
Fig. 3.
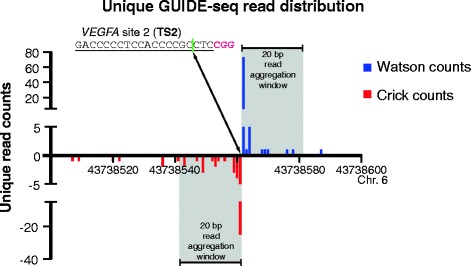
Unique read aggregation into peaks for the identification of potential nuclease cleavage sites. Strand-specific unique reads defined by the GUIDE-seq dsODN integration site and the read 2 genomic reference sequence strand are aggregated over a user-defined window size (20 base default) to define strand-specific peaks. Windows with a read number greater or equal to a user-defined threshold (default = 5) are called peaks. In addition, the signal to noise ratio (SNratio) and a p-value are computed based on the local background window size (defaults 5 kb and Poisson distribution), which can also be employed as filters if desired. For each integration site, the Crick peak should precede the corresponding Watson peak based on the library construction method [19]. Consequently, this order is required to combine counts from the Watson and Crick peaks over a user-defined window size (40 base default). This aggregate “score” is used to rank peaks. The genomic region surrounding each peak (adjustable variables, default 20 bases on each side) is used to search for sequences with homology to the nuclease sequence preference (based on the input guide sequence (gRNA.file) and the PAM sequence (PAM), and the allowed mismatches within each element defined by the parameters: max.mismatch, PAM.pattern and allowed.mismatch.PAM. The GUIDE-seq data shown were generated in house for SpCas9 programmed with a sgRNA to recognize VEGFA site 2 (TS2; protospacer underlined, PAM in red) [11], where the most common dsODN integration site falls at the expected cleavage site within this sequence (green line, hg19)