

Epigenetic alterations involve information heritable during cell division other than the DNA sequence itself. Epigenetic marks were originally thought to involve only unusual phenomena such as position effect variegation in flies (Tartof and Bremer 1990), telomere silencing (Brachmann et al. 1995), mating-type silencing in yeast (Haber 1998), and transgene-induced gene silencing in plants and animals (Dorer and Henikoff 1994). However, it is increasingly clear that epigenetic inheritance plays a central role in defining cellular growth and differentiation. With exceptions such as antibody gene rearrangements and changes in mitochondrial DNA, there is little evidence to support a role for changes in DNA sequence in development, and thus epigenetics is likely to be at the heart of maintaining the differences between stem cells and somatic cells, one cell type and another, and aged versus younger cells.
Epigenetic inheritance involves three interrelated mechanisms: (1) DNA methylation; (2) posttranslational modifications of histones, including methylation, phosphorylation, acetylation, and sumoylation (Strahl and Allis 2000); and (3) chromatin alterations acting over long distances, such as modifications of specific proteins binding to insulator sequences (Chung et al. 1993). Cytosine DNA methylation is a covalent modification of DNA in which a methyl group is transferred from S-adenosylmethionine to the C-5 position of cytosine by a family of cytosine (DNA-5)-methyltransferases (DNMTs). DNA methylation occurs almost exclusively at CpG nucleotides. The pattern of DNA methylation is transmitted through mitosis and maintained after DNA replication by DNMT1, which has a 100-fold greater affinity for hemimethylated DNA (i.e., parental strand methylated, daughter strand unmethylated) than for unmethylated DNA (Gregory 2001). However, developing cells in the zygote and embryo undergo dramatic shifts in DNA methylation, involving both loss of methylation and de novo methylation.
There are two classes of cytosine DNA methylation in the genome. The first occurs throughout the body of genes that show tissue-specific expression, with methylation generally associated with gene silencing (Riggs 1989; Gregory 2001). The second class involves “CpG islands,” or regions rich in CpG dinucleotides (Bird 1986). CpG islands are often described as uniformly unmethylated in normal cells, with the exception of the inactive X chromosome and near imprinted genes (Bird 1986; Riggs and Pfeifer 1992). However, the assumption that autosomal CpG islands (except for imprinted genes) are never methylated is clearly not the case. Strichman-Almashanu demonstrated the presence of normally methylated CpG islands throughout the genome (Strichman-Almashanu et al. 2002). It is also important to note that functionally important methylation information is not always found within CpG islands. For example, the H19 differentially methylated region (DMR) that regulates imprinting of IGF2 and a DMR linked to colorectal cancer risk are not CpG islands (Cui et al. 2002). Thus, epigenome analyses focused solely on these CpG islands would be severely limited in their potential impact.
Epigenetic alteration has two defining characteristics. First is its metastable nature, which involves the capacity for high-frequency alteration, as well as reprogramming in somatic cells and/or specifically through the germ line. The most important consequence of this metastability for human genetics is the apparent high frequency of mutation of affected loci and the ability of large numbers of cells to change the state of their programming, in response to developmental or environmental signals. The second defining characteristic is position effect, in which epigenetic modifications act over a distance along the genome, which in the case of insulators may be in the order of hundreds of kilobases. An important consequence of position effect for human genetics is the ability of regulatory sequences, and epigenetic modifications of them, to act at surprisingly long distances and affect the expression of multiple genes as a group.
Genomic imprinting is a special case of epigenetic modification in which the alteration occurs during germline reprogramming, leading to preferential expression (although generally not absolute in humans) of a specific parental allele in somatic cells of the offspring. At least several hundred genes may show imprinting, and imprinted gene expression appears to be important in a number of rare human genetic disorders as well as common cancer. Both mouse and human chromosomes that undergo uniparental disomy (UPD) often show characteristic phenotypic alterations in the offspring (for review, see Cattanach and Beechey 1990; Ledbetter and Engel 1995). These can include overgrowth in the case of paternal UPD for some chromosomal regions (Cattanach and Beechey 1990; Ledbetter and Engel 1995) and growth retardation in the case of maternal UPD of the same chromosomal regions. There is a strong relationship between imprinted genes and both prenatal and postnatal growth (Moore and Haig 1991). Imprinting is also thought to underlie some quantitative trait loci for growth, with considerable potential commercial application (de Koning et al. 2000). Finally, both imprinted and nonimprinted genes show abnormal expression in animals created by nuclear transfer, and imprinting is thought to be a potential barrier to stem cell transplantation.
Genomics includes a whole-genome approach to genetics. In human disease research, genomics approaches include gene expression arrays, genome scans for sequence variation, and family studies using association tests (transmission test for linkage disequilibrium [Spielman et al. 1993], commonly but erroneously called “transmission disequilibrium test” or TDT).
Epigenomics is defined as a whole-genome approach to epigenetics, similarly advancing epigenetics studies beyond the single-gene level. The field is nascent at present, and efforts to develop it include array-based methylation analysis, array-based hybridization using probes prepared by immunoprecipitation with antibodies to modified histones (so-called “ChIP on chip”), and high-throughput allele-specific expression analysis.
In the following sections, we will describe two efforts to “genomicize” epigenetics. The first is a whole-genome approach to cancer epigenetics, in which array-based gene expression was analyzed after epigenetic modification by combinations of three methods: gene knockout of DNA methyltransferases; treatment with 5-aza-2′-deoxycytidine, an inhibitor of DNA methylation; and treatment with trichostatin A, a histone deacetylase inhibitor. The second approach is an effort to provide a theoretical foundation for a population-based approach to the epigenetic basis of human disease, which we call the “common disease genetic and epigenetic” hypothesis, or CDGE.
CANCER, AN EXAMPLE OF A COMMON DISEASE OF PARTLY EPIGENETIC ORIGIN
Studies of the epigenetics of common human diseases have been generally limited to cancer, and it has not been widely perceived that epigenetics might play a major role in many common complex disease traits. Alterations in DNA methylation were the initial focus (Feinberg and Vogelstein 1983), and epigenetic activation of oncogenes and epigenetic silencing of tumor suppressor genes are both important. Epigenetic activation includes “CT” genes (expressed in cancer and normally only in the testis), e.g., the MAGE gene in melanoma, the PSCA and S100A4 in prostate cancer, and the HPV (human papillomavirus) genome in cervical cancer, to name a few (Feinberg and Tycko 2004). Hypomethylation also leads to chromosomal instability, and this has been shown to promote tumor formation in a mouse model (Gaudet et al. 2003). Hypermethylation is linked to silencing of many tumor suppressor genes, including RB, VHL, and Cadherin. There is some controversy whether the methylation changes initiate silencing, but they at least help to maintain it. Methylation changes are ubiquitous in cancer, affecting all known tumor types at nearly universal frequency and are much more common than genetic changes (Feinberg and Tycko 2004).
