

Abstract
Low pH has a strong stabilising effect on the fibrillar assembly of amyloid β, which is associated with Alzheimer's disease. The stabilising effect is already pronounced at pH 6.0, suggesting that protonation of histidines might mediate this effect. Through the systematic substitution of the three native histidines in Aβ for alanines, we have evaluated their role in fibril stability. Using surface plasmon resonance, we show that at neutral pH the fibrillar forms of all His‐Ala variants are destabilised by a factor of 4–12 compared to wild‐type Aβ. However, none of the His‐Ala Aβ variants impair the stabilising effect of the fibril at low pH.
Keywords: abeta, amyloid, fibril, histidine, stability, surface plasmon resonance
Abbreviations
Aβ, Amyloid β
KD, dissociation constant
Pnf, potentiation factor
SPR, surface plasmon resonance
The self‐assembly of amyloid β (Aβ) into amyloid plaques is strongly linked to the development of Alzheimer's disease, which is the most common form of dementia and afflicts around 35 million people worldwide 1. Aβ is a 39–42 residue long peptide where the two forms Aβ1–40 and Aβ1–42 dominate. Aβ is derived from the proteolytic cleavage of the single transmembrane Aβ precursor protein by β‐ and γ‐secretase activity 2, 3. Depositions of amyloid fibrils seen as plaques as well as intracellular neurofibrillary tangles and neuronal loss are clinical hallmarks of Alzheimer's disease 4.
A delicate balance controls the difference between a native monomeric Aβ peptide and its pathological aggregated state, and just a small shift in this equilibrium can have detrimental effects 5, 6, 7, 8, 9, 10. Elucidation of the mechanisms that mediate the pathological self‐assembly is consequently of interest for developing therapeutic interventions against amyloid diseases.
The formation of Aβ fibrils requires an initial nucleation event, and fibril elongation involves the addition of monomers to an existing fibril. In analogy to all polymerisation processes, Aβ fibril formation is concentration dependent, and at a concentration of free monomers below the dissociation constant (KD), also known as the critical concentration, fibril formation cannot occur 11, 12. We have previously determined that the critical concentration for Aβ at neutral pH is around 100 nm for Aβ1–40 and slightly lower for Aβ1–42 13. A similar value was also previously found by Hasegawa and co‐workers 14. However, the extracellular concentration of Aβ in vivo is around 1 nm for Aβ1–40 and even lower for the Aβ1–42 variant 15. The low concentration in vivo in combination with the significantly higher KD has puzzled the field because fibrillar formation under these conditions should, in theory, be prohibited.
It has recently been shown that Aβ self‐assembly is highly dependent on pH and that a lowering of the pH significantly enhances the stability of the fibrils 11. The KD for Aβ1–40 can be as much as 120 times lower at pH 4.5 than pH 7.4. This has several implications because it shows that Aβ assembly can indeed form at physiological Aβ concentrations and also strongly suggests that it occurs within intracellular compartments having a lower pH, such as endosomes or lysosomes 11. It is therefore of interest to elucidate the stabilising factors associated with fibril stability and the potentiating effect of low pH.
The strong pH dependency is likely caused by a change in charge through the protonation of certain amino acid side chains. The stabilising effect is pronounced already at pH 6.0, and within this range only the histidine side chains, having a pKa around 6.0 are significantly affected. It has also previously been shown that truncating the first 16 amino acids of the N‐terminus of the peptide results in a loss of the stabilising effect at low pH 11, which indicates that the N‐terminal part controls this mechanism. The Aβ sequence contains three histidines; all located in the N‐terminal part of the peptide at positions 6, 13 and 14. In the present work, we have evaluated how the stabilising effect of lowering the pH is affected in the Aβ1–40 variant by the systematic substitution of the three histidines for alanines. Using surface plasmon resonance (SPR), we studied the single mutation variants Aβ1–40 H6A, Aβ1–40 H13A and Aβ1–40 H14A as well as a variant where all three histidines have been exchanged (Aβ1–40 H6A, H13A, H14A).
Amyloid polymerisation follows a template‐dependent polymerisation process where monomers are incorporated onto the fibrillar ends. The stability of an amyloid fibril (in analogy to all binding interactions) is determined by the rate of association (k(on)) and the rate of dissociation (k(off)), which reflects how fast a monomer is incorporated onto the fibrillar end and how long it remains bound.
