

Abstract
Background
Next-Generation Sequencing (NGS) has made genomic mutation-driven therapy feasible for metastatic breast cancer (MBC) patients. We frequently submit tumor tissue from MBC patients for targeted NGS of tumor using the Illumina HiSeq 2000 platform (FoundationOne®, Foundation Medicine, MA). Herein, we report the results and clinical impact of this test in MBC patients.
Patients and Methods
We identified patients with MBC treated at City of Hope from January 2014 to May 2016 who underwent NGS. Patients’ clinical characteristics, response to treatment (clinical assessment of tumor regression), and genomic mutation profiles were reviewed.
Results
Forty-four patients with MBC underwent NGS: 24 triple negative breast cancer, 16 estrogen receptor positive, and 4 human epidermal growth factor receptor 2 positive patients. Twenty-three patients received more than three lines of chemotherapy prior to NGS. Actionable mutations (potentially responsive to targeted therapies that are on the market or in registered clinical trials) were identified in almost all patients (42/44; 95%) and over half of these 42 patients with actionable mutations (23/42; 55%) initiated mutation-driven targeted therapies. Of these 23 patients, 16/23 (70%) had assessable responses, and 7/23 (30%) were not assessable for response due to short exposure (<2 weeks) or hospice transition. The remaining 19/42 (45%) patients did not initiate targeted therapy.
Conclusion
NGS can identify effective targeted therapy options for MBC patients based on actionable mutations that were not previously offered based on pathology type. NGS should be performed early in patients with good performance status and preferably in clinical settings where genomic mutation-driven therapeutic trials are available.
Keywords: metastatic breast cancer (MBC), genomic profiling, genomic mutation, next-generation sequencing (NGS), targeted therapy
INTRODUCTION
Metastatic breast cancer (MBC) is one of the leading causes of cancer mortality in the United States, with over 40,000 deaths annually [1]. Current targeted therapies include everolimus, trastuzumab, lapatinib, and pertuzumab [2, 3]. Identification of actionable targets (those that are potentially responsive to targeted therapy) is urgently needed in patients with MBC who are refractory to standard therapies.
Aberrant mutations are commonly identified in patients with MBC. Screening for targetable genomic mutations and alterations may identify patients who could benefit from specific targeted therapies that may not be typically used in the treatment of breast cancer. As a result of technological advances, high-throughput sequencing, commonly known as “next-generation” sequencing (NGS) is now readily available for clinical use [4]. The success of The Cancer Genome Atlas (TCGA) Project, along with the improved reliability and affordability of NGS, allows the integration of genomic medicine with clinical practice [5].
The concept of using genomic information to guide therapy has become indispensable for precision medicine in cancer treatment. Its feasibility was tested in the SAFIR01/UNICANCER study [6] and in an MD Anderson screening protocol [7]. Both studies concluded that the implementation of genomic mutation/alteration testing is feasible, but only a small percentage of patients with “actionable mutations” were able to enroll onto genotype-matched trials (10.2% and 4.2% in the SAFIR01/UNICANCER and MD Anderson study, respectively) [8].
The concept of genomic-mutation driven therapy is being vigorously tested in the United States. The National Cancer Institute (NCI) recognized the potential of NGS followed by targeted therapy by initiating the Molecular Analysis for Therapy Choice (MATCH) Program. This trial clusters cancers of different types exclusively by genetic mutation and matched targeted therapy. Biopsies from 3000 tumors will undergo NGS to identify individuals whose tumors have genetic abnormalities that may respond to selected targeted drugs. This will be followed by assignment to the genomic-matched phase II study arm [9].
Commercially-available NGS approaches in combination with newer therapeutics targeting genomic mutations has resulted in a paradigm shift to use genomic targeted therapy in routine clinical practice for personalized care of cancer patients. Despite an appreciation for the use of genomics in defining treatment options, genomic mutation-matched clinical trial enrollment remains low [6, 7, 10]. This underscores the value of studying the impact of genomic profiling on treatment of MBC in a real-world setting. At the City of Hope Comprehensive Cancer Center we frequently submit tumor tissue from MBC patients for NGS. Here, we report the results and clinical impact of this test in MBC patients.
RESULTS
Patients
We identified 44 patients with MBC treated at City of Hope from January 2014 to May 2016 who underwent NGS; 24 triple negative breast cancer (TNBC), 16 estrogen receptor positive (ER+), and 4 human epidermal growth factor receptor 2 positive (HER2+) patients. Sites of biopsy varied (14 breast, 9 lymph node, 6 skin, 4 liver, 3 lung, 2 bone, 2 brain, one chest wall, one soft tissue, one adrenal gland, and one other). Patient characteristics, treatment history, presence/absence of actionable mutations, response to treatment, and outcomes associated with genomic testing are show in Table 1. The median age at the time of NGS was 54.5 years (range: 34-78). Of the 44 patients, 21 (47%) were non-Hispanic White, 8 (19%) were Hispanic, 7 (16%) were African American, 7 (16%) were Asian, and one (2%) was unknown. Twenty-three patients received more than three lines of chemotherapy prior to NGS.
