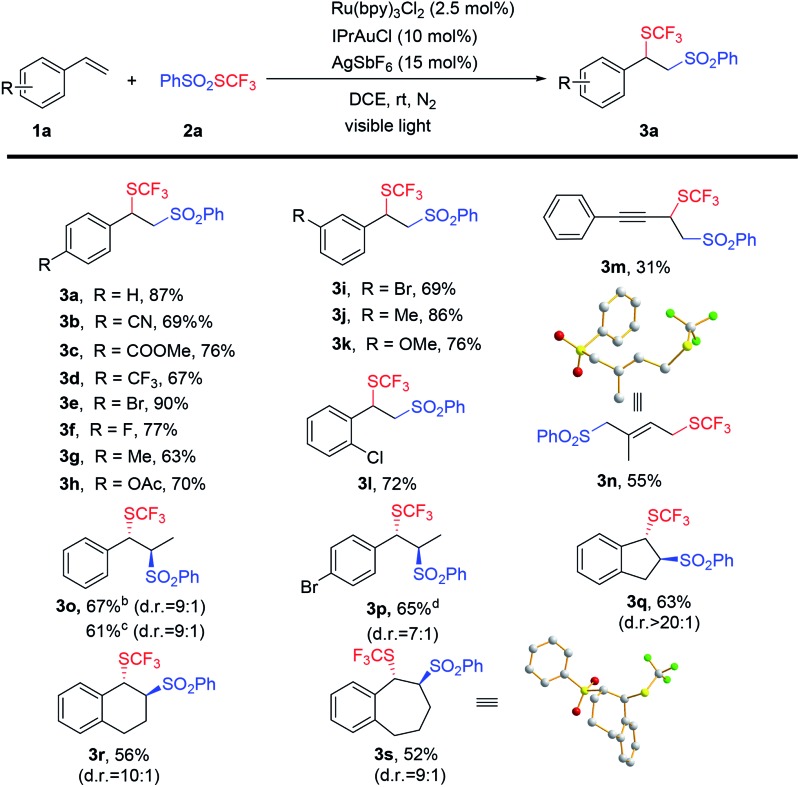
Organosulfur compounds are ubiquitous in the pharmaceutical industry, materials science, and food chemistry.15 The construction of C–S bonds is important but challenging16 because a sulfur atom could coordinate with a metal catalyst, such as Au, leading to catalyst inactivation. When we applied the abovementioned dual catalysis system to the general thiosulfonylation reaction of alkenes using PhSO2SC4H9 (–1.64 V, vs. SCE) as the reagent, which might be more challenging because of its lower oxidative potential compared to that of PhSO2SCF3 (–1.11 V, vs. SCE, Fig. S5, ESI†), to our delight, the reactions were very successful (Table 3). Various alkenes, including styrenes, internal alkenes, and dienes, were all compatible with this thiosulfonylation reaction, giving the corresponding products in good yields (4a–4r). A large variety of alkyl and aromatic thio groups can be easily introduced into a styrene molecule, generating difunctional products generally in very good yields (5a–5h).
Table 3. Substrate scope of alkene thiosulfonylation reactions a .
|
aStandard conditions were employed. Isolated yields were reported. Diastereoselectivities were determined by GC-mass.
bFrom (E)-alkene.
cFrom (Z)-alkene.
dFrom (Z)-alkene : (E)-alkene = 7 : 3.
