

Abstract
Noncoding RNAs (ncRNAs) play important roles in various cellular activities and diseases. In this paper, we presented a comprehensive review on computational methods for ncRNA prediction, which are generally grouped into four categories: (1) homology-based methods, that is, comparative methods involving evolutionarily conserved RNA sequences and structures, (2) de novo methods using RNA sequence and structure features, (3) transcriptional sequencing and assembling based methods, that is, methods designed for single and pair-ended reads generated from next-generation RNA sequencing, and (4) RNA family specific methods, for example, methods specific for microRNAs and long noncoding RNAs. In the end, we summarized the advantages and limitations of these methods and pointed out a few possible future directions for ncRNA prediction. In conclusion, many computational methods have been demonstrated to be effective in predicting ncRNAs for further experimental validation. They are critical in reducing the huge number of potential ncRNAs and pointing the community to high confidence candidates. In the future, high efficient mapping technology and more intrinsic sequence features (e.g., motif and k-mer frequencies) and structure features (e.g., minimum free energy, conserved stem-loop, or graph structures) are suggested to be combined with the next- and third-generation sequencing platforms to improve ncRNA prediction.
1. Background
A noncoding RNA (ncRNA) is a functional RNA that is transcribed from a DNA but does not encode a protein. According to transcriptomic and bioinformatics studies, there are thousands of ncRNAs classified into different categories based on their functions and lengths including transfer RNA (tRNA), ribosomal RNA (rRNA), microRNA (miRNA), and long ncRNA (lncRNA) to name a few [1–3].
These ncRNAs play important roles in various cellular processes. For example, rRNA catalyzes the peptide bond formation between amino acids in translation process [4], miRNA is important in transcription process and performs posttranscriptional regulation of gene expression [5], and lncRNA plays critical diverse roles in X inactivation, imprinting, and regulation of epigenetic marks and gene expression [6–8]. In addition, they also exhibit enormous importance in connection with various diseases. For example, the miR-17-92 cluster functions as oncogenes while the miR-15a–miR-16-1 cluster functions as tumour suppressors [9]. ANRIL, one type of lncRNA, is related to coronary disease, type II diabetes, and intracranial aneurysm [10]. The readers are referred to a review by Esteller [11] and Chen et al. [12] for more information about specific correlations between ncRNAs and human diseases. Specifically, Esteller [11] provides a review on the relationship between dysfunctions of ncRNAs including miRNA, PIWI-interacting RNAs (piRNAs), small nucleolar RNAs (snoRNAs), transcribed ultraconserved regions (T-UCRs), and large intergenic noncoding RNAs (lincRNAs) and a few diseases including tumorigenesis and neurological, cardiovascular, developmental, and other diseases. Chen et al. [12] discussed the roles of lncRNAs in critical biological processes and human diseases like various cancers, diabetes, and AIDS.
Due to the important roles of ncRNAs in cellular processes and disease development, many experimental and bioinformatics methods have been developed to predict ncRNAs and their functions. As for experimental methods, enzymatic and chemical RNA sequencing, parallel cloning of ncRNAs by specialized cDNA libraries, microarray analysis, and genomic SELEX are among the most popular ones. The readers are referred to a review paper for the details of these methods [13]. However, the experimental methods are expensive and time-consuming, and thus hundreds of computational methods have also been developed to prioritize highly confident ncRNA candidates for further experimental validation. In this paper, we present a comprehensive review on these computational methods. We are fully aware that there have already been several review articles on this hot topic [14–17]. However, they either focus on a specific ncRNA category or have been outdated and could not present a panoramic view of the field.
2. Main Text
Generally speaking, there are three major categories of computational methods in predicting ncRNAs, namely, (1) homology-based methods involving evolutionarily conserved RNA sequences and structures, (2) de novo methods using RNA sequence and structure features, and (3) transcriptional sequencing and assembling based methods, according to chronological order of their occurrences. Since miRNA and lncRNA have very specific methods, we reviewed them separately and called these methods RNA family specific methods (Figure 1).
Figure 1.

Four popular categories of computational methods in predicating ncRNAs. (a) Homology-based methods, which compare a query RNA with known ncRNAs deposited in databases based on sequence or structure alignment; (b) de novo methods, which predict ncRNA from primary sequences or structure based on general principles that govern ncRNA folding or statistical tendencies of k-mer features; (c) transcriptional sequencing and assembling based methods, which utilize next-generation sequencing and transcriptome data; and (d) RNA family specific methods, which predict specific ncRNA classes.
2.1. Homology-Based Methods
As probably the earliest ncRNA prediction methods, homology-based methods assume that sequence or structure similar RNAs are evolved from a common ancestor and thus share function similarities [18, 19]. Given a query RNA, these methods usually compare it with known ncRNAs deposited in databases based on sequence or structure alignment. The RNA is predicted to be in a specific ncRNA family if it has sufficient similarity with known ncRNAs in that family (Figure 1(a)). There are a number of ncRNA databases. For example, 2,474 structural families of ncRNAs were cataloged in the database Rfam (version 12.1, April 2016) [20]. We listed a few popular homology-based methods in Table 1, which are further classified into sequence-based methods, structure-based methods, and hybrid methods.
Table 1.
Homology-based ncRNA function prediction methods.
| Name | URL | Feature | Prediction algorithm |
|---|---|---|---|
| BLAST [18] | https://blast.ncbi.nlm.nih.gov/Blast.cgi | Sequence only | BLAST E-value |
|
| |||
| BLAT [21] | https://genome.ucsc.edu/cgi-bin/hgBlat | Sequence only | Pairwise alignment algorithm |
|
| |||
| CSHMM [38] | Structure only | A discriminant function based on likelihood score for a hidden Markov model (CSHMM) | |
|
| |||
| Infernal [20, 24] | http://infernal.janelia.org/ | Sequence and RNA secondary structure | Stochastic context-free grammars called covariance models (CMs), HMM |
|
| |||
| ERPIN [39] | http://rna.igmors.u-psud.fr/Software/erpin.php | Sequence and RNA secondary structure | Profile-based dynamic programming algorithm and E-value |
|
| |||
| QRNA [19] | Sequence only | Pair hidden Markov model | |
|
| |||
| RNAz [23] | https://www.tbi.univie.ac.at/~wash/RNAz/ | RNA secondary structure and thermodynamic stability | Support vector machine regression |
|
| |||
| Evofold [40] | https://github.com/bowhan/kent/blob/master/src/hg/makeDb/trackDb/drosophila/evofold.html | A log-odds score | Phylogenetic stochastic context-free grammars |
|
| |||
| MASTR [25] | http://mastr.binf.ku.dk/ | Mutual information with gap penalty, six canonical base pairs, stacking of adjacent base pairs, and the score combining the log-likelihood of the alignment, a covariation term, and the base-pair probabilities | Sampling approach by Markov chain Monte Carlo in a simulated annealing framework |
2.1.1. Sequence-Based Methods
These methods rely purely on sequence conservations inferred by alignment methods like BLAST [18] and BLAT [21]. They first identify short (gapped) matches called seeds [22] between the query ncRNA and any ncRNA in the database, which are then expanded in both directions to form high-scoring segment pair (HSPs). The statistical significance of a HSP or the joining of several HSPs is evaluated by expected value (called E-value). The query ncRNA is classified into the family containing the ncRNA with the lowest E-value.