Table 1. Characteristics of patients and outcome associated with NGS.
| Outcomes | TotalN=44 | TNBCN=24 | ER+HER2-N=16 | HER2+N=4 | |
|---|---|---|---|---|---|
| Age | Median (Range) | 54.5 (34-78) | |||
| Race | Non-Hispanic White | 21 | 8 | 10 | 3 |
| Hispanic | 8 | 7 | 1 | 0 | |
| African American | 7 | 6 | 1 | 0 | |
| Asian | 7 | 3 | 3 | 1 | |
| Other | 1 | 0 | 1 | 0 | |
| Lines of therapy prior to FM test | Median (Range) | 3 (0-13) | 2 (0-7) | 5 (0-13) | 5 (2-9) |
| 0-2 lines | 21 | 15 | 5 | 1 | |
| ≥ 3 lines | 23 | 9 | 11 | 3 | |
| Actionable mutation | Yes | 42 | 23 | 15 | 4 |
| No | 2 | 1 | 1 | 0 | |
| NGS-driven targeted therapy given | Subtotal | 23 | 13 | 9 | 1 |
| Therapy duration <2 weeks; transition to palliative care | 7 | 4 | 3 | 0 | |
| Therapy duration ≥6 weeks | 16 | 9 | 6 | 1 | |
| No NGS- driven targeted therapy | Subtotal | 19 | 10 | 6 | 3 |
| Transition to palliative care | 7 | 4 | 3 | 1 | |
| Patient's choice | 3 | 3 | 0 | 0 | |
| Treated with conventional chemotherapy | 5 | 3 | 0 | 1 | |
| Used/exhausted FM recommend therapy prior to FM test | 4 | 0 | 3 | 1 | |
| Response to -targeted therapy | Clinical benefit | 8 | 5 | 3 | 0 |
| Disease Progression | 8 | 4 | 3 | 1 | |
| N.A. (could not assess) | 7 | 4 | 3 | 0 |
Actionable mutations (potentially responsive to targeted therapies that are on the market or in registered clinical trials) were identified in almost all patients (42/44; 95%) and over half of these 42 patients with actionable mutations (23/42; 55%) initiated mutation-driven targeted therapies (Figure 1). Of these 23 patients, 16/23 (70%) had assessable responses, and 7/23 (30%) were not assessable for response due to short exposure (<2 weeks) or hospice transition. The remaining 19/42 (45%) patients did not initiate targeted therapy: 7 transitioned to palliative care/hospice, 5 received other chemotherapy by treating physicians, 4 exhausted all recommended targeted therapies, and 3 declined treatment (Figure 1). A total of 14 patients (33%) transitioned to palliative care within 2 months of the genomic test result becoming available.
Figure 1. Patient outcome (n=44).
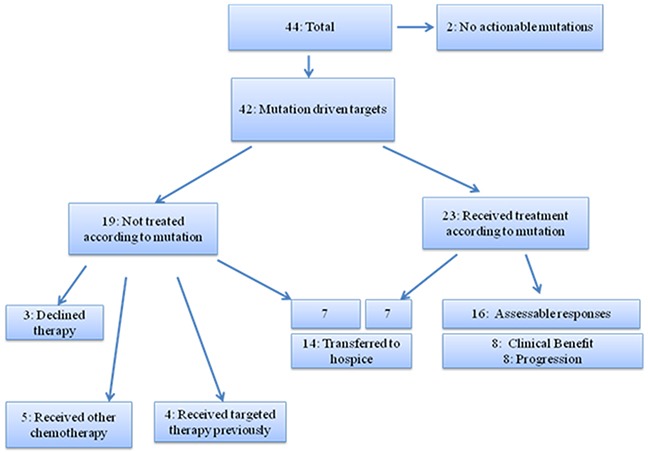
Treatment response
Among the 24 patients with metastatic TNBC, 13 patients received therapies not traditionally approved for TNBC (e.g. everolimus, pazopanib) and 9 had assessable responses (Table 1). Five patients had clinical benefit assessed by the treating clinician; 4 patients had progression of disease. Four patients could not be assessed for response due to limited drug exposure and/or transition to palliative care. Ten patients were not started on targeted therapy (four transitioned to palliative care; 3 chose not to start targeted therapy; and 3 were currently on other therapy). Of the TNBC group, 4 patients enrolled onto a phase I study, which was available at City of Hope. None of the patients were enrolled onto a genomic mutation-matched clinical trial due to lack of access.