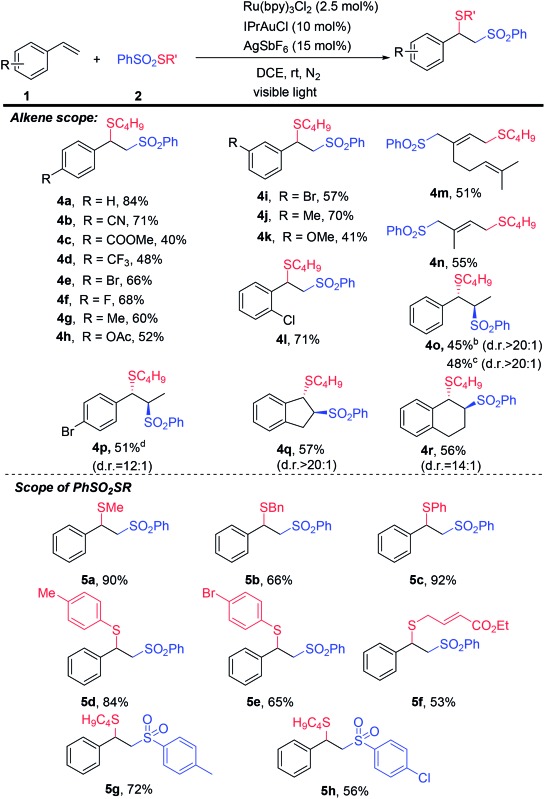
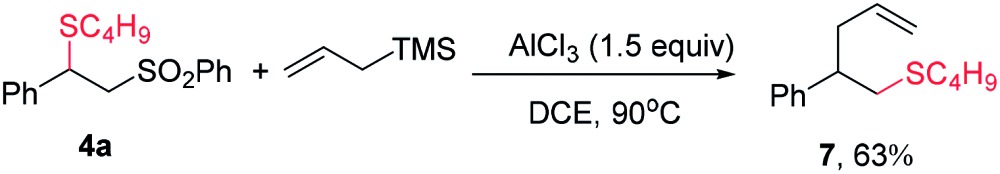
The sulfonyl group (–SO2–) is a useful synthon for further transformations. For example, the reaction of compound 4a with TMSCN or allylsilane in the presence of aluminium chloride provided the sulfur migration substitution products 6 and 7 in good yields (eqn (3) and (4)). This reaction might occur via a neighboring group participating in an SN1 type of reaction.
 |
3 |
 |
4 |
Control experiments were conducted to explore the mechanism of this reaction. When the reaction of 1,1-diphenylethylene 9 was carried out under standard conditions, vinyl sulfone (10) was isolated in 86% isolated yield (Scheme 2b). These results clearly indicate the formation of a benzenesulfonyl radical in the reaction system. The 19F NMR spectrum of the crude mixture from the reaction exhibited an additional signal at –26 ppm, which was later determined to be from IPrAuSCF3 (8). This compound could be independently synthesized by the reaction of IPrAuCl and AgSCF3, which is sufficiently stable to be isolated by column chromatography (Scheme 2a). A stoichiometric reaction between IPrAuCl 2a and the radical scavenger (9) afforded vinyl sulfone (10) in 81% isolated yield and 8 in 32% yield (Scheme 2c). This benzenesulfonyl radical could also be generated by PhSO2Cl (11) in the presence of visible light,3a and the reaction between 8, 11, and 2a afforded the target product (3a) in 98% yield (Scheme 2d). In contrast, without this gold intermediate, the direct atom transfer radical addition adduct 12 was obtained in 81% yield (Scheme 2e). This experiment demonstrated that IPrAuSCF3 is the possible reaction intermediate.17
Scheme 2. Control experiments.