2.1.2. Structure-Based Methods
Sequence-based methods are usually very fast. However, it is commonly believed that ncRNAs are less conserved in sequence. Thus, another category of homology-based methods is introduced based on structure conservations. Instead of sequence alignment, these methods use RNA secondary structure alignment to measure RNA similarity. Popular methods include QRNA [19] and RNAz [23]. Specifically, QRNA compares query RNA with known RNAs using “three probabilistic pair-grammars: a pair stochastic context-free grammar modeling alignments constrained by structural RNA evolution, a pair hidden Markov model modeling alignments constrained by coding sequence evolution, and a pair hidden Markov model modeling a null hypothesis of position-independent evolution” [19], whereas RNAz compares RNAs based on conserved secondary structure and thermodynamic stability [23].
2.1.3. Hybrid Methods
A more robust RNA similarity measure was obtained by incorporating both sequence and structure information. For example, Infernal [24] uses covariance models, which score a combination of sequence consensus and RNA secondary structure consensus to predict ncRNAs homologous to ncRNA families in Rfam [20, 24]. MASTR [25] makes use of simulated annealing method to perform sequence alignment and structural alignment simultaneously.
Though homology-based methods have been extensively used due to their advantages in speed, however, they have a few limitations. First, they compare the query RNA with known ncRNA families and thus are incapable of predicting new ncRNA families. Second, they rely on sequence or structure conservations and thus are inapplicable to predict ncRNAs lacking conservation in sequence and structure. As a result, de novo methods are proposed to solve such dilemma.
2.2. De Novo Methods Using RNA Sequence and Structure Features
Unlike homology methods which require the information of RNAs similar (or homologous) to the query RNA, de novo methods predict ncRNA from primary sequences or structure based on general principles that govern ncRNA folding energetics and/or statistical tendencies of k-mer features that native ncRNA sequences and structures acquire (Figure 1(b)). Based on the source of common features, de novo methods can be divided into sequence feature based methods which only use sequence features, structure feature methods, and hybrid feature methods which use both features.
2.2.1. Sequence Feature Based Methods
One important feature for sequence-based de novo methods is nucleotide composition, which applies for identifying ncRNAs in species with nucleotide compositional biases. For example, by calculating the GC content, Wang et al. identified ncRNA genes with stable secondary structure in an AT-rich extreme hyperthermophile [26]. Another commonly used nucleotide composition is k-mer (nucleotide sequence of length k) frequencies. Methods in this category exploit the finding that the frequencies of many k-mers for ncRNAs in a specific family usually share similar probability distribution. Thus, new ncRNAs can be predicted based on the distribution of their k-mer frequencies. For example, Panwar et al. used the trinucleotide composition (i.e., 3-mer) to predict ncRNA by a support vector machine (SVM) based algorithm [27]. Sun et al. proposed Coding-Non-Coding Index (CNCI), by profiling adjoining nucleotide triplets (i.e., 6-mer) to effectively distinguish protein-coding and noncoding sequences independent of known annotations [28]. In addition, Li et al. developed an algorithm named PLEK to discriminate lncRNAs from mRNAs based on a combination of 1 to 5 mers [29].
Since a single type of sequence feature might be insufficient in effectively identifying ncRNAs, other features have also been proposed in conjunction with nucleotide composition. We summarized a few popular sequence feature based de novo ncRNA identification methods in Table 2. For example, CONC [30] incorporates a few types of features including sequence length, nucleotide composition, and reading frame to characterize ncRNAs. CPC [31] combines the longest reading frame in the three forward frames, log-odds score, coverage of the predicted ORF, and integrity of the predicted ORF, to identify ncRNAs.
Table 2.
De novo ncRNA function prediction methods using RNA sequence features.
| Name | URL | Feature | Prediction algorithm |
|---|---|---|---|
| RNAcon [27] | http://crdd.osdd.net/raghava/rnacon/ | 3-mer of nucleotides | SVM with parameters and kernels optimized by model training |
|
| |||
| CNCI [28] | https://github.com/www-bioinfo-org/CNCI | Frequency of adjoining nucleotide triplets (6-mer), the length and S-score of most-like CDS, length-percentage, score-distance, and codon-bias | SVM using the standard radial basis function kernel |
|
| |||
| PLEK [29] | https://sourceforge.net/projects/plek/ | Normalized frequencies of 1–5 mers of RNA sequences | SVM |
|
| |||
| CONC [30] | Peptide length, amino acid composition, nucleotide frequencies, predicted secondary structure content, predicted percentage of exposed residues, compositional entropy, number of homologs from database searches, and alignment entropy | SVM | |
|
| |||
| CPC [31] | http://cpc.cbi.pku.edu.cn/ | The longest reading frame in the three forward frames, log-odds score, coverage of the predicted ORF, and integrity of the predicted ORF | SVM |
2.2.2. Structure Feature Based Methods
The secondary structures of some kinds of functional RNA are more conserved than their primary sequences [32]. For example, miRNA precursors share common hairpin-like structures and tRNAs share cloverleaf structures. The structure with (or around) the minimum folding energy (MFE) is usually regarded as the most possible fold structure of an RNA. Thus, MFE is extensively used to predict secondary structure of ncRNA sequences. Popular MFE-based methods include RNAfold [33], Mfold [34], and Afold [35]. RNAfold calculates MFE by assigning free energies to both loops and stems, whereas Mfold only assigns free energies to loops. Afold improves the speed in evaluating all possible internal loops by an algorithm constructing sets of conditionally optimal multibranch loop free (MLF) structures. However, it is generally insufficient to use MFE alone for the detection of ncRNAs since different secondary structures of a given RNA sequence may have very similar MFE [36]. As a result, more structure features like thermodynamic stability are also employed in predicting ncRNA [37].
2.2.3. Hybrid Feature Based Methods
As a trend, more and more de novo methods tend to combine both RNA sequence and RNA structure to improve the sensitivity and specificity in predicting ncRNAs.