Genomic imprinting is also important in cancer, first suggested by parent-of-origin specific loss of heterozygosity in several tumor types. Loss of imprinting (LOI) of the autocrine growth factor IGF2, leading to its increased expression, was first observed in Wilms tumor of the kidney and then in many common tumors as well (Rainier et al. 1993; Okamoto et al. 1997). LOI serves as a gatekeeper for some cancers, as methylation changes are found not only in Wilms tumor but in nonneoplastic kidneys surrounding some of the tumors (Moulton et al. 1994; Steenman et al. 1994). Chromatin modifications may also be important in cancer. For example, histone H3 lysine methylation is associated with INK4A tumor supressor gene silencing (Bachman et al. 2003). More importantly, resilencing of INK4 is established by chromatin modification in methylation-deficient DNMT knockout cell lines, suggesting that chromatin alterations rather than DNA methylation initiate silencing (Bachman et al. 2003). Chromatin modifications at a distance are important in regulating normal imprinting of IGF2, which in fetal development is regulated by a DMR between the IGF2 and H19 and which is methylated on the maternal allele only. The insulator protein CTCF binds to this unmethylated DMR, limiting access to an enhancer shared between H19 and IGF2, causing silencing of the maternal IGF2 allele (Ohlsson et al. 2003). In Wilms tumors, LOI appears to be caused by aberrant methylation of the maternal DMR, blocking CTCF binding and causing activation of the normally silent maternal IGF2 allele (Feinberg et al. 2002). Finally, epigenetic factors may act in trans to promote cancer progression. Increased expression of EZH2 is linked to generalized hypermethylation and gene silencing in metastatic prostate cancer (Varambally et al. 2002). EZH2 is an ortholog of the Drosophila chromatin repressor protein enhancer of Zeste, and thus overexpression of this gene could cause epigenetic silencing of multiple genes throughout the genome.
While epigenetic mechanisms are generally accepted as important in cancer initiation and progression, the idea that they might play a role in cancer predisposition is relatively new. However, two epigenetic modifications affecting IGF2 imprinting suggest that may be the case. Beckwith–Wiedemann syndrome (BWS), a disorder of prenatal overgrowth, midline abdominal wall defects, and cancer, is caused by several different genetic and epigenetic mechanisms, which are beyond the scope of this paper and discussed elsewhere (DeBaun et al. 2002). Hypermethylation of the H19 DMR occurs in about 15% of BWS patients, and this alteration is specifically associated with cancer risk. Thus, methylation changes in normal tissue serve as a gatekeeper to cancer development. In addition, hypomethylation of a DMR within IGF2 also is linked to LOI in ~10% of the population (Cui et al. 1998, 2002). This LOI appears to be important in colorectal cancer predisposition as it occurs more frequently in patients with a positive history of colon neoplasms or a positive family history of colorectal cancer (Cui et al. 2003; Woodson et al. 2004). Finally, it was found recently that aberrant methylation of H19 is clustered in families, suggesting the existence of epigenetic polymorphisms in the population (Sandovici et al. 2003).
Malignant transformation requires both the inactivation of genomic fidelity surveillance pathways as well as activation of proproliferative/prosurvival signal transduction cascades. The regulation of DNA methylation and the subsequent chromatin structure are significantly altered in tumor cells suggesting a direct role for altered methylation in the process of in vivo cellular transformation. It has also been demonstrated in vitro that c-fos-induced overexpression of DNMT1, or overexpression of DNMT1 alone, transforms immortalized rat fibroblasts in vitro (Bakin and Curran 1999). In addition, this group also demonstrated that the addition of chemical agents that inhibit methyltransferase activity or chromatin compaction significantly reversed the DNMT1-induced transformed phenotype. The results of these experiments raise an intriguing question: Can the silencing of genes alone by methylation induce transformation without the activation of proproliferative/survival pathways or, more interestingly, does hypermethylation activate the expression of genes regulating cellular proproliferative/survival pathways?
AN EPIGENOMIC APPROACH TO CANCER
It now appears clear that methylation plays a central role in transformation, both in vitro and in vivo; however, the mechanism remains to be fully understood. This is due in part to the significant number of genes altered by changes in intracellular methyltransferase activity and the chemical agent used to modulate gene expression, such as 5-aza-CdR, that like all chemicals undoubtedly has non-specific pharmacological effects. Most previous studies have examined changes at the individual gene level, and a more comprehensive genomic approach would reveal patterns that would otherwise not be apparent. To begin to address these issues we conducted a comprehensive gene expression analysis to reveal the relationship between chemical and genetic manipulation of epigenome. In these studies, HCT116 cells were treated with 5-aza-CdR or TSA followed by microarray analysis to identify changes in gene expression (Gius et al. 2004).
As might be expected, a significant number of genes were increased following exposure to either 5-aza-CdR or TSA, including numerous genes previously shown; however, we also identified multiple genes that are down-regulated following exposure to these agents. A hierarchical cluster analysis identified 231 genes down-regulated at least 1.5-fold and 46 genes at least 2-fold following exposure to 5-aza-CdR and 157 (Table 1) genes down-regulated at least 1.5-fold and 22 genes at least 2-fold following exposure to TSA (Gius et al. 2004). An examination of this microarray analysis demonstrates genes involved in such diverse intracellular processes as cell cycle regulation and growth factors, as well as pro-survival signaling pathways. Interestingly, we observed roughly the same number of genes decreased following chemical exposure as were increased. These preliminary results suggest that in addition to silencing gene expression, hypermethylation is also linked to gene activation. This result would not have been obvious if studies were limited to identifying increases in expression only after demethylation.
Table 1.
Table of Genes Up- and Down-regulated in HCT116 Cells following Treatment with 5-aza-CdR or TSA
| Treatment | Increased
|
Decreased
|
||
|---|---|---|---|---|
| >1.5 | >2.0 | <1.5 | <2.0 | |
| 5-aza-CdR | 280 | 64 | 231 | 46 |
| TSA | 143 | 18 | 157 | 22 |
Total number of genes increased or decreased in HCT116 cells treated with either 5-aza-CdR or TSA. For complete list, see NIH Web page (http://home.ncifcrf.gov/ROSP-Microarray-Lab/DavidGius/ColdSpringHarborl.html).
To further address this idea, a microarray analysis was done on somatic cell HCT116 knockout lines for DNMT1(−/−), DNMT3B(−/−), and double knockout (DKO) cells. Previous studies had shown that both DNMT1 and DNMT3B cooperatively maintain DNA methylation and gene silencing (Rhee et al. 2002), and genetic disruption of both DNMT1 and DNMT3B significantly inhibited methyltransferase activity and reduced genomic DNA methylation by roughly 95% (Rhee et al. 2000, 2002). Similar to the results with 5-aza-CdR and TSA, there were a significant number of genes decreased in cells lacking methyltransferase activity and this number was roughly equal to the number of genes increased in the DNMT1(−/−), DNMT3B(−/−), and DKO cell lines (Table 2). The genes decreased by chemical or genetic inhibition of methyltransferase activity appear to be involved in a diverse range of critical intracellular processes, including cell cycle regulation, DNA repair, programmed cell death, proliferation, and several signaling cascades.