Stern–Volmer fluorescence quenching experiments were performed to gain an insight into the photoredox catalytic cycle (for details, see the ESI (Fig. S1 and S2, ESI†)). The photoluminescence of Ru(bpy)3 2+ was quenched by IPrAuSbF6 with a rate constant of 8.85 × 102 L mol–1. In contrast, PhSO2SCF3 (–1.11 V vs. SCE) and styrene cannot serve as emission quenchers. The cyclic voltammogram of IPrAuSbF6 contains a reversible reduction peak at –0.11 V vs. SCE (Fig. S5, ESI†), indicating that this cationic gold catalyst is easily reduced by the excited state of the photocatalyst Ru(bpy)3 2+ (EIII/*II1/2 = –0.81 V vs. SCE).10b To further characterize this electron transfer reaction, a flash-photolysis study was carried out (Fig. S3†). Upon laser excitation by 355 nm light, the ground state absorption at ∼450 nm was obviously bleached and a characteristic absorption band at ∼360 nm was detected. This is ascribed to the reductive state of bipyridine in Ru(bpy)3(SbF6)2 (Fig. S4-a, ESI†).18 When PhSO2SCF3 was introduced into a solution of Ru(bpy)3(SbF6)2, the transient absorption spectra did not show any difference (Fig. S4-b, ESI†). The lifetime of the excited state of Ru(bpy)3(SbF6)2 slightly decreased from 493 to 473 ns, as observed from the kinetics probed at 450 nm. However, when IPrAuSbF6 was added to a solution of Ru(bpy)3(SbF6)2, a new absorption peak appeared at ∼530 nm. This is characteristic of gold nanoparticles. At the same time, the lifetime of the excited state of Ru(bpy)3(SbF6)2 decreased from 493 to 394 ns (Fig. S4-c, ESI†). All these results suggested that the electron transfer between IPrAuSbF6 and the excited 3MLCT state of Ru(bpy)3(SbF6)2 occurred, generating the active IPrAu(0) species that might aggregate to form gold nanoparticles.19 The reductive IPrAu(0) catalyst formed in situ is highly reactive20 and might react with PhSO2SCF3, providing IPrAuSCF3 (8) and a benzenesulfonyl radical.
On the basis of the abovementioned results and previous reports,12 a tentative proposed mechanism is shown in Scheme 3. Irradiation of Ru(bpy)3 2+ generates a long-lived photoexcited state Ru(bpy)3 2+*, which undergoes a single-electron transfer reaction with cationic IPrAu(i) to initiate the catalytic cycle and provide Ru(bpy)3 3+ and the highly active IPrAu(0), which can reduce PhSO2SCF3 to form IPrAu(i)SCF3 (8) and a benzenesulfonyl radical, which when added to styrene affords an alkyl radical. This radical is able to oxidize 8 to the Au(ii) intermediate A, which is further oxidized to the Au(iii) intermediate (B) by Ru(bpy)3 3+, generating Ru(bpy)3 2+ and thereby completing the photoredox catalytic cycle. Reductive elimination of the Au(iii) intermediate B delivers the product and regenerates the Au(i) catalyst. This alkyl radical directly reacting with another equivalent of PhSO2SCF3 could also afford the target product via a radical chain mechanism. In our reaction, when the internal alkenes were employed as reaction substrates, excellent diastereoselectivities were observed and both trans- and cis-alkenes yielded the same diastereomer, which is not common in a pure radical reaction. The IPrAu(i)SCF3 (8) approached the in situ formed alkyl radical from the less sterically hindered side, which might contribute to the excellent diastereoselectivity of this reaction. The proposed dual gold photoredox catalytic cycle may be the major pathway although the radical chain reaction couldn't be excluded. This gold catalytic cycle from Au(0) to Au(iii) is similar to that reported for nickel participating in photoredox catalytic cycles.
Scheme 3. Proposed mechanism.

Conclusions
In summary, we have reported an intermolecular atom transfer thiosulfonylation reaction of alkenes. Both trifluoromethylthio group and other functionalized thio groups can be introduced into alkenes with excellent regioselectivity and diastereoselectivity. These reactions are promoted by a synergistic combination of gold catalysis and visible light photoredox catalysis. Detailed control experiments and reaction mechanism studies indicate that the gold catalyst experiences four different valencies from Au(0) to Au(iii), which is unprecedented in previously reported Au-catalyzed transformations. This new dual gold and photocatalysis mode holds potential for applications to other important transformations.