For example, Gupta et al. developed a new algorithm ptRNApred to identify and classify posttranscriptional RNA with dinucleotide properties of sequence and secondary structure feature, for example, numbers of loops, bulges, and hairpins or the frequency of nucleotides involved in substructures [45]. It can predict ptRNA-subclasses in eukaryotes including snRNA, snoRNA, RNase P, RNase MRP, Y RNA, and telomerase RNA. We summarized popular de novo ncRNA prediction methods using RNA sequence and structure features in Table 3. For a better view, we also plotted some popular de novo methods and their prediction algorithms in Figure 2. Support vector machine (SVM) is probably the most frequently used method for de novo ncRNA prediction.
Table 3.
De novo ncRNA prediction methods using RNA structure features.
| Name | URL | Feature | Prediction algorithm |
|---|---|---|---|
| RNAfold [33] | Base-pair probabilities and MFE | Partition function and dynamic programming | |
|
| |||
| Mfold [34] | http://unafold.rna.albany.edu/?q=mfold | MFE | Dynamic programming |
|
| |||
| Afold [35] | ftp://ftp.ncbi.nlm.nih.gov/pub/ogurtsov/Afold | Sets of conditionally optimal multibranch loop free structures | Dynamic programming |
|
| |||
| Sfold [41] | http://sfold.wadsworth.org/cgi-bin/index.pl | Internal loops, sets of conditionally optimal MLF structures | Nearest-neighbour model (NNM) |
|
| |||
| Nussinov [42] | http://www.pnas.org/content/77/11/6309 | Individual base pairs and loop structure with the lowest free energy | Dynamic programming |
|
| |||
| Partition function method [43] | http://www.ncbi.nlm.nih.gov/pubmed/1695107 | Full equilibrium partition for secondary structure and the probabilities of various substructures | Dynamic programming |
|
| |||
| Zhang [44] | http://www.ncbi.nlm.nih.gov/pubmed/16395542/ | MFE and GC content | Dynamic programming |
|
| |||
| ptRNApred [45] | http://www.ptrnapred.org/ | 91 features including (1) 7 selected dinucleotide properties as well as their dinucleotide values, (2) 52 properties derived from the secondary structure, for example, the number of loops, and (3) 32 triplet element properties | Random forest and SVM |
|
| |||
| incRNA [46] | http://incrna.gersteinlab.org/ | 9 genomic features including 4 expression features, 3 sequence information, and 2 RNA structure features | Random forest |
Figure 2.
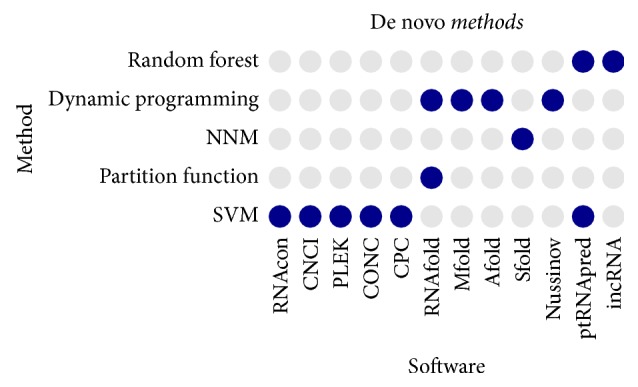
Popular de novo methods and the statistical algorithms applied.
De novo methods are capable of predicting new ncRNA families and classifying ncRNAs lacking conservation with existing ones. They usually have higher sensitivity and lower specificity than homology-based methods. However, this kind of methods depends largely on the features extracted. With the enrichment of biological, chemical, and dynamic knowledge of ncRNA, there might be some further informative features to be extracted, which will greatly benefit de novo ncRNA prediction [46, 68].
2.3. Transcriptional Sequencing and Assembling Based Methods
More recently, with the advances in next-generation sequencing (NGS), especially RNA sequencing (RNA-seq) techniques, more and more transcriptome data are available, which have been utilized to discover novel ncRNAs. A general workflow of transcriptional sequencing and assembling based ncRNA prediction method is described in Figure 1(c). Different from homology-based and de novo methods which require specific RNA sequences, methods in this category usually start from raw single-ended or pair-ended reads. The reads are then mapped into a reference genome and the mapped reads are assembled into transcripts based on overlapping information. After removing protein-coding RNA and known ncRNA transcripts, the remaining transcripts are further assessed for protein-coding potential and novel ncRNAs are reported if the potential is low.
In practice, RNA-seq data are usually combined with other features and methods including tilling array [47], graph-kernel SVM [49], structure features and common motifs [69], differential gene expression (DGE) data [48], and exon array [50] to predict specific ncRNAs. For example, tiling array [47] is used to scan the long and macro non-protein-coding RNAs related to cell-cycle, p53, and STAT3 pathways. DGE is used for discovering novel polyA+noncoding transcripts within human genome [48]. BlockClust [49] tries to predict the ncRNA modified after its transcription by combining the sequence and secondary structure information with a graph-kernel SVM, whose novel thinking lies in a new strategy to formulate expression profiles in compact discrete structures using fast graph-kernel techniques. We summarized some popular sequencing and assembling based ncRNA predication algorithms in Table 4.
Table 4.
Sequencing-assembling based whole ncRNA set methods.
| Name | URL | Feature | Prediction algorithm |
|---|---|---|---|
| Tilling array [47] | http://www.genomebiology.com/2014/15/3/R48 | Synonymous amino acid substitutions, reading frame conservation, and the occurrence of premature stop codons | RNAcode algorithm and biweight kernels |
|
| |||
| DigitagCT [48] | http://cractools.gforge.inria.fr/softwares/digitagct | Genomic sequences, DGE tags, and tiling array expression | Infernal and BLASTN |
|
| |||
| BlockClust [49] | http://toolshed.g2.bx.psu.edu/view/rnateam/blockclust_workflow | (1) The block group: entropy of read starts, entropy of read ends, entropy of read lengths, median of normalized read expressions and normalized read expression levels in first quantile; (2) block: number of multimapped reads, entropy of read lengths, entropy of read expressions, minimum read length and block length, and (3) block edge: contiguity and difference in median read expressions | Graph-kernel SVM |
|
| |||
| Noncoder [50] | http://noncoder.mpi-bn.mpg.de/ | Sequence homology, evolutionary information, the longest reading frame in three forward frames, log-odds score, coverage of the predicted orf, and integrity of the predicted orf | BLAT and PhyloCSF |
|
| |||
| Vicinal [51] | http://nar.oxfordjournals.org/content/42/9/e79.full.pdf+html | Chimeric RNA-cDNA fragments and terminal stem-loop | Bowtie 2 local mapping, filtering, and Vicinal mapping |
|
| |||
| CoRAL [52] | http://nar.oxfordjournals.org/content/41/14/e137.full.pdf+html | Read length, abundance of antisense transcription, 5′ and 3′ positional entropy, four nucleotide frequencies transformed into a log-odds ratio relative to equal base frequencies, and MFE | Multiclass classification random forest |
|
| |||
| FlaiMapper [53] | http://www.ncbi.nlm.nih.gov/pubmed/25338717 | Densities of start and end positions of aligned reads and read lengths | Peak detection on the start and end position densities followed by filtering and a reconstruction process |
As an advantage over homology-based methods and de novo methods, RNA-seq based methods can directly sequence coding and noncoding RNA transcripts with high sensitivity and low false positive rate. It can especially detect new scripts and alternative splicing. However, sometimes it is difficult to tell ncRNAs from protein-coding RNAs and thus other features like sequence conservation [53], deciphering abstract graphical representation [49], designing exon probes [50], finer terminal stem-loop feature [51], or k-mer frequency [52] are often utilized together with RNA-seq analysis to infer ncRNAs. In this sense, one may regard the RNA-seq technology as a platform rather than a certain method.