Using SPR, it is possible to monitor both the k(on) and the k(off) in a polymerisation reaction, such as the formation of amyloid. The method is based on the immobilisation of preformed fibrils onto a surface followed by the subsequent probing of free monomers. The Aβ monomers then incorporate onto the fibrillar ends and a continuous increase in the mass on the SPR surface can be monitored. Upon stopping the injection, while maintaining a flow of the running buffer, fibril dissociation into monomers can selectively be observed. Through monitoring both the association and the dissociation, the strength of the interaction, KD, reflecting the fibril stability, can be evaluated 13, 14, further discussed below.
Using SPR, we show that substituting any of the histidines results in significantly lower stability of the fibrils, at neutral pH, compared to the wild‐type peptide (Aβ1–40 wt). However, no single site mutant impaired the stabilising effect of low pH and, similar to Aβ1–40 wt, they all acquired more than a 200‐fold increase in fibril stability upon shifting the pH from 7.4 to 5.5. This suggests that histidine residues contribute to fibril stability at neutral pH but are not involved in the strong potentiating effect on fibril stability induced by low pH.
Material and methods
Preparation of monomeric Aβ peptides
Recombinant variants of Aβ1–40, Aβ1–40 H6A, Aβ1–40 H13A, Aβ1–40 H14A and Aβ1–40 H6A, H13A, H14A were obtained from AlexoTech (Umeå, Sweden). All lyophilised peptides were solubilised in 20 mm NaOH and separated by size‐exclusion chromatography (Superdex 10/300, GE Life Science, Uppsala, Sweden) in PBS with 2 mm EDTA, 5 mm NaOH and 150 mm NaCl. The pH of all purified peptide solutions was adjusted using a stock solution of phosphate or acetate buffer to give a final buffer concentration corresponding to 20 mm phosphate (pH 7.4) or acetate (pH 5.5) and 150 mm NaCl.
Surface plasmon resonance (SPR)
The interaction study between the fibrillar form of Aβ and its monomeric form was performed using a BIAcore® 3000 biosensor (GE Healthcare®, Uppsala, Sweden) equipped with a CM5 sensor chip (GE Healthcare®). First, the fibrillar form of Aβ was immobilised at a density of 3500 response units (RUs) on the dextran layer of the chip using standard amino coupling reagents at pH 4.0. All SPR experiments were performed at 25 °C at a flow rate of 20 μL·min−1 in either a 20 mm phosphate buffer (pH 7.4) or 20 mm acetate buffer (pH 5.5). All experiments were performed in the presence of 150 mm NaCl, which represents a physiological ion‐strength.
Due to the polymeric nature of amyloid fibrils, the covalent immobilisation on the sensor chip, likely only occurs in minor fraction of the accessible amino groups. This treatment is therefore not anticipated to influence the overall incorporation of monomers in a significant manner. However, the influence from using immobilised fibrils or the dextran layer cannot be fully excluded and should therefore be disclosed.
All sensograms were corrected for nonspecific interactions using a reference surface according to standard procedures 16.
Transmission electron microscopy
A total volume of 4 μL of the different Aβ variants was absorbed for 2 min onto glow‐discharged carbon‐coated copper grids. Samples were washed in water and immediately stained in 50 μL of 1.5% uranyl acetate solution for 30 s. Negatively stained samples were examined on a JEM1230 transmission electron microscope (JEOL) at 80 kV. Transmission electron micrographs were recorded with a Gatan UltraScan 1000 2k × 2k pixel CCD camera using digitalmicropgraph™ software (Gatan, Pleasanton, CA, USA.).
Results
Kinetic analysis of fibril formation using SPR
In analogy to all molecular interactions, the stability of the binding between an Aβ monomer and the fibrillar end is dependent on both the rate of assembly k(on) and the rate of dissociation k(off).
Probing free monomeric Aβ against immobilised fibrils using SPR is a frequently used technique for measuring the intrinsic kinetic properties of Aβ polymerisation 11, 14, 17, 18, 19. The SPR sensograms can be separated into an association phase (k(on)) and a dissociation phase ((k(off)). Figure 1 shows the result when immobilised Aβ1–40 wt fibrils are probed with free Aβ1–40 wt monomers.
Figure 1.

SPR analysis of Aβ fibril formation at pH 7.4. A representative sensogram of monomeric Aβ1–40 wt when probed against immobilised Aβ1–40 wt fibrils. The curve displays both the phase of association, where the end state of association is indicated (arrow 1), and the phase of dissociation‐phase, which frequently is divided into a fast phase (arrow 2) and a slow phase (arrow 3).
The end state of the association phase is indicated by arrow 1 in Fig. 1.