Table 2.
Table of Genes Up- and Down-regulated in Methyltransferase Somatic Cell Knockout Cell Lines versus Control, HCT116 Cells
| Cell Type | Gene expression changes
|
|||
|---|---|---|---|---|
| Increased
|
Decreased
|
|||
| >1.5 | >2.0 | <1.5 | <2.0 | |
| DNMT1(−/−) | 280 | 64 | 231 | 46 |
| DNMT3B(−/−) | 143 | 18 | 157 | 22 |
| DKO | 143 | 18 | 157 | 22 |
Total number of genes increased or decreased in DNMT1(−/−), DNMT3B(−/−), or DKO cells as compared to control, parental HCT116 cells. For complete list, see NIH Web page (http://home.ncifcrf.gov/ROSP-Mcroarray-Lab/DavidGius/ColdSpringHarborl.html).
If genetic inhibition of methyltransferase activity inhibits the activity of specific genes it seems logical that overexpression of DNMT1 would increase expression of genes as well. This idea was recently addressed by the construction of fibroblast cells that constitutively overexpressed DNMT1 and by comparing microarray gene expression patterns between the parent and DNMT1 expression daughter cells. One of the most surprising finds of this study is that following microarray analysis slightly more than 350 genes were increased in the CMVdnmt1 cells while roughly 250 genes were decreased, as compared to the parent cells (Ordway et al. 2004). This result is consistent with our finding that the number of genes that appear to be regulated by hypomethylation is as significant as the number of genes that are silenced by hypermethylation. As such, these results suggest, but do not validate, the idea that methylation may increase the expression of specific genes as well as silence gene expression.
If the idea that methylation can activate gene expression is correct, it would be expected that the decrease in the expression of specific genes in the genetic knockout cells would correspond to promoter hypomethylation. To address this, we examined the promoter region of APM2, which was shown to be silenced in the somatic knockout cell lines (Fig. 1A), and found that the degree of transcriptional silencing closely reflected the degree of hypomethylation observed in the APM2 promoter (Fig. 1B). These results suggest a potential direct mechanism for changes in promoter methylation patterns and the regulation of gene expression.
Figure 1.
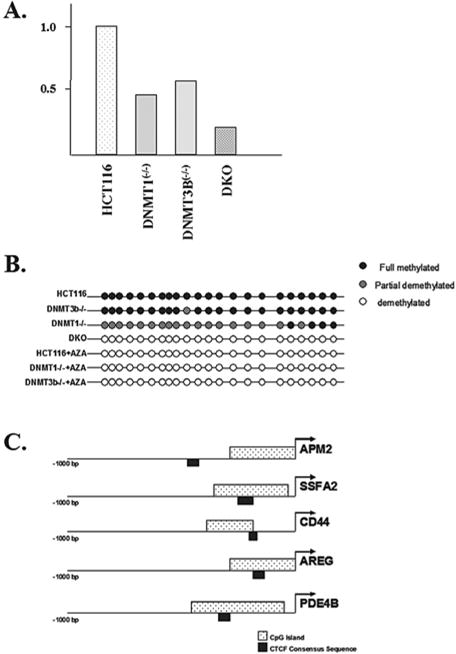
Expression of the APM2 gene is decreased in somatic cell methyltransferase knockout cell lines. (A) The total RNA was isolated from control HCT116, as well as dmnt1(−/−),dnmt3B(−/−), and double knockout (DKO) cells and APM2 RNA levels were determined via real-time RT-PCR (reverse transcriptase polymerase chain reaction). Results represent the average of three independent reactions. (B) The APM2 promoter CpG island is hypomethylated in somatic cell methyltransferase knockout cell lines. The methylation status of the CpG island in the APM2 upstream promoter region was determined in HCT116, DNMT1(−/−), DNMT3B(−/−), and DKO cells. (C) The APM2, SSFA2, CD44, AREG, and PDE4B promoters were examined for the presence of CpG islands and CTCF binding sites using software available at Entrez Genome (http://ncbi.nhn.nih.gov).
One possible mechanism for explaining how hypomethylation might inhibit gene expression involves DNA-binding factors, such as insulator proteins, that may regulate hypomethylation-mediated gene silencing. For example, CTCF, a chromatin insulator protein, binds to unmethylated GC-rich sequences, causing their silencing by inhibiting enhancer elements (Schoenherr et al. 2003; Fedoriw et al. 2004). To address this idea we conducted a detailed examination of the APM2 gene as well as several other genes that were down-regulated by chemical or genetic inhibition of methyltransferase activity. As can be seen, not only do these genes contain CpG islands in their promoter regions but they also contain consensus CTCF DNA-binding sequence in the upstream regulator regions (Fig. 1C). These results suggest a new paradigm for the role of methylation on gene expression suggesting that, in addition to gene silencing, methylation may activate gene expression as well.
One of the most interesting observations from these microarray data is that several prosurvival or proproliferative factors appear to be regulated by methylation status. This raises an interesting idea: Does overexpression of methyltransferase genes both silence tumor suppressor genes and activate prosurvival or antideath genes?
Finally, we observed that a contiguous domain of metallothionein genes was coordinately regulated in these experiments, and this relationship was highly statistically significant (Gius et al. 2004). Thus, drugs that affect chromatin appear to act in unexpected ways, including similarities in effects between classical “demethylation” and “chromatin decondensation,” and to coordinate effects acting over a long distance. These relationships would not be apparent without using an epigenomic approach to their identification.
EPIGENETICS OF RARE HEREDITARY DISORDERS
Two of the known genes involved in DNA methylation have been shown to be involved in rare hereditary diseases. DNA methyltransferase type 3B (DNMT3B) is mutated in the ICF syndrome (immunodeficiency, cytogenetic abnormalities, facial anomalies) (Xu et al. 1999). Centromeric heterochromatin is disrupted in mitogen-treated cells obtained from patients with ICF, suggesting that DNMT3B is involved in this process. However, DNMT3B probably acts more generally in mamtaining repressed chromatin, as ICF also involves activation of genes on the inactive X chromosome. Mutations in MECP2 cause Rett syndrome, an X-linked disorder limited to girls (Amir et al. 1999). This syndrome involves normal development in the first year of life, followed by rapid deterioration of higher brain functions, suggesting that methylation is involved in late postnatal brain development.