Acknowledgments
We are grateful for the financial support received from the Natural Science Foundation of China and Shandong province (no. 21572118 & JQ201505), the fundamental research and subject construction funds (No. 2014JC008, 104.205.2.5), and Zhong-Ying Tang scholar award of Shandong University. We thank Prof. Dr Di Sun who carried out the X-ray crystallographic analysis.
Footnotes
References
- (a) McDonald R. I., Liu G., Stahl S. S. Chem. Rev. 2011;111:2981. doi: 10.1021/cr100371y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jensen K. H., Sigman M. S. Org. Biomol. Chem. 2008;6:4083. doi: 10.1039/b813246a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Zhu H., Chen P., Liu G. J. Am. Chem. Soc. 2014;136:1766. doi: 10.1021/ja412023b. [DOI] [PubMed] [Google Scholar]; (b) Chen C., Chen P., Liu G. J. Am. Chem. Soc. 2015;137:15648. doi: 10.1021/jacs.5b10971. [DOI] [PubMed] [Google Scholar]; (c) Zhu R., Buchwald S. L. J. Am. Chem. Soc. 2015;137:8069. doi: 10.1021/jacs.5b04821. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lu Q., Zhang J., Wei F., Qi Y., Wang H., Liu Z., Lei A. Angew. Chem., Int. Ed. 2013;52:7156. doi: 10.1002/anie.201301634. [DOI] [PubMed] [Google Scholar]
- (a) Wallentin C., Nguyen J. D., Finkbeiner P., Stephenson C. R. J. J. Am. Chem. Soc. 2012;134:8875. doi: 10.1021/ja300798k. [DOI] [PubMed] [Google Scholar]; (b) Nguyen J. D., Tucker J. W., Konieczynska M. D., Stephenson C. R. J. J. Am. Chem. Soc. 2011;133:4160. doi: 10.1021/ja108560e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Tomita R., Yasu Y., Koike T., Akita M. Angew. Chem., Int. Ed. 2014;53:7144. doi: 10.1002/anie.201403590. [DOI] [PubMed] [Google Scholar]; (d) Tomita R., Koike T., Akita M. Angew. Chem., Int. Ed. 2015;54:12923. doi: 10.1002/anie.201505550. [DOI] [PubMed] [Google Scholar]; (e) Qin Q., Ren D., Yu S. Org. Biomol. Chem. 2015;13:10295. doi: 10.1039/c5ob01725d. [DOI] [PubMed] [Google Scholar]; (f) Barton D. H. R., Csiba M. A., Jaszberenyi J. C. Tetrahedron Lett. 1994;35:2869. [Google Scholar]; (g) Bagal D. B., Kachkovskyi G., Knorn M., Rawner T., Bhanage B. M., Reiser O. Angew. Chem., Int. Ed. 2015;54:6999. doi: 10.1002/anie.201501880. [DOI] [PubMed] [Google Scholar]; (h) Courant T., Masson G. J. Org. Chem. 2016;81:6945. doi: 10.1021/acs.joc.6b01058. [DOI] [PubMed] [Google Scholar]
- Hansch C., Leo A., Unger S. H., Kim K. H., Nikaitani D., Lien E. J. J. Med. Chem. 1973;16:1207. doi: 10.1021/jm00269a003. [DOI] [PubMed] [Google Scholar]
- (a) Giudicelli J. F., Richer C., Berdeaux A. Br. J. Clin. Pharmacol. 1976;3:113. doi: 10.1111/j.1365-2125.1976.tb00578.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Harder A., Haberkorn A. Parasitol. Res. 1989;76:8. doi: 10.1007/BF00931064. [DOI] [PubMed] [Google Scholar]