Note that the dissociation phase exposes a biphasic curvature. This is highly representative and frequently observed when performing these kinds of experiments. The suggested mechanism behind this phenomenon is further discussed below. However, the initial fast phase of dissociation (indicated by arrow 2, Fig. 1) is much weaker than the second phase (indicated by arrow 3, Fig. 1) and does not contribute significantly to the fibril stability. All analyses of decay rates within this work are therefore exclusively focused on the slower second phase of dissociation.
Through a systematic exchange of the three histidine residues of Aβ, we will expose below their role within fibril formation with respect to association, dissociation and fibril stability.
Kinetic analysis of the association rate, k(on)
Fibrils from recombinant variants of Aβ1–40, Aβ1–40 H6A, Aβ1–40 H13A, Aβ1–40 H14A and Aβ1–40 H6A, H13A, H14A were prepared by prolonged incubation in either PBS (pH 7.4) or acetate buffer pH 5.5 containing 150 mm NaCl. Fibrils immobilised on a CM5 chip were then probed with their monomeric counterpart at different concentrations of Aβ monomers and analysed by SPR. Samples were injected for exactly 60 s and the maximal value at the end state of each injection (indicated by arrow 1 in Fig. 1A) was plotted as a function of the monomeric concentration, Fig. 2. This generates an association rate at each point of measurement. A guideline is given in the figures to illustrate the linearity.
Figure 2.

SPR analysis of the Aβ association rate as function of monomer concentration at pH 7.4 and 5.5. The monomeric Aβ variants at the indicated concentrations were probed towards their corresponding immobilised fibrils. The rates were acquired by measuring the end state of association after exactly 1 min of injection. pH 7.4 (filled squares), pH 5.5 (open squares). (A) Aβ1–40 wt. (B) Aβ1–40 H6A. (C) Aβ1–40 H13A. (D) Aβ1–40 H14A. (E) Aβ1–40 H6A, H13A, H14A.
By comparing the rate between the different Aβ variants, it is possible to determine relative differences in k(on).
At pH 7.4, all histidine mutants of Aβ had a k(on) slightly slower than Aβ1–40 wt. The results are shown in Fig. 2 and the relative value for k(on), (RA) for the different mutants versus Aβ1–40 wt varies between 0.22 and 0.45 of Aβ1–40 wt shown in Table 1.
Table 1.
Relative change of association and dissociation rates of Aβ1–40 wt and the His‐Ala Aβ variants, and the potentiation of Aβ fibril formation as a function of lowering the pH. RA, Relative association; RD, Relative dissociation; PnF, Potentiation factor KD(pH7.4)/KD(pH5.5)
| Aβ Variants | pH 7.4 | pH 5.5 | PnF | ||||||
|---|---|---|---|---|---|---|---|---|---|
| RA | RD | RD/RA | KD (nm) | RA | RD | RD/RA | KD (nm) | ||
| Aβ1–40 wt | 1 | 1 | 1 | 100 | 1 | 1 | 1 | 1 | 100 |
| Aβ1–40 H6A | 0.45 | 1.92 | 4.27 | 427 | 1.94 | 2.78 | 1.43 | 1.43 | 299 |
| Aβ1–40 H13A | 0.31 | 2.22 | 7.16 | 716 | 1.01 | 2.13 | 2.11 | 2.11 | 339 |
| Aβ1–40 H14A | 0.34 | 2.70 | 7.94 | 794 | 1.01 | 2.22 | 2.19 | 2.19 | 363 |
| Aβ1–40 H6A, H13A, H14A | 0.22 | 2.70 | 12.27 | 1227 | 1.57 | 8.33 | 5.31 | 5.31 | 231 |
Interestingly, the corresponding k(on) at pH 5.5 revealed that all Aβ His‐Ala variants, in analogy to Aβ1–40 wt, displayed a much faster rate of association as a function of a decrease in pH to 5.5. The degree of potentiation, indicated in Table 1, is similar to Aβ1–40 wt and a significant difference in RA was only noted regarding Aβ1–40 H6A and Aβ1–40 H6A, H13A, H14A, which displayed a 1.94 and 1.57 times higher RA when compared to Aβ1–40 wt.
Kinetic analysis of dissociation rate
In contrast to the association rate, kon, the rate of dissociation, koff, is independent of the concentration of monomers. Subsequent to probing, the immobilised fibrils with free monomers, the rate of dissociation can be seen as a decrease in the signal over time. Figure 3A shows a comparison between the rate of dissociation between the different fibrillar forms at pH 7.4. The result shows that all Aβ1–40 His‐Ala variants exhibit a higher rate of dissociation than Aβ1–40 wt. Figure 3B–F shows the intrinsic difference regarding each variant as a function of changing the pH from 7.4 to 5.5. The results show that Aβ1–40 wt, in accordance with previous results 11, is significantly stabilised by the decrease in pH. The results, however, also expose a significant stabilisation for all Aβ His‐Ala variants and a significantly slower phase of dissociation is observed upon lowering the pH to 5.5.