Actionable mutations were identified in 15/16 (94%) patients with ER+/HER2- cancers (Table 1). Nine patients were started on targeted therapy of whom 6 had assessable response (3 clinical benefit, 3 progression of disease), and 3 did not have assessable response due to short exposure of <2 weeks and transition to hospice. Six patients did not receive targeted therapy as 3 had exposure to targeted therapies prior to genomic testing and 3 transitioned to palliative care.
Only one of the 4 patients with HER2+ MBC was treated with targeted therapy based on the NGS result and had progression of disease after enrolling onto two clinical trials. Three patients were not started on targeted therapy; one transitioned to hospice, one exhausted therapy options, and one is currently on other chemotherapy.
Mutations based on tumor type
Among three distinctive molecular subtypes, the most common shared alterations identified were TP53, PIK3CA, FGFR1, ZNF703, and CCND1 (Figure 2). TP53 and PIK3CA mutations were the most common genomic alterations observed (Figure 3): TP53 mutations (n=30; 68%); PIK3CA mutation or amplification (n=18; 41%); MYC amplification (n =12; 27%); PTEN loss (n=10; 23%); MYST3 (n=8; 18%); FGFR1 (n=8; 18%); ZNF703 (n=8; 18%); ERBB2 mutation or amplification (n=6; 14%); CDH1 (n=6; 14%); CCND1, FGF3, FGF4, and FGF 19 (n=5; 11%); GATA3 (n=5; 11%); and ESR1 (n=5; 11%). Consistent with other reports, FGF3, FGF4, and FGF19 were consistently co-amplified (often with CCND1), which can be attributed to the fact that they reside on the same amplicon on chromosome 11 [11, 12]. The number of patients with genetic alterations classified by cell signaling pathways such as RAS/MAPK, RTK/GFs, cell cycling, PI3K/mTOR, and p53 are shown in Figure 4.
Figure 2. Venn diagram showing number of patients (in parentheses) with specific molecular alterations based on receptor status (TNBC, ER+HER2-, and HER2+) (n=44).
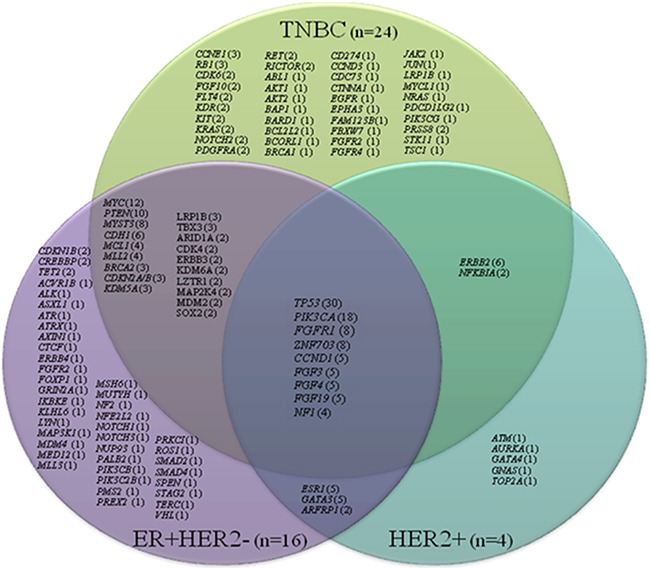
Figure 3. Number of patients with genomic alterations based on receptor status (TNBC, ER+HER2-, and HER2+) (n=44).
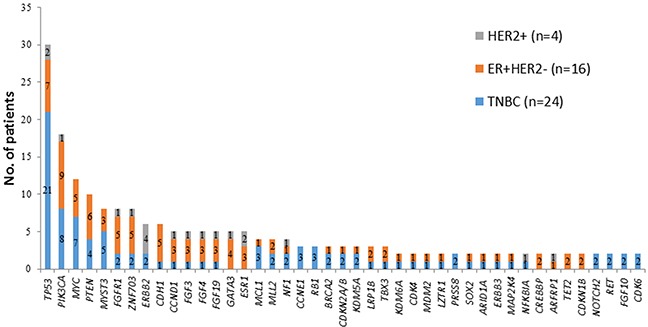
Figure 4. Number of breast cancer patients (n=44) with genetic alterations classified by cell signaling pathways: RAS/MAPK, RTK/GFs, cell cycling, PI3K/mTOR, and P53.