2.4. RNA Family Specific Methods
Since miRNA and lncRNA are two special and important ncRNAs, we reviewed a few computational methods related to them separately (Figure 1(d)).
2.4.1. miRNA Specific Methods
miRNAs are very short in length, usually around 22 nt. The short length and relatively low conservation of pre-miRNA sequences restrict the usage of sequence-based methods in identifying miRNAs. Fortunately, it is known that miRNAs are mostly derived from regions of RNA transcripts that fold back on themselves to form short hairpins, which make this RNA relatively conserved in secondary structure. Thus, a few methods exploit more secondary features for new miRNA gene detection instances. For example, as a homology-based method, miRAlign employs sequence alignment, secondary structure alignment, and miRNA's position on the stem-loop structure to identify RNA homologs. It has higher sensitivity and comparable specificity than other homology-based methods [70]. MiPred adopts the local contiguous structure sequence composition, MFE, and P value of randomization test to predict miRNA precursor with a random forest algorithm [54]. We summarized popular methods for predicting miRNA in Table 5.
Table 5.
Methods to predict miRNA.
| Name | URL | Feature | Prediction algorithm |
|---|---|---|---|
| CSHMM [38] | Structure only | A discriminant function based on likelihood score for a hidden Markov model | |
|
| |||
| MiPred [54] | 32 possible combinations of the middle nucleotide among the triplet elements, local contiguous structure sequence composition, MFE, and P value of randomization test | Random forest | |
|
| |||
| PlantMiRNAPred [55] | http://nclab.hit.edu.cn/PlantMiRNAPred/ | 115 features including (1) 17 primary sequence-related features, (2) 64 secondary structure-related features, and (3) 34 energy- and thermodynamics-related features | SVM |
|
| |||
| miRdentify [56] | http://www.ncrnalab.dk/#mirdentify/mirdentify.php | 5′ heterogeneity, overhangs, negative numbers indicating 5′ overhang, thermodynamics, entropy, tailing, and multimapping | Mapping and seeking duplex-forming reads within 46-80nt distance with the guide strand |
|
| |||
| CID-miRNA [57] | https://github.com/alito/CID-miRNA | Secondary structure likelihood | Stochastic context-free grammar model, Chomsky normal form; Cocke-Young-Kasami algorithm, and Classification tree |
|
| |||
| miRank [58] | https://omictools.com/mirank-tool | 36 global and local intrinsic features, including the normalized MFE of folding, the normalized base pairing propensities of both arms, and the normalized loop length | Belief propagation on a weighted graph, random walks-based ranking algorithm |
|
| |||
| miRCat [59] | http://srna-workbench.cmp.uea.ac.uk/tools/analysis-tools/mircat/ | E-value of alignment and MFE of secondary structure | Dynamic programming |
|
| |||
| mirTool [60] | http://centre.bioinformatics.zj.cn/mirtools/ | miRNA/miRNA, absolute/relative reads count, and the most abundant tag | Folding the flanking genomic sequence using the miRDeep program |
|
| |||
| miRanalyzer [61] | http://bioinfo5.ugr.es/miRanalyzer/miRanalyzer.php | Number of bindings in read cluster sequence, normalized mean free energy of precursor sequence, number of bindings in precursor, length of read cluster, the corresponding putative mature star sequence, number of bindings in read cluster divided by the read cluster length, number of reads in read cluster, mean free energy of precursor sequence, degree of bulb asymmetry in precursor, and the number of bulbs in precursor secondary structure | Random forest |
|
| |||
| sRNAbench [62] | http://bioinfo5.ugr.es/sRNAbench/ | Within cluster ratio, 5′ fluctuations, most frequent to all ratio, minimum number of hairpin bindings, minimum number of mature bindings, most frequent read, length interval, and minimum reads | Hierarchical clustering |
2.4.2. lncRNA Specific Methods
Long noncoding RNAs (lncRNAs) are ncRNAs longer than 200 nt, including long intronic noncoding RNA and intergenic noncoding RNA. lncRNAs are believed to regulate gene expression through changing chromatin state and correlate with cancer pathogenesis and various clinical traits [63–66, 71]. In fact, lncRNA prediction is a very challenging task, because many lncRNAs exhibit low sequence and structure conservation; moreover, they are often capped and spliced. Some databases like lncRNAdb [72] provide comprehensive annotations of specific lncRNAs, for example, eukaryotic lncRNAs. A general flow to identify lncRNA is as follows: first the transcriptome data are annotated and the protein-coding sequences are filtered; then sequences shorter than 200 nt are removed and the remaining ones are viewed as candidate lncRNAs [63]; finally, the candidate lncRNAs are evaluated based on features like secondary structures [73, 74], protein-coding ability [28, 29], conserved splicing sites [75], DGE+RNA-seq, conserved promoters [66], and chromatin signatures such as “K4–K36” domain [67], and only those that pass certain significance levels are inferred to be lncRNAs. We summarized popular lncRNA prediction methods in Table 6.
Table 6.
Methods to predict lncRNAs.
| Name | Feature | Prediction algorithm |
|---|---|---|
| Estimating lincRNome size for human [63] | lincRNA numbers validated experimentally in human and mouse, and their overlap lincRNA number | System of nonlinear equations |
|
| ||
| Classifying human lncRNA [64] | RNA sequence-structure patterns (RSSPs) describing 42 highly structured families, motif binding sites extracted as 1314 Position-Weight Matrices (PWMs), all k-words of length k = 2,3, 4,5, 6,7, 8, the sequence complexity | Classifying human lncRNA by being able (or disable) to bind the polycomb repressive complex (PRC2), SVM with linear kernel |
|
| ||
| Identify, classify, and localize maize lncRNAs [65] | Transcript length, open reading frame (ORF) size, and homology with known proteins | SVM |
|
| ||
| The GENCODE v7 catalog of human lncRNA [66] | Lack of homology with known proteins, no reasonable-sized open reading frame (ORF), and no high conservation, confirmed by PhyloCSF through the majority of exons conserved promoters | Manual annotation and pattern recognition |
|
| ||
| Highly conserved large noncoding RNAs [67] | Chromatin signatures “K4–K36” domain | Maximum CSF score observed across the entire genomic locus |
Besides the above two RNA families, some specific classification and prediction methods have been developed for ncRNAs with strong conservation information, for example, tRNA [76–78], snoRNA [79–81], and rRNA [82]. Recently, the largest ncRNA set, piRNA, can be predicted by an improved Fisher algorithm with 1364-D vectors representing RNA sequences [83, 84].