- (a) Shao X., Wang X., Yang T., Lu L., Shen Q. Angew. Chem., Int. Ed. 2013;52:3457. doi: 10.1002/anie.201209817. [DOI] [PubMed] [Google Scholar]; (b) Teverovskiy G., Surry D. S., Buchwald S. L. Angew. Chem., Int. Ed. 2011;50:7312. doi: 10.1002/anie.201102543. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhang C., Vicic D. A. J. Am. Chem. Soc. 2012;134:183. doi: 10.1021/ja210364r. [DOI] [PubMed] [Google Scholar]; (d) Ferry A., Billard T., Langlois B. R., Bacque E. Angew. Chem., Int. Ed. 2009;48:8551. doi: 10.1002/anie.200903387. [DOI] [PubMed] [Google Scholar]; (e) Danoun G., Bayarmagnai B., Gruenberg M. F., Goossen L. J. Chem. Sci. 2014;5:1312. [Google Scholar]; (f) Xiao Q., He Q., Li J., Wang J. Org. Lett. 2015;17:6090. doi: 10.1021/acs.orglett.5b03116. [DOI] [PubMed] [Google Scholar]; (g) Luo J., Zhu Z., Liu Y., Zhao X. Org. Lett. 2015;17:3620. doi: 10.1021/acs.orglett.5b01727. [DOI] [PubMed] [Google Scholar]
- (a) Zhu L., Wang G., Guo Q., Xu Z., Zhang D., Wang R. Org. Lett. 2014;16:5390. doi: 10.1021/ol502624z. [DOI] [PubMed] [Google Scholar]; (b) Yin F., Wang X. Org. Lett. 2014;16:1128. doi: 10.1021/ol403739w. [DOI] [PubMed] [Google Scholar]; (c) Zhang K., Liu J., Qing F. Chem. Commun. 2014;50:14157. doi: 10.1039/c4cc07062c. [DOI] [PubMed] [Google Scholar]; (d) Hu M., Rong J., Miao W., Ni C., Han Y., Hu J. Org. Lett. 2014;16:2030. doi: 10.1021/ol500612n. [DOI] [PubMed] [Google Scholar]; (e) Zeng Y., Hu J. Org. Lett. 2016;18:856. doi: 10.1021/acs.orglett.6b00142. [DOI] [PubMed] [Google Scholar]; (f) Honeker R., Garza-Sanchez R. A., Hopkinson M. N., Glorius F. Chem.–Eur. J. 2016;22:4395. doi: 10.1002/chem.201600190. [DOI] [PubMed] [Google Scholar]
- Zhao F., Wang J., Ding X., Shu S., Liu H. Chin. J. Org. Chem. 2016;36:490. [Google Scholar]
- Najera C., Yus M. Tetrahedron. 1999;55:10547. [Google Scholar]
- For reviews of visible-light photoredox catalysis, see: ; (a) Xuan J., Zhang Z., Xiao W. Angew. Chem., Int. Ed. 2015;54:15632. doi: 10.1002/anie.201505731. [DOI] [PubMed] [Google Scholar]; (b) Prier C. K., Rankic D. A., MacMillan D. W. C. Chem. Rev. 2013;113:5322. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Xuan J., Xiao W. Angew. Chem., Int. Ed. 2012;51:6828. doi: 10.1002/anie.201200223. [DOI] [PubMed] [Google Scholar]; (d) Narayanam J. M. R., Stephenson C. R. J. Chem. Soc. Rev. 2011;40:102. doi: 10.1039/b913880n. [DOI] [PubMed] [Google Scholar]; (e) Yoon T. P., Ischay M. A., Du J. Nat. Chem. 2010;2:527. doi: 10.1038/nchem.687. [DOI] [PubMed] [Google Scholar]; (f) Chatterjee T., Iqbal N., You Y., Cho E. J. Acc. Chem. Res. 2016;49:2284. doi: 10.1021/acs.accounts.6b00248. [DOI] [PubMed] [Google Scholar]