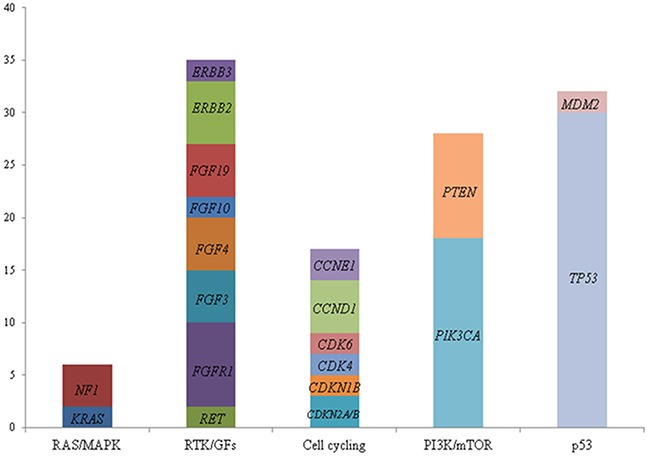
The ordinate indicates number of patients with alterations. RAS/MAPK: Ras GTPase/MAP kinase; RTK/GFs: Receptor tyrosine kinase/growth factors; PI3K/mTOR: phosphatidylinositol-3-kinase/mammalian target of rapamycin; P53: tumor protein p53.
The genomic mutations and clinical characteristics of the 7 patients who derived clinical benefit are listed in Table 2. The detailed information regarding these genes including function, frequency, and potential therapy, is listed in Table 3. Based on FGFR1 or FGFR2 amplifications, 2 patients received pazopanib. One had to stop therapy after 8 weeks due to elevated liver enzymes. Based on mutations in PIK3CA, 4 patients received an everolimus-containing regimen. Durable response (>32 weeks) was observed in 3 patients. Interestingly, these 3 patients carry different mutations: C420R, E545K-subclonal and N345K-subclonal, and E545K. PIK3CA C420R is in the tensin-type C2 domain and shows constitutive activation of lipid kinase activity and phosphorylation of Akt. PIK3CA E545K is a mutation in exon 9 that has shown poor prognosis. FGFR1 amplification has been correlated with mRNA over-expression, positive ER status, expression of P53, and poor prognosis (Table 3).
Table 2. Characteristics of the patients with clinical benefit from genomic mutation driven targeted therapy.
| Pt | Diagnosis | Prior lines of therapy | Mutation/alteration | Therapy(weeks) | Clinical course/Response | |
|---|---|---|---|---|---|---|
| 1 | TNBC | 3 | FGFR2 amplificationRET amp-equivocalCCND3 amplificationMCL1 amplification | MYC amplificationTP53 G266ECDC73 rearrangement intron 3 | Pazopanib (8) | Therapy discontinued due to elevated liver enzymes/ poor appetite after several dose reductions. Best response/duration: 12 weeks |
| 2 | TNBC | 6 | PIK3CA E726KPIK3CA H1047RNF1 Q1447*CCND1 amplificationBRCA2 E97*FGF19 amplification | MDM2 amplificationBCL2L2 amp–equivocalCDH1 splice site 531+1G>TFGF3 amplificationFGF4 amplification | Everolimusplus eribulin trial (8) | Inflammatory breast cancer with chest wall involvement. Response was quick but not durable. |
| 3 | TNBC | 1 | PIK3CA C420RCDK6 amplificationMYCL1 amplification | TP53 P190delJUN amplification | Everolimus (34) | Durable response of 34 weeks. |
| 4 | TNBC | 2 | TSC1 L628*TSC1 L628fs*45TP53 T256fs*8 | CDKN2A/B lossMLL2 S952fs*38 | Everolimus (13) | Significant improvement of systemic disease for 12 weeks. Then brain metastases and transition to hospice after whole brain radiation therapy. |
| 5 | ER+HER2- | 2 | PIK3CA E545K-subclonalPIK3CA N345K-subclonalFGFR1 amplificationARID1A G87fs*24 | ARID1A R356fs*44CDH1 D433NZNF703 amplification | Everolimusplus exemestane (24) | Progression on letrozole plus palbociclib prior switching to exemestane plus everolimus. Still on treatment after 32 weeks |
| 6 | ER+HER2- | 5 | FGFR1 amplificationMYC amplificationCREBBP Q943* | MYST3 amplificationZNF703 amplification | Pazopanib (32) | Duration of clinical benefit: 8 months on pazopanib with SD. 7/22/14 – 3/16/2015 –mastectomy. Post-op endocrine therapy till 8/21/15 liposomal doxorubicin till 9/14/15. Then eribulin 10/12/15 till now |
| 7 | ER+HER2- | 2 | PIK3CA E545KMAP2K4 T78fs*10 | Everolimusplus exemestane (32) | Progressed after durable response of 32 weeks | |
Bold: Actionable mutations.