The number of known mutations in various factors related to chromatin structure and remodeling have increased in number in the last several years and are discussed in several recent reviews (Hendrich and Bickmore 2001; Huang et al. 2003). These include ATRX syndrome, which involves mental retardation and dysmorphic features and is associated with altered DNA methylation and relaxation of repressed chromatin. Rubinstein–Taybi syndrome involves short stature, dysmorphology, and mental retardation and is caused by mutations of CREB binding protein (CBP) (Murata et al. 2001), a histone acetyltransferase. Fascioscapulohumoral dystrophy is associated with contraction of a DNA repeat, which often is associated with hypomethylation of the repeat and expression changes in a gene cluster upstream of the repeat. The epigenetic abnormality is present even in patients with a normal repeat size, indicating a causative role for the epigenetic abnormality (van Overveld et al. 2003).
Several human diseases are known to involve imprinted genes. These include Prader–Willi syndrome and Angelman syndrome, both of which are characterized by short stature, mental retardation, and behavioral disorders, in which the phenotype depends on the parent of origin. Another well known example involving imprinted genes is BWS, a cause of prenatal overgrowth and predisposition to Wilms tumor, hepatoblastoma, and neuroblastoma and of pseudohypoparathyroidism type IA, which involves osteodystrophy and gonadal dysfunction. The common mechanism of BWS is loss of imprinting of the normally silent maternal allele of either insulin-like growth factor II (IGF2) or the antisense RNA LIT1 (DeBaun et al. 2002).
THE COMMON DISEASE GENETIC AND EPIGENETIC HYPOTHESIS (CDGE)
In a recent review we have made the case that epigenetics might play a role in the etiology of common human disease (Bjornsson et al. 2004). The elements of CDGE are as follows.
1. Epigenetic information maintains a developmentally specific pattern, but shows alteration over time
We argue that the stability of epigenetic marks over time is a major factor in the development of common disease. This idea is predicated in part on studies suggesting that an important step in development is the stabilization of patterns of transcriptional activation and silencing by chromatin proteins such as trithorax and polycomb, respectively (Orlando 2003). A recent paper by Ruden has shown a great deal of epigenetic variation in Drosophila, which may account for masking of latent mutations, and this masking is environmentally sensitive (e.g., through heat shock [Sollars et al. 2003]). Historically, however, more attention has been focused on the role of DNA methylation in maintaining developmental states. Riggs (1975) and Holliday (Holliday and Pugh 1975) originally suggested that expression patterns were established by DNA methylation. DNA methylation is an attractive candidate as each parental strand remains hemimethylated during replication, thus allowing the methylation to be preserved through replication. This hypothesis has been challenged (Bestor 2000), however, but still remains the dominant hypothesis of the epigenetic literature. Epigenetic modifications are tissue specific, and abnormal epigenetic patterns lead to abnormal development (BWS, Fragile X syndrome, ICF syndrome). Furthermore, the DNA of germ cells goes through extensive demethylation and remethylation stages during gametogenesis and development, thus adding plausability to the idea (Reik et al. 2001).
2. There are several sources of epigenetic variation
These include genetic factors, the individual’s environment, the parental environment, the parental genotype and epigenotype, aging, and stochastic events. The fact that epigenetic systems may be influenced by so many events allows a certain developmental “elasticity” for long-lived multicellular organisms. A combined epigenetic and genetic pathogenesis (CDGE model) might provide for a plausible explanation for several presently unexplained features of common complex diseases like late onset, the environmental sensitivity of common human disease, and the apparent tissue specificity of common human disease.
Adding to epigenetic variation may be DNA methylation itself. Genomic DNA methylation is the leading endogenous mutagen accounting for about 30% of germ cell mutations (Krawczak et al. 1998). It is likely that the same phenomenon occurs throughout the somatic life of a cell in that there might be a constant loss of methylated CpG nucleotides, either through point mutation or loss of methyl groups. Many somatic tissues actually have higher total levels of methylation than sperm (Ehrlich et al. 1982) and in those tissues the effect might even be more pronounced than in germ cells. A recent study from mice suggests that the frequency of epimutation might be as much as an order of a magnitude higher than that of genetic mutation (Bennett-Baker et al. 2003).
3. Human disease occurs in part when developmental patterns are lost
Epigenetic modifications could influence disease occurrence either directly or by modifying penetrance. Epigenetic abnormalities predisposing to cancer constitute the best known example of both types. Another example where the epigenetic modification appears to predispose to human disease is that of transient neonatal diabetes, which is a rare disorder with an early diabetes that patients recover from before 18 months of age. This syndrome has recently been shown to be associated with loss of imprinting of two imprinted genes, ZAC and HYMAI (Mackay et al. 2002). Although this is a rare disease, a large proportion of individuals (40%) go on to develop a common adult onset type 2 diabetes mellitus later in life (Mackay et al. 2002). This neonatal form might be an important lead into the pathogenesis of type 2 adult onset diabetes mellitus, a disease that presently is reaching epidemic proportions.
4. The CDGE approach might explain some features of common complex diseases
A combinatorial set of single nucleotide polymorphisms is neither sufficient nor necessary to cause complex diseases, because complex diseases do not occur at birth but usually need a 40-year lag period before the actual disease occurs. To date, the mechanisms through which the environment causes permanent detrimental effects on tissues that act in an additive manner over time have largely remained elusive. Only two models of adult onset disease and progression provide an obvious explanation for such a phenomenon—a mitochondrial model and an epigenetic model. An epigenetic explanation would be that the normal pattern of epigenetic marks is temporally degraded and when a certain threshold is reached the symptoms of the disease are expressed. Indeed, when a simulation is performed that takes this degeneration of epigenetic patterns into account, the population attributable risk goes up with age, a result that is similar to what is seen for actual common human disease (Figs. 2 and 3) (Bjornsson et al. 2004).
Figure 2.
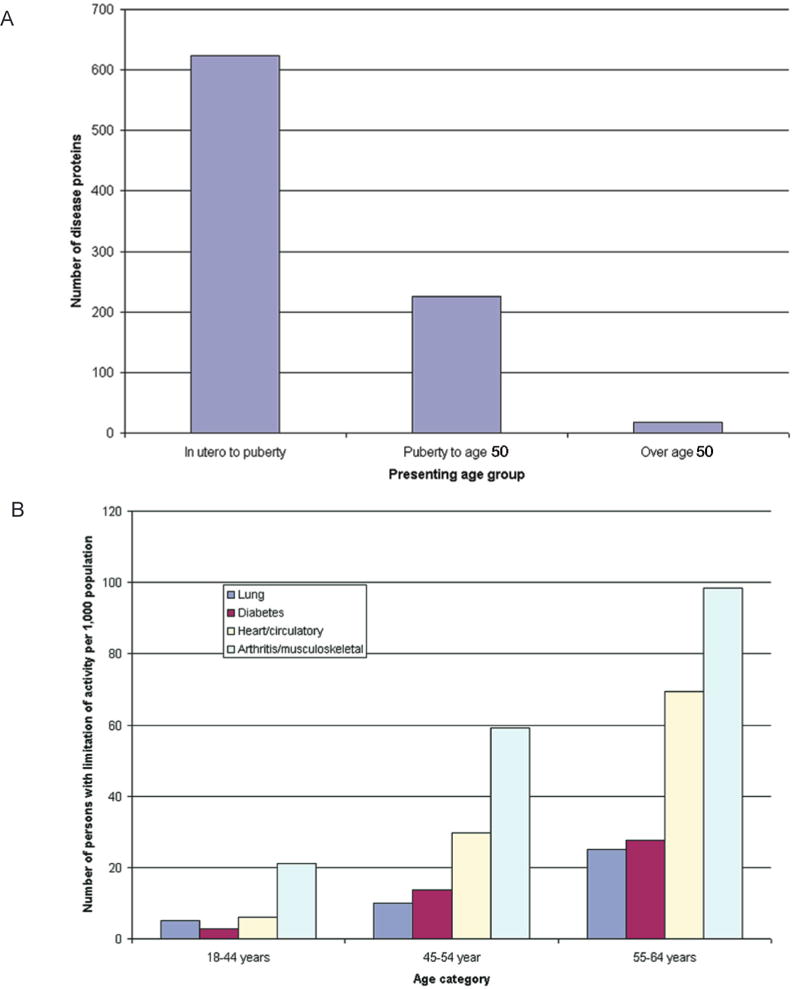
Age of presentation of conventional (A) Mendelian disorders and (B) common complex traits causing limitation of activity among working-age adults, 1999–2001. Data for panel A are from Jimenez-Sanchez et al. (2001), and data for panel B are from the U.S. National Center for Health Statistics (Statistics 2003). (Reprinted, with permission, from Bjornsson et al. 2004 [©Elsevier].)