Figure 3.
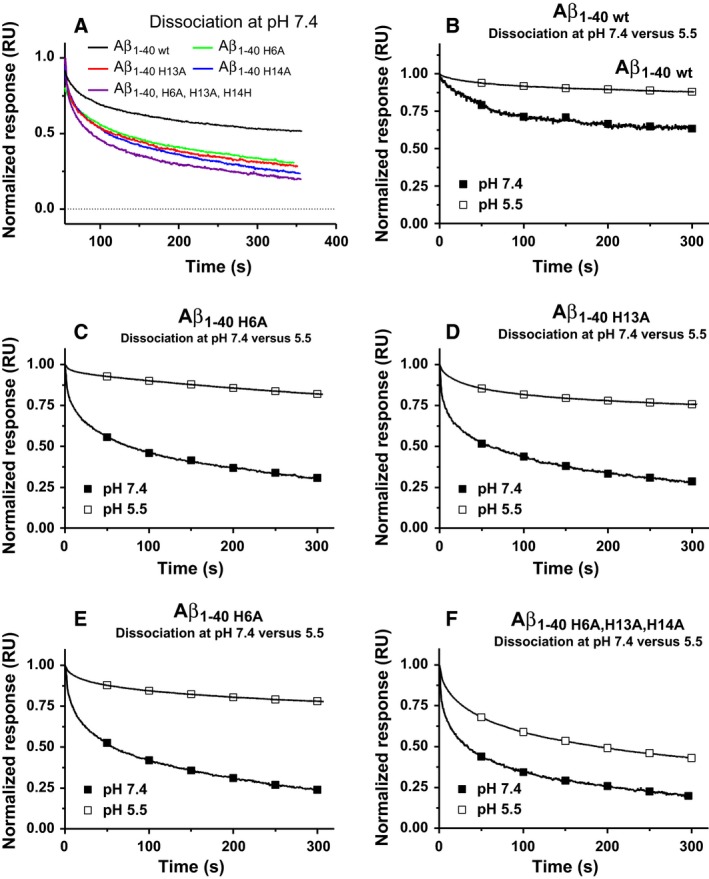
SPR analysis of Aβ fibrillar dissociation (k(off)). Sensograms showing the dissociation phase acquired subsequent to the injection of the monomeric Aβ variants at 2 μm onto their corresponding immobilised fibrils. (A) An overlay of all Aβ His‐Ala variants and Aβ1–40 wt at pH 7.4. (B) Aβ1–40 wt at pH 7.4 and 5.5. (C) Aβ1–40 H6A at pH 7.4 and 5.5. (D) Aβ1–40 H13A at pH 7.4 and 5.5. (E) Aβ1–40 H14A at pH 7.4 and 5.5. (F) Aβ1–40 H6A, H13A, H14A at pH 7.4 and 5.5.
Because the latter part of the curve, representing the slower phase of dissociation (indicated by arrow 3 Fig. 1), strongly dominates the interaction strength, we have exclusively focused on this part, and no points before 100 s were included in the curve. The dissociation curve could then be fitted to a one exponential decay. The relative dissociation rate in relation to Aβ1–40 wt (RD) for the different mutants is given in Table 1.
Determination of the relative fibril stability
While a simple interaction between, for example, a receptor and ligand can easily be determined through saturation experiments, this cannot be done with polymers since they, per definition, cannot be saturated.
This problem can, however, be circumvented by defining the concentration of free monomers where the fibrils are at steady state, i.e. the concentration of free monomers where neither association nor dissociation can be detected. We have previously shown that the KD for Aβ1–40 wt at neutral pH is around 100 nm and around 1 nm at pH 5.5 11, 13. The KD at neutral pH has also been investigated by Hasegawa and co‐workers 14 with similar results. All figures used here are therefore given as relative values to the KD for Aβ1–40 wt corresponding to 100 nm at neutral pH and 1 nm at pH 5.5.
Multiplying the relative RD/RA with the KD for Aβ 1–40 wt will then give the KD for the histidine derivatives.