Table 3. Actionable mutations and genomic mutation driven targeted therapy†.
| Mutations | Gene Function | Frequency in breastcancer and prognosis | Potential treatment |
|---|---|---|---|
| FGFR1 amplification Fibroblast growth factor receptor 1 (Fgfr1) | Cell cycle and angiogenesis; upstream regulator of Ras, MAPK, and Akt signaling pathways | 10-17%; poor prognosis | FGFR family inhibitors (pazopanib, regorafenib, ponatinib). Overexpression may be a mechanism of acquired resistance to gefitinib. FGFR1 amplification may contribute to resistance to hormonal therapy in ER+ breast cancers |
| FGFR2 amplification Fibroblast growth factor receptor 2 (Fgfr2) | Tyrosine kinase cell surface receptor (cell differentiation, growth, angiogenesis) | 1-11.5%; associated with resistance to chemotherapy | FGFR family of inhibitors (pazopanib, regorafenib, ponatinib) |
| PIK3CA E726K, PIK3CA H1047R Protein p110-alpha, catalytic subunit of PI3K | Cell signaling regulating cell growth, proliferation, differentiation, motility, and survival | 25-40%; H1047R associated with better prognosis than E542K, E545K mutations | PI3k/AKT/mTOR inhibitors (everolimus and temsirolimus); other mTOR inhibitors; PI3K and Akt inhibitors alone or in combination; PI3K alpha-specific BYL719. CDK4/6 inhibitors may sensitize PIK3CA mutant breast cancer to PI3K inhibitors. |
| PIK3CA C420R Protein p110-alpha, catalytic subunit of PI3K | Cell signaling regulating cell growth, proliferation, differentiation, motility, and survival | 26-38.5% | PI3k/AKT/mTOR inhibitors (everolimus and temsirolimus); other mTOR inhibitors; PI3K and Akt inhibitors alone or in combination; PI3K alpha-specific BYL719 CDK4/6 inhibitors may sensitize PIK3CA mutant breast cancer to PI3K inhibitors. |
| PIK3CA E545K – subclonal PIK3CA N345K – subclonal Protein p110-alpha, catalytic subunit of PI3K | Cell signaling regulating cell growth, proliferation, differentiation, motility, and survival. | 25-40% | PI3k/AKT/mTOR inhibitors (everolimus and temsirolimus); other mTOR inhibitors; PI3K and Akt inhibitors alone or in combination; PI3K alpha-specific BYL719 CDK4/6 inhibitors may sensitize PIK3CA mutant breast cancer to PI3K inhibitors; combined HER2+ PI3K pathways may be necessary in tumors with ERBB2 amplification and PIK3CA mutation. |
| TSC1 L628*, TSC1 L628fs*45 Protein hamartin | TSC1 forms heterodimer with TSC2 that acts as a GTPase activating protein for Rheb, a potent activator of the mammalian target of rapamycin (mTOR). | <1% | mTOR inhibitors (everolimus, temsirolimus, exemestane). Loss of TSC1 leads to activation of mTOR and therefore may predict sensitivity to mTOR inhibitors. |
†Information provided through Foundation Medicine®
DISCUSSION
In this single-center, retrospective analysis, we have shown that a genomic mutation profiling-based approach is feasible in identifying targetable genomic alterations and mutations in patients with advanced MBC. Genomic profiling led to treatment with molecular targeted therapy in 23 of 44 patients (52%). Of those, 7 had clinical response, highlighting the potential utility of the genomic profiling tool in this population. Unfortunately, 14 of 42 (33%) patients deteriorated quickly prior to initiation of targeted therapy or had a very short exposure to the targeted therapy (<2 weeks). None of the patients were enrolled onto matched genomic mutation-targeted therapeutic trials due to lack of access. The most commonly prescribed targeted therapy in this study was everolimus, which largely reflects the prevalence of phosphatidylinositol 3-kinase/mammalian targets of rapamycin (PI3K/mTOR) pathway alterations in the breast cancer population. This is a major pathway involved in the regulation of cell survival and proliferation, and it is the most frequently altered pathway in breast cancer [13]. Despite the encouraging findings, few other genomic mutations were targeted due to lack of access. These findings suggest that genomic testing must be offered early in the course of MBC treatment to allow access to targeted therapy and the opportunity to assess response. In addition, access to genomic mutation-driven clinical trials is most critical in utilizing genomic driven therapy. This finding is supported by other studies, and adds to the body of literature applying genomic medicine to real-world practice in breast cancer.
Lack of access to genotype-matched targeted therapy trials is a recurring theme across several studies. Parker et al. reported on the utilization of a multidisciplinary molecular tumor board in optimizing the management of 43 patients with advanced heavily-pretreated breast cancer undergoing genomic testing [11]. Seventeen of the 43 patients (40%) were treated with targeted therapy; 7 (16%) had stable disease for ≥6 months (n=2) or partial remission (n=5) [11]. Lack of access to targeted therapy was the main reason that patients could not be treated.