Figure 3.
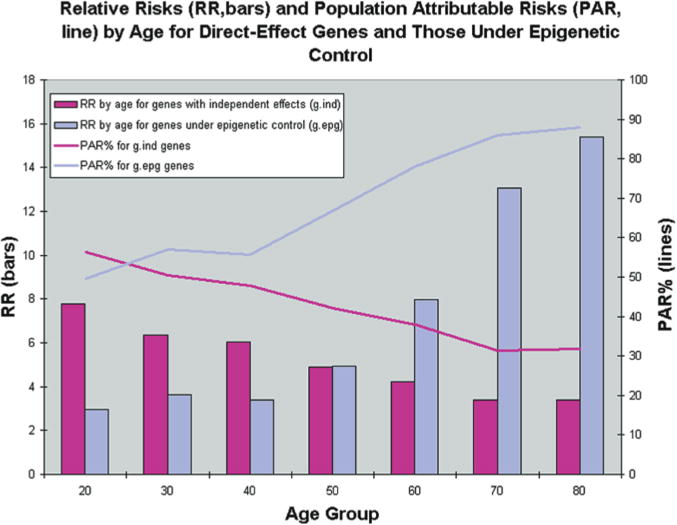
Results from simulations of 40 populations. Simulations were used to create 40 populations containing affected and unaffected individuals for a genetic epidemiologic analysis. Age, environmental status, and genotypes for three different genes were first simulated at random according to specified frequencies. Disease status was then simulated according to CDGE. From these simulated populations, relative risks for gind and gdep genes were estimated in cross-sectional analysis by each age decade, and averaged over 40 populations (bars). Because this risk reflects only the magnitude of a genetic effect, and not the importance of that genotype with respect to all cases in the population, we also estimated population attributable risk percentage (PAR%) at each decade. This reflects the proportion of cases in the population that can be explained by the particular genetic effect. (Reprinted, with permission, from Bjornsson et al. 2004 [©Elsevier].)
An interesting experimental finding that adds some weight to this idea comes from nuclear transfer experiments. Mice conceived by nuclear transfer develop obesity (Tamashiro et al. 2002), which is not transmitted to their offspring, suggesting an epigenetic role. However, the cloned animals are normal at birth and abnormalities do not present in these mice until 8–10 weeks of age (Tamashiro et al. 2002). The life span of cloned mice that are apparently normal at birth is significantly decreased, but this does not become evident until about 311 days after birth (Ogonuki et al. 2002). Both these cases display a lag time and both are models of epigenetic disease, as the phenotype is not transmitted to offspring.
Environmental factors play a major role in the causation of complex diseases. For some diseases, such as cardiovascular disease, a partial cause is known. But for most cases of complex disease a mechanism through which an effect of the environment could act is unknown. Perhaps environmental factors interact with the organism at the transcriptional level in a semipermanent manner, possibly through long-term modification of epigenetic marks. A possible reason for a link between permanent changes in gene expression and nutrition could simply be the need for sufficient metabolites for the maintenance of various epigenetic modifications (Cooney 1993). The nutrients needed for maintenance of epigenetic marks are versatile, including methyl donors, many of which are folate dependent for maintenance of methylation of both DNA and histones. Availability of acetyl groups and phosphoryl groups might also in the same way link metabolic intermediates to the control of gene expression and therefore offer a simple explanation of how gene expression is affected by various metabolic states. A recent study found that uremic patients have abnormalities of normal methylation and allele-specific expression patterns that are reversible by folate supplementation (Ingrosso et al. 2003).
5. CDGE provides a theoretical foundation for integrating epigenetic and genetic interaction
While a great deal is now known about chromatin biochemistry, this knowledge has not previously been linked to population genetics. We suggest that epigenetic modifications of some, but certainly not all, genes have an impact on disease penetrance. We refer to such genes as gdep, and the epigenetic modifications themselves may occur directly or may be mediated by variations in the sequence of genes that encode epigenetic modifiers (referred to as gepg). Genes in which epigenetic modification does not play a role in disease are referred to as gind, even though such genes nevertheless may be epigenetically modified (see Fig. 4).
Figure 4.
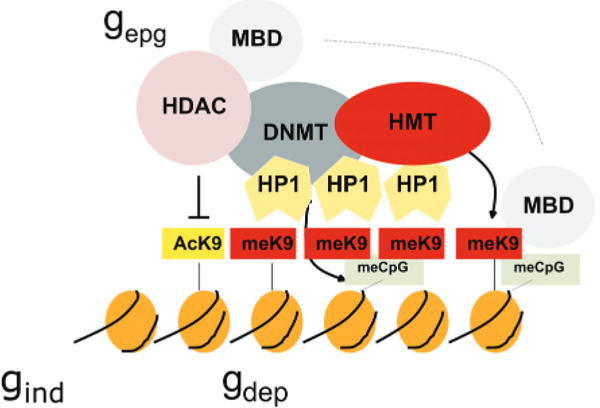
Cooperative and self-reinforcing organization of the chromatin- and DNA-modifying machinery responsible for gene silencing in normal and malignant cells. Histone (H3) modifications include lysine (K) acetylation (Ac) and lysine methylation (Me). Lysines at other positions are also modified. The HP1 protein recognizes MeK9 and, as this protein also binds the histone methyltransferase (HMT), heterochromatin can spread. Histone deacetylases (HDAC) deacetylate lysine residues as a prerequisite for their subsequent methylation. DNA methyltransferases (DNMT) participate in multiprotein complexes that contain HDACs and HMTs, and methyl-C binding proteins (MBD) can be loaded onto methylated DNA through their interactions with both HDACs and HMTs. Much of the evidence comes from studies of constitutive heterochromatin, but recent studies indicate similar interactions of genes silenced de novo in cancer cells. (Reprinted, with permission, from Feinberg and Tycko 2004 [©Nature Publishing Group; http://www.nature.com].)