For all of the evaluated His‐Ala Aβ variants, the substitutions destabilised the fibrillar conformation at neutral pH as indicated by a more rapid decay compared to Aβ 1–40 wt (Fig. 3A). The overall stability was decreased 4–12‐fold in all of the His‐Ala variants compared to Aβ1–40 wt. At low pH, all of the histidine to alanine substitutions displayed a pronounced stabilisation. The relative stabilisation was even stronger than observed in Aβ1–40 wt and the potentiation ranged between 238 and 358 times simply by shifting the pH from 7.4 to 5.5. The dissociation relative to Aβ1–40 wt and the overall potentiation factor (PnF) considering both the kon and koff is indicated in Table 1.
Transmission electron microscopy verifies a fibrillar morphology
To verify the fibrillar morphology of the assembled peptides, we evaluated the acquired aggregates using negative‐stain transmission electron microscopy. Assemblies of Aβ1–40 wt and the different Aβ His‐Ala variants, formed at pH 7.4 as well as 5.5, were analysed. The results showed that none of the variants were impaired in forming fibrils and all displayed a similar morphology as Aβ1–40 wt fibrils, Fig. 4A–J.
Figure 4.

Negative stain transmission electron microscopy of Aβ assemblies. Evaluation of the morphology of the formed aggregates at pH 7.4 (A–E) and pH 5.5 (F–J). (A) Aβ1–40 wt. (B) Aβ1–40 H6A. (C) Aβ1–40 H13A. (D) Aβ1–40 H14A. (E) Aβ1–40 H6A, H13A, H14A. (F) Aβ1–40 wt. (G) β1–40 H6A. (H) Aβ1–40 H13A. (I) Aβ1–40 H14A. (J) Aβ1–40 H6A, H13A, H14A.
Discussion
A very delicate balance controls the self‐assembly of Aβ monomers in vivo, and also a very small shift in the equilibrium can have detrimental effects 6, 7, 8, 9, 20. Today, major efforts are being directed towards identifying methods that can prevent either Aβ excision from APP 3, 21, 22 or impair Aβ assembly 23, 24, 25, 26.
It is therefore of interest to understand the factors controlling fibril formation, as well as the properties of the acquired assemblies.
It has recently been shown how low pH has a strong potentiating effect on fibril stability 11. This finding also provides a possible explanation to how fibrils may form at the very low Aβ concentrations found in the human brain, which had previously been considered to be far below the critical concentration for Aβ fibril formation. This finding also strongly indicates that fibril formation occurs within the low pH compartments of the cell, such as endosomes and lysosomes.
From a mechanistic perspective, the effects of a change in pH on fibril formation are most likely due to protonation of amino acid side chains. Interestingly, a moderate lowering of the pH also has a strong effect where the potentiation factor already at pH 6.5 is around 20 times 11. This suggests that some functional groups, having a pKa, within this pH range, likely mediate the effect of pH. The histidine side chains in Aβ have a pKa corresponding to 6.0 27 and their protonation may therefore possibly represent a part of the mechanism.
In this work, we show how a single exchange of any of the three histidines in Aβ1–40 decreases the fibril stability at neutral pH relative to Aβ1–40 wt. Substitution of either His13 or His14 results in an approximately seven‐ to eightfold lower stability of the fibril, while the exchange of His6 results in an approximately fourfold decrease in fibril stability. Exchange of all three histidines results in an overall 12 times less stable fibrillar assembly. In all cases, we show that the destabilising effect is dependent on both a decreased rate of association and an increased rate of dissociation.
Somewhat surprisingly, all His‐Ala variants displayed a strong potentiation in binding strength upon lowering the pH from 7.4 to 5.5 and increased the fibrillar stability from 240–350 times. The result suggests that the histidine side chains do not mediate the potentiating effect.
It should also be noted that the relatively higher potentiation regarding the Aβ His‐Ala variants is mostly an effect of the decreased stability at neutral pH and does not imply a stronger fibril than Aβ1–40 wt at low pH. At pH 5.5, all single‐substitution variants had 1.42–2.22‐fold lower stability compared to Aβ1–40 wt, while the fibrillar fold of the triple‐substituted variant had a 5.26‐fold lower stability.
Notably, the initial rapid phase of decay is also much more pronounced in the His‐Ala substitution variants. A biphasic behaviour of the dissociation curve has been reported previously and is suggested to be an effect of a dock and lock mechanism 28, 29, 30. This phenomenon implies that an initial poor association of the free monomer onto the fibrillar end first occurs, which is defined as the ‘dock’ phase. The peptide then adopts a more optimal orientation, with a stronger interaction between the newly incorporated monomer and the templating fibril, which is defined as the ‘lock’ phase.
A recent publication, however, suggests an alternative explanation where the initial low affinity interaction is due to a lateral assembly of monomers along already formed fibrils 31 and consequently does not reflect the interaction within the fibrillar end.