3. Conclusions
It is very important to predict ncRNAs since they are related to many diseases [85, 86]. Many ncRNA sequences are stored in databases such as fRNAbd [87], NONCODE [88], and Rfam [20] and grouped into classes based on their structures. The popular software Infernal [24] can predict 2,474 families of ncRNA. However, there are still ncRNAs that cannot be predicted by Infernal, including piRNA, Air, BC200, mature miRNA, gRNA, mRNA-like RNA, BC1 RNA, BM1 RNA, and so on. The major issue is that these ncRNAs lack sequence and structure conservation. To thoroughly predict the ncRNA classes and whole ncRNA set, we need to construct a series of new methods, including extracting new features and developing novel algorithms.
Homology search has become much faster with the development of bioinformatics tools, for example, from Smith-Waterman dynamic programming algorithm to BLAST or GMAP [89] based on simplified consecutive k-mer match or gapped k-mer (also called spaced seeds) techniques [22, 90]. However, these methods are less sensitive in ncRNA identification. On the other hand, de novo algorithms try to retrieve significant intrinsic features from RNA sequences, structures, energy, stability, and even deep-sequencing mapping profile. They use the features to discriminate a certain class of ncRNAs from other RNA sequences. However, de novo algorithms have high false positive rate. At present, how to combine these features and select a proper classifying machine is another hotspot to improve the sensitivity and specificity of ncRNA identification. With the rapid increasing of second- and third-generation sequencing (TGS) data, the information derived from deep-sequencing and single-molecule long-read sequencing may provide a great opportunity to enhance the efficiency in ncRNA prediction.
In addition, it has become central for understanding biological process by studying RNA globally. However, methods like microarrays and short-read sequencing are incapable of describing the entire RNA molecule from 5′ to 3′ end. Scientists use single-molecule long-read sequencing technology from Pacific Biosciences to sequence the polyadenylated RNA complement for human, without the need for fragmentation or amplification [91]. TGS can get full-length RNA molecules of up to 1.5 kb with little sequence loss at the 5′ ends. In total, ~14,000 spliced GENCODE genes of human were identified [91], but >10% of the alignments are mapped to unannotated regions; these transcripts are novel noncoding RNAs. Obviously, TGS may give more power to lncRNA discovery.
Finally, in order to assemble and correct long transcripts, one can integrate reads sequenced by five sequencing platforms including Illumina HiSeq, Life Technologies' PGM and Proton, Pacific Biosciences RS, and Roche's 454 [92]. Software programs like TMAP (PGM and Proton), GSRM (454), and GMAP (PacBio) are the best in mapping the sequencing reads to a reference genome. It has been shown that the integration results showed high concordance in both intraplatform and interplatform studies [92]. In addition, the integrated data also performed effectively in analyzing degraded RNA samples. Thus, platform integration is very promising for improvement of RNA-seq as well as ncRNA identification in the future.
Acknowledgments
The study was funded by the National Science Foundation (no. 11171088 to Yi Zhang and no. 61300120 to Jing Qiu); the Fundamental Research Funds for the Central Universities (no. 2016BC021 to Dahan Zhang); the Natural Science Foundation of Hebei Province (no. A2015208108 to Yi Zhang); the Educational Commission of Hebei Province on Humanities and Social Sciences (no. SZ16180 to Haiyun Huang); Science and Technology Plan Project of Hebei Province (no. 15210341 to Haiyun Huang); the research project of University Libraries in Hebei Province (201503Z to Haiyun Huang); the Science Fund of the Hebei University of Science and Technology Foundation (no. 2014PT67 to Yi Zhang); and the Hebei Province Foundation for Advanced Talents (no. A201400121 to Yi Zhang).
Abbreviations
- ncRNA:
Noncoding RNA
- RT-PCR:
Reverse transcription polymerase chain reaction
- tRNA:
Transfer RNA
- rRNA:
Ribosomal RNA
- snoRNA:
Small nucleolar RNA
- miRNA:
microRNA
- siRNA:
Small interfering RNA
- snRNA:
Small nuclear RNA
- exRNA:
Extracellular RNA
- piRNA:
Piwi-interacting RNA
- lncRNA:
Long noncoding RNA
- Xist:
X-inactive specific transcript
- HOTAIR:
HOX transcript antisense RNA
- ceRNA:
Competing endogenous RNA
- MALAT-1:
Metastasis-associated lung adenocarcinoma transcript 1
- HSP:
High-scoring segment pair
- HMM:
Hidden Markov model
- MFE:
Minimum folding energy
- SVM:
Support vector machine
- DGE:
Differential gene expression.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Authors' Contributions
Jialiang Yang and Yi Zhang conceived and designed the study. Yi Zhang, Jialiang Yang, Haiyun Huang, Dahan Zhang, Jing Qiu, Jiasheng Yang, Kejing Wang, Lijuan Zhu, and Jingjing Fan were involved in literature mining and summary. Jialiang Yang and Yi Zhang wrote the paper. All authors reviewed and approved the final manuscript. Yi Zhang, Haiyun Huang, and Dahan Zhang contributed equally to this work.