The implications of this model are that it distinguishes between four possibilities for epigenetic–genetic combinations at a disease-causing locus: (1) At a gind locus, disease is caused by a genetic variation and is not affected by epigenetics (Fig. 5a), i.e., the special case of CDCV independent of epigenetic variation. Here, the disease phenotype is affected by genetic variation (V) without epigenetic modification playing a role. (2) A locus may exist in which the particular phenotypic contribution at this locus is directly caused by epigenetic variation alone (epg; Fig. 5b). Of course, genetic variation at other loci may still contribute in an additive way to the total disease phenotype, but not at this particular locus. Interestingly, epg can lead to continuously quantitative variation even at a single locus, because epg can be quantitative rather than discrete. (3) A locus with a genetic variant contributing to disease phenotype (like possibility 1 above) may be modified in its penetrance by epigenetic factors (gdep; Fig. 5c). We think this is likely to be a common mechanism for integrating genetic and epigenetic variation. Finally, (4) genetic variation at a distant locus (gepg) can affect both gdep and epg (Fig. 5d).
Figure 5.
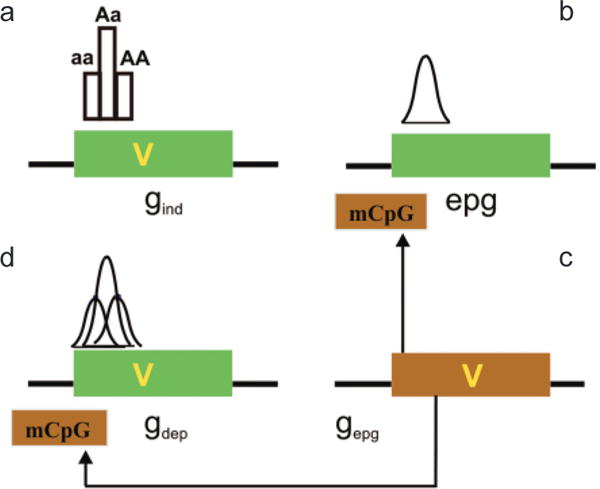
Interaction of genetic and epigenetic variation at a genomic level. (a) A gene (green box) can contain a sequence variant (V) that contributes to disease. Gene variants that are not influenced by epigenetic modification in their disease contribution (although epigenetics can contribute to normal function) are epigenetic independent (gind). The distribution of phenotype of a single locus will not be Gaussian in the absence of other factors, e.g., environmental. (b) Epigenetic variation can contribute to disease phenotype directly, independent of a genetic variation in the target gene (epg). Epigenetic variation itself is quantitative and thus can impart the quantitative nature to a trait, even at a single locus. (c) If the penetrance of a gene sequence variant is affected by epigenetic modification (mCpG), the gene is “epigenetic dependent” (gdep). In this case, the genetic and epigenetic variation together could contribute to a Gaussian distribution, even at a single locus. Note that the epigenetic modification is drawn on the gene but it could be at some distance upstream or downstream from that gene. The epigenetic modification need not be methylation, which is drawn here for convenience. (d) A genetic variant that can influence this epigenetic modification (e.g., encoding a chromatin-modifying protein) is referred to as gepg, and its influence is denoted by arrows. (Reprinted, with permission, from Bjornsson et al. 2004 [©Elsevier].)
CONCLUSION
We chose as our contribution to this meeting some recent efforts of our own group to try to advance the field of epigenomics, because of the history of the Cold Spring Harbor Symposium itself. The symposia were created in an effort to unite disparate fields both within and outside of biology, and also to infuse them with a quantitative approach (http://www.cshl.org/public/history.htm). This series partially spawned contemporary molecular biology. Until recently, epigenetics research, our own included, has been a somewhat eccentric stepchild of this molecular biology. However, recent advances in genomic technology should allow us to approach some of the great questions of human epigenetics, similar to what has been accomplished over the last two decades in human genetics. As occurred in “genomicizing” genetics, the initial stages were method intensive and relatively slow, but that should not deter us in similarly developing human epigenomics technology. Given the vast resources expended toward cancer genomics, the clear importance of epigenomics to cancer research, and the promise of epigenomics for other disorders, we cannot afford to wait.
Acknowledgments
This work was supported by NIH grants HG003233 and CA65145 to A.P.F.
References
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Bachman KE, Park BH, Rhee I, Rajagopalan H, Herman JG, Baylin SB, Kinzler KW, Vogelstein B. Histone modifications and silencing prior to DNA methylation of a tumor suppressor gene. Cancer Cell. 2003;3:89. doi: 10.1016/s1535-6108(02)00234-9. [DOI] [PubMed] [Google Scholar]
- Bakin AV, Curran T. Role of DNA 5-methylcytosine transferase in cell transformation by fos. Science. 1999;283:387. doi: 10.1126/science.283.5400.387. [DOI] [PubMed] [Google Scholar]
- Bennett-Baker PE, Wilkowski J, Burke DT. Age-associated activation of epigenetically repressed genes in the mouse. Genetics. 2003;165:2055. doi: 10.1093/genetics/165.4.2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bestor TH. The DNA methyltransferases of mammals. Hum Mol Genet. 2000;9:2395. doi: 10.1093/hmg/9.16.2395. [DOI] [PubMed] [Google Scholar]
- Bird AP. CpG-rich islands and the function of DNA methylation. Nature. 1986;321:209. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- Bjornsson HT, Fallin MD, Feinberg AP. An integrated epigenetic and genetic approach to common human disease. Trends Genet. 2004;20:350. doi: 10.1016/j.tig.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Brachmann CB, Sherman JM, Devine SE, Cameron EE, Pillus L, Boeke JD. The SIR2 gene family, conserved from bacteria to humans, functions in silencing, cell cycle progression, and chromosome stability. Genes Dev. 1995;9:2888. doi: 10.1101/gad.9.23.2888. [DOI] [PubMed] [Google Scholar]
- Cattanach BM, Beechey CV. Autosomal and X-chromosome imprinting. Dev Suppl. 1990;1990:63. [PubMed] [Google Scholar]
- Chung JH, Whiteley M, Felsenfeld G. A 5′ element of the chicken beta-globin domain serves as an insulator in human erythroid cells and protects against position effect in Drosophila. Cell. 1993;74:505. doi: 10.1016/0092-8674(93)80052-g. [DOI] [PubMed] [Google Scholar]