- For review, see: ; (a) Skubi K. L., Blum T. R., Yoon T. P. Chem. Rev. 2016;116:10035. doi: 10.1021/acs.chemrev.6b00018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kalyani D., McMurtrey K. B., Neufeldt S. R., Sanford M. S. J. Am. Chem. Soc. 2011;133:18566. doi: 10.1021/ja208068w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Neufeldt S. R., Sanford M. S. Adv. Synth. Catal. 2012;354:3517. doi: 10.1002/adsc.201200738. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zoller J., Fabry D. C., Ronge M. A., Rueping M. Angew. Chem., Int. Ed. 2014;53:13264. doi: 10.1002/anie.201405478. [DOI] [PubMed] [Google Scholar]; (e) Xuan J., Zeng T., Feng Z., Deng Q., Chen J., Lu L., Xiao W., Alper H. Angew. Chem., Int. Ed. 2015;54:1625. doi: 10.1002/anie.201409999. [DOI] [PubMed] [Google Scholar]; (f) Zuo Z., Ahneman D. T., Chu L., Terrett J. A., Doyle A. G., MacMillan D. W. C. Science. 2014;345:437. doi: 10.1126/science.1255525. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Tellis J. C., Primer D. N., Molander G. A. Science. 2014;345:433. doi: 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Terrett J. A., Cuthbertson J. D., Shurtleff V. W., MacMillan D. W. C. Nature. 2015;524:330. doi: 10.1038/nature14875. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Zuo Z., Cong H., Li W., Choi J., Fu G. C., MacMillan D. W. C. J. Am. Chem. Soc. 2016;138:1832. doi: 10.1021/jacs.5b13211. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Ye Y., Sanford M. S. J. Am. Chem. Soc. 2012;134:9034. doi: 10.1021/ja301553c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For combination of photoredox with gold catalysis, see: ; (a) Hopkinson M. N., Tlahuext-Aca A., Glorius F. Acc. Chem. Res. 2016;49:2261. doi: 10.1021/acs.accounts.6b00351. [DOI] [PubMed] [Google Scholar]; (b) Sahoo B., Hopkinson M. N., Glorius F. J. Am. Chem. Soc. 2013;135:5505. doi: 10.1021/ja400311h. [DOI] [PubMed] [Google Scholar]; (c) Hopkinson M. N., Sahoo B., Glorius F. Adv. Synth. Catal. 2014;356:2794. [Google Scholar]; (d) Shu X., Zhang M., He Y., Frei H., Toste F. D. J. Am. Chem. Soc. 2014;136:5844. doi: 10.1021/ja500716j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) He Y., Wu H., Toste F. D. Chem. Sci. 2015;6:1194. doi: 10.1039/c4sc03092c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Kim S., Rojas-Martin J., Toste F. D. Chem. Sci. 2016;7:85. doi: 10.1039/c5sc03025k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Um J., Yun H., Shin S. Org. Lett. 2016;18:484. doi: 10.1021/acs.orglett.5b03531. [DOI] [PubMed] [Google Scholar]; (h) Huang L., Rudolph M., Rominger F., Hashmi A. S. K. Angew. Chem., Int. Ed. 2016;55:4808. doi: 10.1002/anie.201511487. [DOI] [PubMed] [Google Scholar]; (i) Cornilleau T., Hermange P., Fouquet E. Chem. Commun. 2016;52:10040. doi: 10.1039/c6cc04239b. [DOI] [PubMed] [Google Scholar]; (j) Gauchot V., Lee A.-L. Chem. Commun. 2016;52:10163. doi: 10.1039/c6cc05078f. [DOI] [PubMed] [Google Scholar]; (k) Tlahuext-Aca A., Hopkinson M. N., Daniliuc C. G., Glorius F. Chem.–Eur. J. 2016;22:11587. doi: 10.1002/chem.201602649. [DOI] [PubMed] [Google Scholar]; (l) Tlahuext-Aca A., Hopkinson M. N., Sahoo B., Glorius F. Chem. Sci. 2016;7:89. doi: 10.1039/c5sc02583d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Tlahuext-Aca A., Hopkinson M. N., Garza-Sanchez R. A., Glorius F. Chem.