Based on recent large center experiences, genomic mutation-driven therapy has not yet been shown to improve patient's quality of life or clinical outcome. Recently Meric-Bernstam et al. reported a genomic mutation screening study of 2000 patients with advanced cancer who underwent a genomic testing protocol. Thirty nine percent of patients had ≥1 actionable mutation, but only 11% (n=83) of those were enrolled onto genotype-matched clinical trials. This translates to 4.1% of total patients screened [14]. Similarly, the SAFIR01/UNICANCER trial was designed as a multi-center molecular screening study to identify genomic mutations/alterations in breast cancer patients to provide matched targeted therapy [6]. Of 423 patients enrolled, genomic analyses led to potential matched targeted therapy in only 55 (13%) patients due to limited availability of therapeutic agents. Forty three (10%) patients received targeted therapy, 4 had an objective response, and 9 had stable disease for over 4 months [6]. Collectively, these studies underscore the urgent need to discover more effective molecularly targeted agents, refine treatment algorithms to take drug combinations into account, and investigate this approach at an earlier clinical time point.
Mutation profiles of refractory breast cancers in the current study were heterogeneous and none of the tumors carried the same genomic mutation profile; this is consistent with previous studies [15, 16]. Of the mutations identified, TP53 loss andPIK3CA mutation were the most common genomic alterations observed in this cohort of patients. Hyperactivation of the PI3K pathway occurs in 70% of breast cancers; and approximately 30% of breast cancers have mutations in PIK3CA [13]. PIK3CA mutation in TNBC has been reported with variable frequency, from 13% to 23.7% across several studies [8, 17–19]. Comparing the genomic mutation profiling with TCGA database, which mainly analyzed primary TNBC at the time of initial surgery, our heavily pretreated TNBC tumors carried a higher percentage of PIK3CA mutations (29% vs. 8%, p<0.01). In contrast, there were only moderate changes identified in the ER+HER2- population comparing metastatic/resistant tumors with primary tumors in the TCGA database. It is suspected that in refractory metastatic tumors, the mutation burden of this pathway may increase due to selection pressure from chemotherapies. The significance of these findings remains to be determined but may be attributed to small sample size and tumor heterogeneity.
Increasing evidence has demonstrated that the mutational landscape of tumors can change upon treatment with chemotherapy and/or targeted therapies with both gain and loss of actionable alterations. When alterations not detected in the original biopsy are present in a subsequent biopsy, it remains unclear whether these represent new mutations or selection for rare sub-clones already present in the primary tumor [7]. In order to understand individual tumor evolution with treatment selection pressure, we are currently conducting a paired tumor tissue genomic analysis study comparing primary and recurrent/refractory TNBCs. We will test if targeted exome sequencing can capture the heterogeneity in a tumor and the sub-clones that contribute to therapy resistance.
Our study is limited by small sample size and retrospective approach. This study used a targeted exome sequencing method without normal breast tissue control. A recent study by Jones et al. emphasized the importance of paired tumor-normal tissue analysis for precise identification and interpretation of somatic and germline alterations, which may impact management of patients [20]. In addition, genomic mutation-driven targeted therapies were recommended by each individual treating physician. Ideally, a molecular tumor board discussion with consensus agreement should be implemented for this approach. Additionally, during the study period, there were no genomic mutation-driven clinical trial options available. Lastly, one third of patients could not be treated or evaluated because of transition to palliative care or hospice, which limited the accessibility of the targeted agents in the metastatic setting.
METHODS
Patients
A team of physicians identified patients with MBC treated at City of Hope from January 2014 to May 2016. Archival tumor samples obtained from standard diagnostic or therapeutic procedures were tested with Clinical Laboratory Improvement Amendment-certified targeted NGS (FoundationOne®, Foundation Medicine, MA) using the Illumina HiSeq 2000 platform. FoundationOne® is a comprehensive genomic profile that applies NGS to identify base substitutions, insertions and deletions (indels), copy number alterations (CNAs), and rearrangements using formalin-fixed paraffin-embedded (FFPE) samples. FoundationOne® applies NGS across all genes known to be unambiguous drivers of solid tumors with high accuracy by sequencing the coding regions of 315 cancer-related genes plus introns from 28 genes often rearranged or altered in cancer to a typical median depth of coverage of greater than 500x. Eligible patients had stage IV breast cancer requiring systemic therapy. Patients’ age, demographics, tumor pathology types, stage and treatment histories, response to treatment, and genomic mutation profiles were collected by electronic medical chart review. FoundationOne® reports were reviewed.