- Cooney CA. Are somatic cells inherently deficient in methylation metabolism? A proposed mechanism for DNA methylation loss, senescence and aging. Growth Dev Aging. 1993;57:261. [PubMed] [Google Scholar]
- Cui H, Horon IL, Ohlsson R, Hamilton SR, Feinberg AP. Loss of imprinting in normal tissue of colorectal cancer patients with microsatellite instability. Nat Med. 1998;4:1276. doi: 10.1038/3260. [DOI] [PubMed] [Google Scholar]
- Cui H, Onyango P, Brandenburg S, Wu Y, Hsieh CL, Feinberg AP. Loss of imprinting in colorectal cancer linked to hypomethylation of H19 and IGF2. Cancer Res. 2002;62:6442. [PubMed] [Google Scholar]
- Cui H, Cruz-Correa M, Giardiello FM, Hutcheon DF, Kafonek DR, Brandenburg S, Wu Y, He X, Powe NR, Feinberg AP. Loss of IGF2 imprinting: A potential marker of colorectal cancer risk. Science. 2003;299:1753. doi: 10.1126/science.1080902. [DOI] [PubMed] [Google Scholar]
- DeBaun MR, Niemitz EL, McNeil DE, Brandenburg SA, Lee MP, Feinberg AP. Epigenetic alterations of H19 and LIT1 distinguish patients with Beckwith-Wiedemann syndrome with cancer and birth defects. Am J Hum Genet. 2002;70:604. doi: 10.1086/338934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Koning DJ, Rattink AP, Harlizius B, van Arendonk JA, Brascamp EW, Groenen MA. Genome-wide scan for body composition in pigs reveals important role of imprinting. Proc Natl Acad Sci. 2000;97:7947. doi: 10.1073/pnas.140216397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorer DR, Henikoff S. Expansions of transgene repeats cause heterochromatin formation and gene silencing in Drosophila. Cell. 1994;77:993. doi: 10.1016/0092-8674(94)90439-1. [DOI] [PubMed] [Google Scholar]
- Ehrlich M, Gama-Sosa MA, Huang LH, Midgett RM, Kuo KC, McCune RA, Gehrke C. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res. 1982;10:2709. doi: 10.1093/nar/10.8.2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedoriw AM, Stein P, Svoboda P, Schultz RM, Bartolomei MS. Transgenic RNAi reveals essential function for CTCF in H19 gene imprinting. Science. 2004;303:238. doi: 10.1126/science.1090934. [DOI] [PubMed] [Google Scholar]
- Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4:143. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983;301:89. doi: 10.1038/301089a0. [DOI] [PubMed] [Google Scholar]
- Feinberg AP, Cui H, Ohlsson R. DNA methylation and genomic imprinting: Insights from cancer into epigenetic mechanisms. Semin Cancer Biol. 2002;12:389. doi: 10.1016/s1044-579x(02)00059-7. [DOI] [PubMed] [Google Scholar]
- Gaudet F, Hodgson JG, Eden A, Jackson-Grusby L, Dausman J, Gray JW, Leonhardt H, Jaenisch R. Induction of tumors in mice by genomic hypomethylation. Science. 2003;300:489. doi: 10.1126/science.1083558. [DOI] [PubMed] [Google Scholar]
- Gius D, Cui H, Bradbury CM, Cook J, Smart DK, Zhao S, Young L, Brandenburg SA, Hu Y, Bisht KS, Ho AS, Mattson D, Sun L, Munson PJ, Chuang EY, Mitchell JB, Feinberg AP. Distinct effects on gene expression of chemical and genetic manipulation of the cancer epigenome revealed by a multimodality approach. Cancer Cell. 2004;6:361. doi: 10.1016/j.ccr.2004.08.029. [DOI] [PubMed] [Google Scholar]
- Gregory PD. Transcription and chromatin converge: Lessons from yeast genetics. Curr Opin Genet Dev. 2001;11:142. doi: 10.1016/s0959-437x(00)00171-4. [DOI] [PubMed] [Google Scholar]
- Haber JE. Mating-type gene switching in Saccharomyces cerevisiae. Annu Rev Genet. 1998;32:561. doi: 10.1146/annurev.genet.32.1.561. [DOI] [PubMed] [Google Scholar]
- Hendrich B, Bickmore W. Human diseases with underlying defects in chromatin structure and modification. Hum Mol Genet. 2001;10:2233. doi: 10.1093/hmg/10.20.2233. [DOI] [PubMed] [Google Scholar]
- Holliday R, Pugh JE. DNA modification mechanisms and gene activity during development. Science. 1975;187:226. [PubMed] [Google Scholar]
- Huang C, Sloan EA, Boerkoel CF. Chromatin remodeling and human disease. Curr Opin Genet Dev. 2003;13:246. doi: 10.1016/s0959-437x(03)00054-6. [DOI] [PubMed] [Google Scholar]
- Ingrosso D, Cimmino A, Perna AF, Masella L, De Santo NG, De Bonis ML, Vacca M, D’Esposito M, D’Urso M, Galletti P, Zappia V. Folate treatment and unbalanced methylation and changes of allelic expression induced by hyperhomocysteinaemia in patients with uraemia. Lancet. 2003;361:1693. doi: 10.1016/S0140-6736(03)13372-7. [DOI] [PubMed] [Google Scholar]
- Jimenez-Sanchez G, Childs B, Valle D. Human disease genes. Nature. 2001;409:853. doi: 10.1038/35057050. [DOI] [PubMed] [Google Scholar]
- Krawczak M, Ball EV, Cooper DN. Neighboring-nucleotide effects on the rates of germ-line single-base-pair substitution in human genes. Am J Hum Genet. 1998;63:474. doi: 10.1086/301965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledbetter DH, Engel E. Uniparental disomy in humans: Development of an imprinting map and its implications for prenatal diagnosis. Hum Mol Genet. 1995;4:1757. doi: 10.1093/hmg/4.suppl_1.1757. (Spec No) [DOI] [PubMed] [Google Scholar]
- Mackay DJ, Coupe AM, Shield JP, Storr JN, Temple IK, Robinson DO. Relaxation of imprinted expression of ZAC and HYMAI in a patient with transient neonatal diabetes mellitus. Hum Genet. 2002;110:139. doi: 10.1007/s00439-001-0671-5. [DOI] [PubMed] [Google Scholar]
- Moore T, Haig D. Genomic imprinting in mammalian development: A parental tug-of-war. Trends Genet. 1991;7:45. doi: 10.1016/0168-9525(91)90230-N. [DOI] [PubMed] [Google Scholar]
- Moulton T, Crenshaw T, Hao Y, Moosikasuwan J, Lin N, Dembitzer F, Hensle T, Weiss L, McMorrow L, Loew T, et al. Epigenetic lesions at the H19 locus in Wilms’ tumour patients. Nat Genet. 1994;7:440. doi: 10.1038/ng0794-440. [DOI] [PubMed] [Google Scholar]