References
- 1.Cheng J., Kapranov P., Drenkow J., et al. Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science. 2005;308(5725):1149–1154. doi: 10.1126/science.1108625. [DOI] [PubMed] [Google Scholar]
- 2.Birney E., Stamatoyannopoulos J. A., Dutta A., et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447(7146):799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Washietl S., Pedersen J. S., Korbel J. O., et al. Structured RNAs in the ENCODE selected regions of the human genome. Genome Research. 2007;17(6):852–864. doi: 10.1101/gr.5650707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nissen P., Hansen J., Ban N., Moore P. B., Steitz T. A. The structural basis of ribosome activity in peptide bond synthesis. Science. 2000;289(5481):920–930. doi: 10.1126/science.289.5481.920. [DOI] [PubMed] [Google Scholar]
- 5.Chen K., Rajewsky N. The evolution of gene regulation by transcription factors and microRNAs. Nature Reviews Genetics. 2007;8(2):93–103. doi: 10.1038/nrg1990. [DOI] [PubMed] [Google Scholar]
- 6.Pandey R. R. A., Kanduri C. Transcriptional and posttranscriptional programming by long noncoding RNAs. Progress in Molecular and Subcellular Biology. 2011;51:1–27. doi: 10.1007/978-3-642-16502-3_1. [DOI] [PubMed] [Google Scholar]
- 7.Heo J. B., Sung S. Vernalization-mediated epigenetic silencing by a long intronic noncoding RNA. Science. 2011;331(6013):76–79. doi: 10.1126/science.1197349. [DOI] [PubMed] [Google Scholar]
- 8.Plessy C., Pascarella G., Bertin N., et al. Promoter architecture of mouse olfactory receptor genes. Genome Research. 2012;22(3):486–497. doi: 10.1101/gr.126201.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calin G. A., Croce C. M. MicroRNA signatures in human cancers. Nature Reviews Cancer. 2006;6(11):857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 10.Broadbent H. M., Peden J. F., Lorkowski S., et al. Susceptibility to coronary artery disease and diabetes is encoded by distinct, tightly linked SNPs in the ANRIL locus on chromosome 9p. Human Molecular Genetics. 2008;17(6):806–814. doi: 10.1093/hmg/ddm352. [DOI] [PubMed] [Google Scholar]
- 11.Esteller M. Non-coding RNAs in human disease. Nature Reviews Genetics. 2011;12(12):861–874. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 12.Chen X., Yan C. C., Zhang X., You Z. Long non-coding RNAs and complex diseases: from experimental results to computational models. Briefings in Bioinformatics. 2016:1–19. doi: 10.1093/bib/bbw060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hüttenhofer A., Vogel J. Experimental approaches to identify non-coding RNAs. Nucleic Acids Research. 2006;34(2):635–646. doi: 10.1093/nar/gkj469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ilott N. E., Ponting C. P. Predicting long non-coding RNAs using RNA sequencing. Methods. 2013;63(1):50–59. doi: 10.1016/j.ymeth.2013.03.019. [DOI] [PubMed] [Google Scholar]
- 15.Machado-Lima A., del Portillo H. A., Durham A. M. Computational methods in noncoding RNA research. Journal of Mathematical Biology. 2008;56(1-2):15–49. doi: 10.1007/s00285-007-0122-6. [DOI] [PubMed] [Google Scholar]
- 16.Wang C., Wei L., Guo M., Zou Q. Computational approaches in detecting non- coding RNA. Current Genomics. 2013;14(6):371–377. doi: 10.2174/13892029113149990005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krzyzanowski P. M., Muro E. M., Andrade-Navarro M. A. Computational approaches to discovering noncoding RNA. Wiley Interdisciplinary Reviews RNA. 2012;3(4):567–579. doi: 10.1002/wrna.1121. [DOI] [PubMed] [Google Scholar]
- 18.Altschul S. F., Gish W., Miller W., Meyers E. W., Lipman D. J. Basic local alignment search tool. Journal of Molecular Biology. 1990;215(3):403–410. doi: 10.1006/jmbi.1990.9999. [DOI] [PubMed] [Google Scholar]
- 19.Rivas E., Eddy S. R. Noncoding RNA gene detection using comparative sequence analysis. BMC Bioinformatics. 2001;2, article 8 doi: 10.1186/1471-2105-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gardner P. P., Daub J., Tate J. G., et al. Rfam: updates to the RNA families database. Nucleic Acids Research. 2009;37(1):D136–D140. doi: 10.1093/nar/gkn766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kent W. J. BLAT—the BLAST-like alignment tool. Genome Research. 2002;12(4):656–664. doi: 10.1101/gr.229202.articlepublishedonlinebeforemarch2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang J., Zhang L. Run probabilities of seed-like patterns and identifying good transition seeds. Journal of Computational Biology. 2008;15(10):1295–1313. doi: 10.1089/cmb.2007.0209. [DOI] [PubMed] [Google Scholar]
- 23.Washietl S., Hofacker I. L., Stadler P. F. Fast and reliable prediction of noncoding RNAs. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(7):2454–2459. doi: 10.1073/pnas.0409169102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nawrocki E. P., Kolbe D. L., Eddy S. R. Infernal 1.0: inference of RNA alignments. Bioinformatics. 2009;25(10):1335–1337. doi: 10.1093/bioinformatics/btp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lindgreen S., Gardner P. P., Krogh A. MASTR: multiple alignment and structure prediction of non-coding RNAs using simulated annealing. Bioinformatics. 2007;23(24):3304–3311. doi: 10.1093/bioinformatics/btm525. [DOI] [PubMed] [Google Scholar]
- 26.Wang X.-J., Reyes J. L., Chua N.-H., Gaasterland T. Prediction and identification of Arabidopsis thaliana microRNAs and their mRNA targets. Genome Biology. 2004;5(9, article R65) doi: 10.1186/gb-2004-5-9-r65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Panwar B., Arora A., Raghava G. P. S. Prediction and classification of ncRNAs using structural information. BMC Genomics. 2014;15(1, article 127) doi: 10.1186/1471-2164-15-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun L., Luo H., Bu D., et al. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Research. 2013;41(17, article e166) doi: 10.1093/nar/gkt646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li A., Zhang J., Zhou Z. PLEK: a tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinformatics. 2014;15(1, article 311) doi: 10.1186/1471-2105-15-311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu J., Gough J., Rost B. Distinguishing protein-coding from non-coding RNAs through support vector machines. PLoS Genetics. 2006;2(4, article e29) doi: 10.1371/journal.pgen.0020029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kong L., Zhang Y., Ye Z.-Q., et al. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Research. 2007;35(2):W345–W349. doi: 10.1093/nar/gkm391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Washietl S., Hofacker I. L., Lukasser M., Hüttenhofer A., Stadler P. F. Mapping of conserved RNA secondary structures predicts thousands of functional noncoding RNAs in the human genome. Nature Biotechnology. 2005;23(11):1383–1390. doi: 10.1038/nbt1144. [DOI] [PubMed] [Google Scholar]