Assessment of treatment response
The treatment response was based on retrospective review of restaging imaging using x-ray computerized tomography (CT) and bone scans, and/or treating physician's clinical assessment (not response evaluation criteria in solid tumor (RECIST) criteria).
Analysis of genomic mutation data
We generated a database of all genomic alterations based on the FoundationOne® reports. Descriptive statistics were used for this study.
CONCLUSION
Targeted genomic sequencing tools such as NGS can identify alterations that may respond to targeted therapies that have not generally been used based on pathology type alone. NGS should be performed early in patients with good performance status. We predict an increased use of the test in the community, although we recognize this approach should ideally be utilized in a setting where genomic mutation-driven therapeutic trials are available. In addition, customized combinations of targeted therapy, along with novel clinical trial design, may be needed to make genomic mutation-driven cancer medicine feasible in order to overcome tumor heterogeneity.
Acknowledgments
This study was supported by the National Cancer Institute of the National Institutes of Health under award number K12CA001727 (Joanne Mortimer MD) and the STOP Cancer Foundation (Yuan Yuan MD PhD).
Abbreviations
- ER+
Estrogen receptor positive
- HER2+
Human epidermal growth factor receptor 2 positive
- MATCH
Molecular Analysis for Therapy Choice
- MBC
Metastatic breast cancer
- NCI
National Cancer Institute
- NGS
Next-Generation Sequencing
- TCGA
The Cancer Genome Atlas
- TNBC
Triple negative breast cancer
Footnotes
CONFLICTS OF INTEREST
Dr. Yuan Yuan has contracted research sponsored by Merck, Eisai, Novartis and Pfizer. The other authors indicated no financial relationships.
FUNDING
This study was supported by the National Cancer Institute of the National Institutes of Health under award number K12CA001727 (Joanne Mortimer MD) and the STOP Cancer Foundation (Yuan Yuan MD PhD).
Author contributions
All of the authors made substantial contributions to data analysis, interpretation, and final approval of the manuscript. The first and last authors were responsible for the conception and design, acquisition of data, analysis and interpretation of data, and manuscript preparation.
Presentation
This study was presented as an abstract at the 2016 American Society of Clinical Oncology Annual Meeting; J Clin Oncol 34, 2016 (suppl; abstr e12558).
REFERENCES
- 1.Carneiro BA, Costa R, Taxter T, Chandra S, Chae YK, Cristofanilli M, Giles FJ. Is personalized medicine here? Oncology (Williston Park) 2016;30:293–303. 307. [PubMed] [Google Scholar]
- 2.Carneiro BA, Elvin JA, Kamath SD, Ali SM, Paintal AS, Restrepo A, Berry E, Giles FJ, Johnson ML. FGFR3-TACC3: A novel gene fusion in cervical cancer. Gynecologic Oncology Reports. 2015;13:53–56. doi: 10.1016/j.gore.2015.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chae YK, Pan A, Davis AA, Raparia K, Mohindra NA, Matsangou M, Giles FJ. Biomarkers for PD-1/PD-L1 Blockade Therapy in Non-Small-cell Lung Cancer: Is PD-L1 Expression a Good Marker for Patient Selection? Clinical Lung Cancer. 2016;S1525-7304:30057–30052. doi: 10.1016/j.cllc.2016.03.011. [DOI] [PubMed] [Google Scholar]
- 4.Emily Marcinkowski E, Luu T, Yuan Y, Mortimer J, Leong L, Portnow J, Xing Q, Wen W, Yim JH. The combination of eribulin and everolimus results in enhanced suppression of tumors in mouse models of triple negative breast cancer. SABCS. 2015 P6-13-17. [Google Scholar]
- 5.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andre F, Bachelot T, Commo F, Campone M, Arnedos M, Dieras V, Lacroix-Triki M, Lacroix L, Cohen P, Gentien D, Adelaide J, Dalenc F, Goncalves A, et al. Comparative genomic hybridisation array and DNA sequencing to direct treatment of metastatic breast cancer: a multicentre, prospective trial (SAFIR01/UNICANCER) The Lancet Oncology. 2014;15:267–274. doi: 10.1016/S1470-2045(13)70611-9. [DOI] [PubMed] [Google Scholar]