- Murata T, Kurokawa R, Krones A, Tatsumi K, Ishii M, Taki T, Masuno M, Ohashi H, Yanagisawa M, Rosenfeld MG, Glass CK, Hayashi Y. Defect of histone acetyltransferase activity of the nuclear transcriptional coactivator CBP in Rubinstein-Taybi syndrome. Hum Mol Genet. 2001;10:1071. doi: 10.1093/hmg/10.10.1071. [DOI] [PubMed] [Google Scholar]
- Ogonuki N, Inoue K, Yamamoto Y, Noguchi Y, Tanemura K, Suzuki O, Nakayama H, Doi K, Ohtomo Y, Satoh M, Nishida A, Ogura A. Early death of mice cloned from somatic cells. Nat Genet. 2002;30:253. doi: 10.1038/ng841. [DOI] [PubMed] [Google Scholar]
- Ohlsson R, Kanduri C, Whitehead J, Pfeifer S, Lobanenkov V, Feinberg AP. Epigenetic variability and the evolution of human cancer. Adv Cancer Res. 2003;88:145. doi: 10.1016/s0065-230x(03)88306-9. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Morison IM, Taniguchi T, Reeve AE. Epigenetic changes at the insulin-like growth factor II/H19 locus in developing kidney is an early event in Wilms tumorigenesis. Proc Natl Acad Sci. 1997;94:5367. doi: 10.1073/pnas.94.10.5367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordway JM, Williams K, Curran T. Transcription repression in oncogenic transformation: Common targets of epigenetic repression in cells transformed by Fos, Ras or Dnmt1. Oncogene. 2004;23:3737. doi: 10.1038/sj.onc.1207483. [DOI] [PubMed] [Google Scholar]
- Orlando V. Polycomb, epigenomes, and control of cell identity. Cell. 2003;112:599. doi: 10.1016/s0092-8674(03)00157-0. [DOI] [PubMed] [Google Scholar]
- Rainier S, Johnson LA, Dobry CJ, Ping AJ, Grundy PE, Feinberg AP. Relaxation of imprinted genes in human cancer. Nature. 1993;362:747. doi: 10.1038/362747a0. [DOI] [PubMed] [Google Scholar]
- Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- Rhee I, Jair KW, Yen RW, Lengauer C, Herman JG, Kinzler KW, Vogelstein B, Baylin SB, Schuebel KE. CpG methylation is maintained in human cancer cells lacking DNMT1. Nature. 2000;404:1003. doi: 10.1038/35010000. [DOI] [PubMed] [Google Scholar]
- Rhee I, Bachman KE, Park BH, Jair KW, Yen RW, Schuebel KE, Cui H, Feinberg AP, Lengauer C, Kinzler KW, Baylin SB, Vogelstein B. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature. 2002;416:552. doi: 10.1038/416552a. [DOI] [PubMed] [Google Scholar]
- Riggs AD. X inactivation, differentiation, and DNA methylation. Cytogenet Cell Genet. 1975;14:9. doi: 10.1159/000130315. [DOI] [PubMed] [Google Scholar]
- Riggs AD. DNA methylation and cell memory. Cell Biophys. 1989;15:1. doi: 10.1007/BF02991574. [DOI] [PubMed] [Google Scholar]
- Riggs AD, Pfeifer GP. X-chromosome inactivation and cell memory. Trends Genet. 1992;8:169. doi: 10.1016/0168-9525(92)90219-t. [DOI] [PubMed] [Google Scholar]
- Sandovici I, Leppert M, Hawk PR, Suarez A, Linares Y, Sapienza C. Familial aggregation of abnormal methylation of parental alleles at the IGF2/H19 and IGF2R differentially methylated regions. Hum Mol Genet. 2003;12:1569. doi: 10.1093/hmg/ddg167. [DOI] [PubMed] [Google Scholar]
- Schoenherr CJ, Levorse JM, Tilghman SM. CTCF maintains differential methylation at the locus. Nat Genet. 2003;33:66. doi: 10.1038/ng1057. [DOI] [PubMed] [Google Scholar]
- Sollars V, Lu X, Xiao L, Wang X, Garfinkel MD, Ruden DM. Evidence for an epigenetic mechanism by which Hsp90 acts as a capacitor for morphological evolution. Nat Genet. 2003;33:70. doi: 10.1038/ng1067. [DOI] [PubMed] [Google Scholar]
- Spielman RS, McGinnis RE, Ewens WJ. Transmission test for linkage disequilibrium: The insulin gene region and insulin-dependent diabetes mellitus (IDDM) Am J Hum Genet. 1993;52:506. [PMC free article] [PubMed] [Google Scholar]
- Statistics (National Center for Health Statistics) Health United States, 2003. 2003 http://www.cdc.gov/nchs/hushtm.
- Steenman MJ, Rainier S, Dobry CJ, Grundy P, Horon IL, Feinberg AP. Loss of imprinting of IGF2 is linked to reduced expression and abnormal methylation of H19 in Wilms’ tumour. Nat Genet. 1994;7:433. doi: 10.1038/ng0794-433. [DOI] [PubMed] [Google Scholar]
- Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- Strichman-Almashanu LZ, Lee RS, Onyango PO, Perlman E, Flam F, Frieman MB, Feinberg AP. A genome-wide screen for normally methylated human CpG islands that can identify novel imprinted genes. Genome Res. 2002;12:543. doi: 10.1101/gr.224102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamashiro KL, Wakayama T, Akutsu H, Yamazaki Y, Lachey JL, Wortman MD, Seeley RJ, D’Alessio DA, Woods SC, Yanagimachi R, Sakai RR. Cloned mice have an obese phenotype not transmitted to their offspring. Nat Med. 2002;8:262. doi: 10.1038/nm0302-262. [DOI] [PubMed] [Google Scholar]
- Tartof KD, Bremer M. Mechanisms for the construction and developmental control of heterochromatin formation and imprinted chromosome domains. Dev Suppl. 1990;1990:35–45. [PubMed] [Google Scholar]
- van Overveld PG, Lemmers RJ, Sandkuijl LA, Enthoven L, Winokur ST, Bakels F, Padberg GW, van Ommen GJ, Frants RR, van der Maarel SM. Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nat Genet. 2003;35:315. doi: 10.1038/ng1262. [DOI] [PubMed] [Google Scholar]
- Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RG, Otte AP, Rubin MA, Chinnaiyan AM. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- Woodson K, Flood A, Green L, Tangrea JA, Hanson J, Cash B, Schatzkin A, Schoenfeld P. Loss of insulin-like growth factor-II imprinting and the presence of screen-detected colorectal adenomas in women. J Natl Cancer Inst. 2004;96:407. doi: 10.1093/jnci/djh042. [DOI] [PubMed] [Google Scholar]
- Xu GL, Bestor TH, Bourc’his D, Hsieh CL, Tommerup N, Bugge M, Hulten M, Qu X, Russo JJ, Viegas-Pequignot E. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature. 1999;402:187. doi: 10.1038/46052. [DOI] [PubMed] [Google Scholar]