However, in terms of binding affinities, the initial rapid phase of dissociation only provides a small contribution to the overall binding. Upon analysis of the overall binding strength, the first phase has therefore been disregarded.
It is also of interest to discuss the role of histidines from a pathology perspective of AD. The role of fibrils as inert bystanders or active participants of the pathology is still a matter of debate and it is well known that prefibrillar assemblies, such as oligomers, exhibit a much higher cytotoxic effect than mature fibrils 32, 33, 34, 35. Factors that partly impair the assembly into fibrils may, instead, facilitate the formation of alternative assemblies. Aβ also has metal‐binding properties and the binding of both Zn2+ and Cu2+ has been shown 36, 37, 38, 39 which are both potent inducers of Aβ assembly 40, 41. However, the structure of the acquired aggregates differs significantly as compared to fibrils acquired in the absence of metals 36. Interestingly, a dysregulation of Zn2+, in the AD brain has also been assigned a pathophysiological role within AD where the increased levels of Zn2+ results in alternative Aβ assemblies 42. The binding of both Zn2+ and Cu2+ to Aβ involve all three histidines 43, 44 and, given present results showing that histidine residues affect the fibrillar stability, may suggest that metals may have the same effect.
Taken together, it can be concluded that the histidine residues of Aβ contribute to fibrillar stability at neutral pH but that their protonation is not a key event that can explain the strong potentiation in Aβ fibril stability that is induced as a function of low pH.
Author contributions
KB performed the SPR experiments and LS performed the TEM analysis. AO, TI and KB directed the research and wrote the manuscript.
Acknowledgement
This work was supported by Insamlingsstiftelsen at Umeå University, Alzheimerfonden, Åhlen‐stiftelsen, J.C. Kempes stiftelse, the Swedish Research Council, Magn. Bergvalls stiftelse, Parkinsonfonden, Centre‐UCEM, Hjärnfonden, Torsten Söderbergs stiftelse, and the Medical Faculty of Umeå University.
Edited by Prof Miguel De la Rosa
References
- 1. Maresova P, Mohelska H, Dolejs J and Kuca K (2015) Socio‐economic aspects of Alzheimer's disease. Curr Alzheimer Res 12, 903–911. [DOI] [PubMed] [Google Scholar]
- 2. Fernandez MA, Biette KM, Dolios G, Seth D, Wang R and Wolfe MS (2016) Transmembrane substrate determinants for gamma‐secretase processing of APP CTFbeta. Biochemistry 55, 5675–5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stromberg K, Eketjall S, Georgievska B, Tunblad K, Eliason K, Olsson F, Radesater AC, Klintenberg R, Arvidsson PI, von Berg S et al (2015) Combining an amyloid‐beta (Abeta) cleaving enzyme inhibitor with a gamma‐secretase modulator results in an additive reduction of Abeta production. FEBS J 282, 65–73. [DOI] [PubMed] [Google Scholar]
- 4. Eisele YS and Duyckaerts C (2016) Propagation of Ass pathology: hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol 131, 5–25. [DOI] [PubMed] [Google Scholar]
- 5. Tarasoff‐Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E, Axel L, Rusinek H, Nicholson C, Zlokovic BV et al (2015) Clearance systems in the brain‐implications for Alzheimer disease. Nat Rev Neurol 11, 457–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Benilova I, Karran E and De Strooper B (2012) The toxic Abeta oligomer and Alzheimer's disease: an emperor in need of clothes. Nat Neurosci 15, 349–357. [DOI] [PubMed] [Google Scholar]
- 7. Wilcock DM and Griffin WS (2013) Down's syndrome, neuroinflammation, and Alzheimer neuropathogenesis. J Neuroinflammation 10, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K et al (1995) Cloning of a gene bearing missense mutations in early‐onset familial Alzheimer's disease. Nature 375, 754–760. [DOI] [PubMed] [Google Scholar]
- 9. Levy‐Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K et al (1995) Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science 269, 973–977. [DOI] [PubMed] [Google Scholar]