- 33.Hofacker I. L. Vienna RNA secondary structure server. Nucleic Acids Research. 2003;31(13):3429–3431. doi: 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Research. 2003;31(13):3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ogurtsov A. Y., Shabalina S. A., Kondrashov A. S., Roytberg M. A. Analysis of internal loops within the RNA secondary structure in almost quadratic time. Bioinformatics. 2006;22(11):1317–1324. doi: 10.1093/bioinformatics/btl083. [DOI] [PubMed] [Google Scholar]
- 36.Rivas E., Eddy S. R. Secondary structure alone is generally not statistically significant for the detection of noncoding RNAs. Bioinformatics. 2000;16(7):583–605. doi: 10.1093/bioinformatics/16.7.583. [DOI] [PubMed] [Google Scholar]
- 37.Tran T. T., Zhou F., Marshburn S., Stead M., Kushner S. R., Xu Y. De novo computational prediction of non-coding RNA genes in prokaryotic genomes. Bioinformatics. 2009;25(22):2897–2905. doi: 10.1093/bioinformatics/btp537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Agarwal S., Vaz C., Bhattacharya A., Srinivasan A. Prediction of novel precursor miRNAs using a context-sensitive hidden Markov model (CSHMM) BMC Bioinformatics. 2010;11(supplement 1, article S29) doi: 10.1186/1471-2105-11-s1-s29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gautheret D., Lambert A. Direct RNA motif definition and identification from multiple sequence alignments using secondary structure profiles. Journal of Molecular Biology. 2001;313(5):1003–1011. doi: 10.1006/jmbi.2001.5102. [DOI] [PubMed] [Google Scholar]
- 40.Pedersen J. S., Bejerano G., Siepel A., et al. Identification and classification of conserved RNA secondary structures in the human genome. PLoS Computational Biology. 2006;2(4, article e33) doi: 10.1371/journal.pcbi.0020033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ding Y., Chan C. Y., Lawrence C. E. Sfold web server for statistical folding and rational design of nucleic acids. Nucleic Acids Research. 2004;32:W135–W141. doi: 10.1093/nar/gkh449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nussinov R., Jacobson A. B. Fast algorithm for predicting the secondary structure of single-stranded RNA. Proceedings of the National Academy of Sciences of the United States of America. 1980;77(11):6903–6913. doi: 10.1073/pnas.77.11.6309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McCaskill J. S. The equilibrium partition function and base pair binding probabilities for RNA secondary structure. Biopolymers. 1990;29(6-7):1105–1119. doi: 10.1002/bip.360290621. [DOI] [PubMed] [Google Scholar]
- 44.Zhang B. H., Pan X. P., Cox S. B., Cobb G. P., Anderson T. A. Evidence that miRNAs are different from other RNAs. Cellular and Molecular Life Sciences. 2006;63(2):246–254. doi: 10.1007/s00018-005-5467-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gupta Y., Witte M., Möller S., et al. PtRNApred: computational identification and classification of post-transcriptional RNA. Nucleic Acids Research. 2014;42(22, article e167) doi: 10.1093/nar/gku918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu Z. J., Yip K. Y., Wang G., et al. Prediction and characterization of noncoding RNAs in C. elegans by integrating conservation, secondary structure, and high-throughput sequencing and array data. Genome Research. 2011;21(2):276–285. doi: 10.1101/gr.110189.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hackermüller J., Reiche K., Otto C., et al. Cell cycle, oncogenic and tumor suppressor pathways regulate numerous long and macro non-protein-coding RNAs. Genome Biology. 2014;15(3, article R48) doi: 10.1186/gb-2014-15-3-r48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Philippe N., Bou Samra E., Boureux A., et al. Combining DGE and RNA-sequencing data to identify new polyA+ non-coding transcripts in the human genome. Nucleic Acids Research. 2014;42(5):2820–2832. doi: 10.1093/nar/gkt1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Videm P., Rose D., Costa F., Backofen R. BlockClust: efficient clustering and classification of non-coding RNAs from short read RNA-seq profiles. Bioinformatics. 2014;30(12):I274–I282. doi: 10.1093/bioinformatics/btu270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gellert P., Ponomareva Y., Braun T., Uchida S. Noncoder: a web interface for exon array-based detection of long non-coding RNAs. Nucleic Acids Research. 2013;41(1, article e20) doi: 10.1093/nar/gks877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lu Z., Matera A. G. Vicinal: a method for the determination of ncRNA ends using chimeric reads from RNA-seq experiments. Nucleic Acids Research. 2014;42(9, article e79) doi: 10.1093/nar/gku207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Leung Y. Y., Ryvkin P., Ungar L. H., Gregory B. D., Wang L.-S. CoRAL: predicting non-coding RNAs from small RNA-sequencing data. Nucleic Acids Research. 2013;41(14, article e137) doi: 10.1093/nar/gkt426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hoogstrate Y., Jenster G., Martens-Uzunova E. S. FlaiMapper: computational annotation of small ncRNA-derived fragments using RNA-seq high-throughput data. Bioinformatics. 2015;31(5):665–673. doi: 10.1093/bioinformatics/btu696. [DOI] [PubMed] [Google Scholar]
- 54.Jiang P., Wu H., Wang W., Ma W., Sun X., Lu Z. MiPred: classification of real and pseudo microRNA precursors using random forest prediction model with combined features. Nucleic Acids Research. 2007;35(2):W339–W344. doi: 10.1093/nar/gkm368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xuan P., Guo M., Liu X., Huang Y., Li W., Huang Y. PlantMiRNAPred: efficient classification of real and pseudo plant pre-miRNAs. Bioinformatics. 2011;27(10):1368–1376. doi: 10.1093/bioinformatics/btr153. [DOI] [PubMed] [Google Scholar]
- 56.Hansen T. B., Venø M. T., Kjems J., Damgaard C. K. miRdentify: high stringency miRNA predictor identifies several novel animal miRNAs. Nucleic Acids Research. 2014;42(16, article e124) doi: 10.1093/nar/gku598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tyagi S., Vaz C., Gupta V., et al. CID-miRNA: a web server for prediction of novel miRNA precursors in human genome. Biochemical and Biophysical Research Communications. 2008;372(4):831–834. doi: 10.1016/j.bbrc.2008.05.134. [DOI] [PubMed] [Google Scholar]
- 58.Xu Y., Zhou X., Zhang W. MicroRNA prediction with a novel ranking algorithm based on random walks. Bioinformatics. 2008;24(13):i50–i58. doi: 10.1093/bioinformatics/btn175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moxon S., Schwach F., Dalmay T., MacLean D., Studholme D. J., Moulton V. A toolkit for analysing large-scale plant small RNA datasets. Bioinformatics. 2008;24(19):2252–2253. doi: 10.1093/bioinformatics/btn428. [DOI] [PubMed] [Google Scholar]
- 60.Zhu E., Zhao F., Xu G., et al. mirTools: microRNA profiling and discovery based on high-throughput sequencing. Nucleic Acids Research. 2010;38(2):W392–W397. doi: 10.1093/nar/gkq393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hackenberg M., Sturm M., Langenberger D., Falcón-Pérez J. M., Aransay A. M. miRanalyzer: a microRNA detection and analysis tool for next-generation sequencing experiments. Nucleic Acids Research. 2009;37(2):W68–W76. doi: 10.1093/nar/gkp347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Barturen G., Rueda A., Hamberg M., et al. sRNAbench: profiling of small RNAs and its sequence variants in single or multi-species high-throughput experiments. Methods in Next Generation Sequencing. 2014;1(1):21–31. doi: 10.2478/mngs-2014-0001. [DOI] [Google Scholar]