–Eur. J. 2016;22:5909. doi: 10.1002/chem.201600710. [DOI] [PubMed] [Google Scholar]; (n) Zhang Q., Zhang Z., Fu Y., Yu H. ACS Catal. 2016;6:798. [Google Scholar]; (o) Patil D. V., Yun H., Shin S. Adv. Synth. Catal. 2015;357:2622. [Google Scholar]; (p) Alcaide B., Almendros P., Busto E., Luna A. Adv. Synth. Catal. 2016;358:1526. [Google Scholar]; (q) Huang L., Rominger F., Rudolpha M., Hashmi A. S. K. Chem. Commun. 2016;52:6435. doi: 10.1039/c6cc02199a. [DOI] [PubMed] [Google Scholar]
- Gold(i) as photocatalyst, see: ; (a) Revol G., MaCallum T., Morin M., Gagosz F., Barriault L. Angew. Chem., Int. Ed. 2013;52:13342. doi: 10.1002/anie.201306727. [DOI] [PubMed] [Google Scholar]; (b) Xie J., Shi S., Zhang T., Mehrkens N., Rudolph M., Hashmi A. S. K. Angew. Chem., Int. Ed. 2015;54:6046. doi: 10.1002/anie.201412399. [DOI] [PubMed] [Google Scholar]
- (a) Zhang G., Cui L., Wang Y., Zhang L. J. Am. Chem. Soc. 2010;132:1474. doi: 10.1021/ja909555d. [DOI] [PubMed] [Google Scholar]; (b) Brenzovich W. E., Benitez D., Lackner A. D., Shunatona H. P., Tkatchouk E., Goddard III W. A., Toste F. D. Angew. Chem., Int. Ed. 2010;49:5519. doi: 10.1002/anie.201002739. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) de Haro T., Nevado C. Angew. Chem., Int. Ed. 2011;50:906. doi: 10.1002/anie.201005763. [DOI] [PubMed] [Google Scholar]; (d) Peng H., Xi Y., Ronaghi N., Dong B., Akhmedov N. G., Shi X. J. Am. Chem. Soc. 2014;136:13174. doi: 10.1021/ja5078365. [DOI] [PubMed] [Google Scholar]
- (a) Ilardi E. A., Vitaku E., Njardarson J. T. J. Med. Chem. 2014;57:2832. doi: 10.1021/jm401375q. [DOI] [PubMed] [Google Scholar]; (b) Feng M., Tang B., Liang S. H., Jiang X. Curr. Top. Med. Chem. 2016;16:1200. doi: 10.2174/1568026615666150915111741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Liu H., Jiang X. Chem.–Asian J. 2013;8:2546. doi: 10.1002/asia.201300636. [DOI] [PubMed] [Google Scholar]; (b) Qiao Z., Jiang X. Org. Lett. 2016;18:1550. doi: 10.1021/acs.orglett.6b00324. [DOI] [PubMed] [Google Scholar]; (c) Wang W., Peng X., Wei F., Tung C., Xu Z. Angew. Chem., Int. Ed. 2016;55:649. doi: 10.1002/anie.201509124. [DOI] [PubMed] [Google Scholar]
- Hashmi A. S. K. Angew. Chem., Int. Ed. 2010;49:5232. doi: 10.1002/anie.200907078. [DOI] [PubMed] [Google Scholar]
- Gao X., Meng Q., Li J., Zhong J., Lei T., Li X., Tung C., Wu L. ACS Catal. 2015;5:2391. [Google Scholar]
- For N-heterocyclic carbene modified gold surfaces, see: Zhukhovitskiy A. V., Mavros M. G., Voorhis T. V., Johnson J. A., J. Am. Chem. Soc., 2013, 135 , 7418 . [DOI] [PubMed] [Google Scholar]
- Gold nanoparticle could reduce thiol to form Au–S bond and release H2, see: Matthiesen J. E., Jose D., Sorensen C. M., Klabunde K. J., J. Am. Chem. Soc., 2012, 134 , 9376 . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.