- 10. Garai K, Verghese PB, Baban B, Holtzman DM and Frieden C (2014) The binding of apolipoprotein E to oligomers and fibrils of amyloid‐beta alters the kinetics of amyloid aggregation. Biochemistry 53, 6323–6331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brannstrom K, Ohman A, Nilsson L, Pihl M, Sandblad L and Olofsson A (2014) The N‐terminal region of amyloid beta controls the aggregation rate and fibril stability at low pH through a gain of function mechanism. J Am Chem Soc 136, 10956–10964. [DOI] [PubMed] [Google Scholar]
- 12. Williams AD, Shivaprasad S and Wetzel R (2006) Alanine scanning mutagenesis of Abeta(1‐40) amyloid fibril stability. J Mol Biol 357, 1283–1294. [DOI] [PubMed] [Google Scholar]
- 13. Brannstrom K, Ohman A, Lindhagen‐Persson M and Olofsson A (2013) Ca(2+) enhances Abeta polymerization rate and fibrillar stability in a dynamic manner. Biochem J 450, 189–197. [DOI] [PubMed] [Google Scholar]
- 14. Hasegawa K, Ono K, Yamada M and Naiki H (2002) Kinetic modeling and determination of reaction constants of Alzheimer's beta‐amyloid fibril extension and dissociation using surface plasmon resonance. Biochemistry 41, 13489–13498. [DOI] [PubMed] [Google Scholar]
- 15. Jarrett JT, Berger EP and Lansbury PT Jr (1993) The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease. Biochemistry 32, 4693–4697. [DOI] [PubMed] [Google Scholar]
- 16. Myszka DG (1999) Improving biosensor analysis. J Mol Recognit 12, 279–284. [DOI] [PubMed] [Google Scholar]
- 17. Brannstrom K, Ohman A and Olofsson A (2011) Abeta peptide fibrillar architectures controlled by conformational constraints of the monomer. PLoS One 6, e25157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Aguilar MI and Small DH (2005) Surface plasmon resonance for the analysis of beta‐amyloid interactions and fibril formation in Alzheimer's disease research. Neurotox Res 7, 17–27. [DOI] [PubMed] [Google Scholar]
- 19. Hu J, Geng M, Li J, Xin X, Wang J, Tang M, Zhang J, Zhang X and Ding J (2004) Acidic oligosaccharide sugar chain, a marine‐derived acidic oligosaccharide, inhibits the cytotoxicity and aggregation of amyloid beta protein. J Pharmacol Sci 95, 248–255. [DOI] [PubMed] [Google Scholar]
- 20. Head E, Lott IT, Wilcock DM and Lemere CA (2016) Aging in Down Syndrome and the development of Alzheimer's disease neuropathology. Curr Alzheimer Res 13, 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Teranishi Y, Inoue M, Yamamoto NG, Kihara T, Wiehager B, Ishikawa T, Winblad B, Schedin‐Weiss S, Frykman S and Tjernberg LO (2015) Proton myo‐inositol cotransporter is a novel gamma‐secretase associated protein that regulates Abeta production without affecting Notch cleavage. FEBS J 282, 3438–3451. [DOI] [PubMed] [Google Scholar]
- 22. Kennedy ME, Stamford AW, Chen X, Cox K, Cumming JN, Dockendorf MF, Egan M, Ereshefsky L, Hodgson RA, Hyde LA et al (2016) The BACE1 inhibitor verubecestat (MK‐8931) reduces CNS beta‐amyloid in animal models and in Alzheimer's disease patients. Sci Transl Med 8, 363ra150. [DOI] [PubMed] [Google Scholar]
- 23. Tabassum S, Sheikh AM, Yano S, Ikeue T, Handa M and Nagai A (2015) A carboxylated Zn‐phthalocyanine inhibits fibril formation of Alzheimer's amyloid beta peptide. FEBS J 282, 463–476. [DOI] [PubMed] [Google Scholar]
- 24. Liu J, Yang B, Ke J, Li W and Suen WC (2016) Antibody‐based drugs and approaches against amyloid‐beta species for Alzheimer's disease immunotherapy. Drugs Aging 33, 685–697. [DOI] [PubMed] [Google Scholar]
- 25. Sevigny J, Chiao P, Bussiere T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S, Chen T, Ling Y et al (2016) The antibody aducanumab reduces Abeta plaques in Alzheimer's disease. Nature 537, 50–56. [DOI] [PubMed] [Google Scholar]
- 26. Palhano FL, Lee J, Grimster NP and Kelly JW (2013) Toward the molecular mechanism(s) by which EGCG treatment remodels mature amyloid fibrils. J Am Chem Soc 135, 7503–7510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang S and Lee JP (2000) Selectively 2H‐labeled Glu/Asp: application to pKa measurements in Abeta amyloid peptides. J Pept Res 55, 1–6. [DOI] [PubMed] [Google Scholar]