- 63.Managadze D., Lobkovsky A. E., Wolf Y. I., Shabalina S. A., Rogozin I. B., Koonin E. V. The vast, conserved mammalian lincRNome. PLoS Computational Biology. 2013;9(2) doi: 10.1371/journal.pcbi.1002917.e1002917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Glazko G. V., Zybailov B. L., Rogozin I. B. Computational prediction of polycomb-associated long non-coding RNAs. PLoS ONE. 2012;7(9) doi: 10.1371/journal.pone.0044878.e44878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Boerner S., McGinnis K. M. Computational identification and functional predictions of long noncoding RNA in Zea mays. PLoS ONE. 2012;7(8) doi: 10.1371/journal.pone.0043047.e43047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Derrien T., Johnson R., Bussotti G., et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Research. 2012;22(9):1775–1789. doi: 10.1101/gr.132159.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Guttman M., Amit I., Garber M., et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458(7235):223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Saeys Y., Inza I., Larrañaga P. A review of feature selection techniques in bioinformatics. Bioinformatics. 2007;23(19):2507–2517. doi: 10.1093/bioinformatics/btm344. [DOI] [PubMed] [Google Scholar]
- 69.Liu T.-T., Zhu D., Chen W., et al. A global identification and analysis of small nucleolar RNAs and possible intermediate-sized non-coding RNAs in or. Molecular Plant. 2013;6(3):830–846. doi: 10.1093/mp/sss087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang X., Zhang J., Li F., et al. MicroRNA identification based on sequence and structure alignment. Bioinformatics. 2005;21(18):3610–3614. doi: 10.1093/bioinformatics/bti562. [DOI] [PubMed] [Google Scholar]
- 71.Chisholm K. M., Wan Y., Li R., Montgomery K. D., Chang H. Y., West R. B. Detection of long non-coding RNA in archival tissue: correlation with polycomb protein expression in primary and metastatic breast carcinoma. PLoS ONE. 2012;7(10) doi: 10.1371/journal.pone.0047998.e47998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Quek X. C., Thomson D. W., Maag J. L. V., et al. lncRNAdb v2.0: expanding the reference database for functional long noncoding RNAs. Nucleic Acids Research. 2015;43(1):D168–D173. doi: 10.1093/nar/gku988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ponjavic J., Oliver P. L., Lunter G., Ponting C. P. Genomic and transcriptional co-localization of protein-coding and long non-coding RNA pairs in the developing brain. PLoS Genetics. 2009;5(8) doi: 10.1371/journal.pgen.1000617.e1000617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mercer T. R., Dinger M. E., Mattick J. S. Long non-coding RNAs: insights into functions. Nature Reviews Genetics. 2009;10(3):155–159. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- 75.Rose D., Hiller M., Schutt K., Hackermüller J., Backofen R., Stadler P. F. Computational discovery of human coding and non-coding transcripts with conserved splice sites. Bioinformatics. 2011;27(14):1894–1900. doi: 10.1093/bioinformatics/btr314. [DOI] [PubMed] [Google Scholar]
- 76.Zou Q., Guo J., Ju Y., Wu M., Zeng X., Hong Z. Improving tRNAscan-SE annotation results via ensemble classifiers. Molecular Informatics. 2015;34(11-12):761–770. doi: 10.1002/minf.201500031. [DOI] [PubMed] [Google Scholar]
- 77.Lowe T. M., Eddy S. R. TRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Research. 1997;25(5):955–964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chan P. P., Lowe T. M. GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Research. 2009;37(1):D93–D97. doi: 10.1093/nar/gkn787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hertel J., Hofacker I. L., Stadler P. F. SnoReport: computational identification of snoRNAs with unknown targets. Bioinformatics. 2008;24(2):158–164. doi: 10.1093/bioinformatics/btm464. [DOI] [PubMed] [Google Scholar]
- 80.Schattner P., Brooks A. N., Lowe T. M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Research. 2005;33(2):W686–W689. doi: 10.1093/nar/gki366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lestrade L., Weber M. J. snoRNA-LBME-db, a comprehensive database of human H/ACA and C/D box snoRNAs. Nucleic Acids Research. 2006;34:D158–D162. doi: 10.1093/nar/gkj002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lagesen K., Hallin P., Rødland E. A., Stærfeldt H.-H., Rognes T., Ussery D. W. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Research. 2007;35(9):3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Betel D., Sheridan R., Marks D. S., Sander C. Computational analysis of mouse piRNA sequence and biogenesis. PLoS Computational Biology. 2007;3(11, article e222) doi: 10.1371/journal.pcbi.0030222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang Y., Wang X., Kang L. A k-mer scheme to predict piRNAs and characterize locust piRNAs. Bioinformatics. 2011;27(6):771–776. doi: 10.1093/bioinformatics/btr016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zou Q., Li J., Hong Q., et al. Prediction of microRNA-disease associations based on social network analysis methods. BioMed Research International. 2015;2015:9. doi: 10.1155/2015/810514.810514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zou Q., Li J., Song L., Zeng X., Wang G. Similarity computation strategies in the microRNA-disease network: a survey. Briefings in Functional Genomics. 2016;15(1):55–64. doi: 10.1093/bfgp/elv024. [DOI] [PubMed] [Google Scholar]
- 87.Kin T., Yamada K., Terai G., et al. fRNAdb: a platform for mining/annotating functional RNA candidates from non-coding RNA sequences. Nucleic Acids Research. 2007;35(1):D145–D148. doi: 10.1093/nar/gkl837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu C., Bai B., Skogerbø G., et al. NONCODE: an integrated knowledge database of non-coding RNAs. Nucleic Acids Research. 2005;33:D112–D115. doi: 10.1093/nar/gki041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wu T. D., Watanabe C. K. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005;21(9):1859–1875. doi: 10.1093/bioinformatics/bti310. [DOI] [PubMed] [Google Scholar]
- 90.Langmead B., Trapnell C., Pop M., Salzberg S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology. 2009;10(3, article R25) doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sharon D., Tilgner H., Grubert F., Snyder M. A single-molecule long-read survey of the human transcriptome. Nature Biotechnology. 2013;31(11):1009–1014. doi: 10.1038/nbt.2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li S., Tighe S. W., Nicolet C. M., et al. Erratum: multi-platform assessment of transcriptome profiling using RNA-seq in the ABRF next-generation sequencing study. Nature Biotechnology. 2014;32(11):p. 1166. doi: 10.1038/nbt.2972. [DOI] [PMC free article] [PubMed] [Google Scholar]