- 7.Meric-Bernstam F, Johnson A, Holla V, Bailey AM, Brusco L, Chen K, Routbort M, Patel KP, Zeng J, Kopetz S, Davies MA, Piha-Paul SA, Hong DS, et al. A decision support framework for genomically informed investigational cancer therapy. Journal of the National Cancer Institute. 2015:107. doi: 10.1093/jnci/djv098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cossu-Rocca P, Orru S, Muroni MR, Sanges F, Sotgiu G, Ena S, Pira G, Murgia L, Manca A, Uras MG, Sarobba MG, Urru S, De Miglio MR. Analysis of PIK3CA Mutations and Activation Pathways in Triple Negative Breast Cancer. PloS one. 2015;10:e0141763. doi: 10.1371/journal.pone.0141763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McNeil C. NCI-MATCH Launch Highlights New Trial Design in Precision-Medicine Era. Journal of the National Cancer Institute. 2015:107. doi: 10.1093/jnci/djv193. [DOI] [PubMed] [Google Scholar]
- 10.Le Tourneau C, Delord JP, Gonçalves A, Gavoille C, Dubot C, Isambert N, Campone M, Trédan O, Massiani MA, Mauborgne C, Armanet S, Servant N, Bièche I, et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. The Lancet Oncology. 16:1324–1334. doi: 10.1016/S1470-2045(15)00188-6. [DOI] [PubMed] [Google Scholar]
- 11.Parker BA, Schwaederlé M, Scur MD, Boles SG, Helsten T, Subramanian R, Schwab RB, Kurzrock R. Breast Cancer Experience of the Molecular Tumor Board at the University of California, San Diego. Journal of Oncology Practice. 2015;11:442–449. doi: 10.1200/JOP.2015.004127. [DOI] [PubMed] [Google Scholar]
- 12.Schwaederle M, Daniels GA, Piccioni DE, Fanta PT, Schwab RB, Shimabukuro KA, Parker BA, Kurzrock R. Cyclin alterations in diverse cancers: Outcome and co-amplification network. Oncotarget. 2015;6:3033–3042. doi: 10.18632/oncotarget.2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leroy C, Ramos P, Cornille K, Bonenfant D, Fritsch C, Voshol H, Bentires-Alj M. Activation of IGF1R/p110β/AKT/mTOR confers resistance to α-specific PI3K inhibition. Breast Cancer Research. 2016;18:1–13. doi: 10.1186/s13058-016-0697-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meric-Bernstam F, Brusco L, Shaw K, Horombe C, Kopetz S, Davies MA, Routbort M, Piha-Paul SA, Janku F, Ueno N, Hong D, De Groot J, Ravi V, et al. Feasibility of Large-Scale Genomic Testing to Facilitate Enrollment Onto Genomically Matched Clinical Trials. Journal of clinical oncology. 2015;33:2753–2762. doi: 10.1200/JCO.2014.60.4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wheler JJ, Parker BA, Lee JJ, Atkins JT, Janku F, Tsimberidou AM, Zinner R, Subbiah V, Fu S, Schwab R, Moulder S, Valero V, Schwaederle M, et al. Unique molecular signatures as a hallmark of patients with metastatic breast cancer: Implications for current treatment paradigms. Oncotarget. 2014;5:2349–2354. doi: 10.18632/oncotarget.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santarpia L, Qi Y, Stemke-Hale K, Wang B, Young EJ, Booser DJ, Holmes FA, O'Shaughnessy J, Hellerstedt B, Pippen J, Vidaurre T, Gomez H, Valero V, et al. Mutation profiling identifies numerous rare drug targets and distinct mutation patterns in different clinical subtypes of breast cancers. Breast cancer research and treatment. 2012;134:333–343. doi: 10.1007/s10549-012-2035-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arsenic R, Lehmann A, Budczies J, Koch I, Prinzler J, Kleine-Tebbe A, Schewe C, Loibl S, Dietel M, Denkert C. Analysis of PIK3CA mutations in breast cancer subtypes. Applied immunohistochemistry & molecular morphology. 2014;22:50–56. doi: 10.1097/pdm.0b013e318297afea. [DOI] [PubMed] [Google Scholar]
- 18.Arsenic R, Treue D, Lehmann A, Hummel M, Dietel M, Denkert C, Budczies J. Comparison of targeted next-generation sequencing and Sanger sequencing for the detection of PIK3CA mutations in breast cancer. BMC clinical pathology. 2015;15:20. doi: 10.1186/s12907-015-0020-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Millis SZ, Gatalica Z, Winkler J, Vranic S, Kimbrough J, Reddy S, O'Shaughnessy JA. Predictive Biomarker Profiling of > 6000 Breast Cancer Patients Shows Heterogeneity in TNBC, With Treatment Implications. Clinical Breast Cancer. 2015;15:473–481.e473. doi: 10.1016/j.clbc.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 20.Jones S, Anagnostou V, Lytle K, Parpart-Li S, Nesselbush M, Riley DR, Shukla M, Chesnick B, Kadan M, Papp E. Personalized genomic analyses for cancer mutation discovery and interpretation. Science translational medicine. 2015;7:283ra253–283ra253. doi: 10.1126/scitranslmed.aaa7161. [DOI] [PMC free article] [PubMed] [Google Scholar]