- 28. Stravalaci M, Beeg M, Salmona M and Gobbi M (2011) Use of surface plasmon resonance to study the elongation kinetics and the binding properties of the highly amyloidogenic Abeta(1‐42) peptide, synthesized by depsi‐peptide technique. Biosens Bioelectron 26, 2772–2775. [DOI] [PubMed] [Google Scholar]
- 29. Nguyen PH, Li MS, Stock G, Straub JE and Thirumalai D (2007) Monomer adds to preformed structured oligomers of Abeta‐peptides by a two‐stage dock‐lock mechanism. Proc Natl Acad Sci USA 104, 111–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Esler WP, Stimson ER, Jennings JM, Vinters HV, Ghilardi JR, Lee JP, Mantyh PW and Maggio JE (2000) Alzheimer's disease amyloid propagation by a template‐dependent dock‐lock mechanism. Biochemistry 39, 6288–6295. [DOI] [PubMed] [Google Scholar]
- 31. Saric A, Buell AK, Meisl G, Michaels TCT, Dobson CM, Linse S, Knowles TPJ and Frenkel D (2016) Physical determinants of the self‐replication of protein fibrils. Nat Phys 12, 874–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brannstrom K, Lindhagen‐Persson M, Gharibyan AL, Iakovleva I, Vestling M, Sellin ME, Brannstrom T, Morozova‐Roche L, Forsgren L and Olofsson A (2014) A generic method for design of oligomer‐specific antibodies. PLoS One 9, e90857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lindhagen‐Persson M, Brannstrom K, Vestling M, Steinitz M and Olofsson A (2010) Amyloid‐beta oligomer specificity mediated by the IgM isotype–implications for a specific protective mechanism exerted by endogenous auto‐antibodies. PLoS One 5, e13928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL et al (1998) Diffusible, nonfibrillar ligands derived from Abeta1‐42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA 95, 6448–6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW and Glabe CG (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489. [DOI] [PubMed] [Google Scholar]
- 36. Olofsson A, Lindhagen‐Persson M, Vestling M, Sauer‐Eriksson AE and Ohman A (2009) Quenched hydrogen/deuterium exchange NMR characterization of amyloid‐beta peptide aggregates formed in the presence of Cu2+ or Zn2+. FEBS J 276, 4051–4060. [DOI] [PubMed] [Google Scholar]
- 37. Istrate AN, Kozin SA, Zhokhov SS, Mantsyzov AB, Kechko OI, Pastore A, Makarov AA and Polshakov VI (2016) Interplay of histidine residues of the Alzheimer's disease Abeta peptide governs its Zn‐induced oligomerization. Sci Rep 6, 21734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lim KH, Kim YK and Chang YT (2007) Investigations of the molecular mechanism of metal‐induced Abeta (1‐40) amyloidogenesis. Biochemistry 46, 13523–13532. [DOI] [PubMed] [Google Scholar]
- 39. Zirah S, Kozin SA, Mazur AK, Blond A, Cheminant M, Segalas‐Milazzo I, Debey P and Rebuffat S (2006) Structural changes of region 1‐16 of the Alzheimer disease amyloid beta‐peptide upon zinc binding and in vitro aging. J Biol Chem 281, 2151–2161. [DOI] [PubMed] [Google Scholar]
- 40. Isaacs AM, Senn DB, Yuan M, Shine JP and Yankner BA (2006) Acceleration of amyloid beta‐peptide aggregation by physiological concentrations of calcium. J Biol Chem 281, 27916–27923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. House E, Collingwood J, Khan A, Korchazkina O, Berthon G and Exley C (2004) Aluminium, iron, zinc and copper influence the in vitro formation of amyloid fibrils of Abeta42 in a manner which may have consequences for metal chelation therapy in Alzheimer's disease. J Alzheimers Dis 6, 291–301. [DOI] [PubMed] [Google Scholar]
- 42. Li LB and Wang ZY (2016) Disruption of brain zinc homeostasis promotes the pathophysiological progress of Alzheimer's disease. Histol Histopathol 31, 623–627. [DOI] [PubMed] [Google Scholar]
- 43. Warmlander S, Tiiman A, Abelein A, Luo J, Jarvet J, Soderberg KL, Danielsson J and Graslund A (2013) Biophysical studies of the amyloid beta‐peptide: interactions with metal ions and small molecules. ChemBioChem 14, 1692–1704. [DOI] [PubMed] [Google Scholar]
- 44. Ghalebani L, Wahlstrom A, Danielsson J, Warmlander SK and Graslund A (2012) pH‐dependence of the specific binding of Cu(II) and Zn(II) ions to the amyloid‐beta peptide. Biochem Biophys Res Commun 421, 554–560. [DOI] [PubMed] [Google Scholar]