


Abstract
Experimental evidence indicates that about 60% of miRNA-binding activity does not follow the canonical rule about the seed matching between miRNA and target mRNAs, but rather a non-canonical miRNA targeting activity outside the seed or with a seed-like motifs. Here, we propose a new unbiased method to identify canonical and non-canonical miRNA-binding sites from peaks identified by Ago2 Cross-Linked ImmunoPrecipitation associated to high-throughput sequencing (CLIP-seq). Since the quality of peaks is of pivotal importance for the final output of the proposed method, we provide a comprehensive benchmarking of four peak detection programs, namely CIMS, PIPE-CLIP, Piranha and Pyicoclip, on four publicly available Ago2-HITS-CLIP datasets and one unpublished in-house Ago2-dataset in stem cells. We measured the sensitivity, the specificity and the position accuracy toward miRNA binding sites identification, and the agreement with TargetScan. Secondly, we developed a new pipeline, called miRBShunter, to identify canonical and non-canonical miRNA-binding sites based on de novo motif identification from Ago2 peaks and prediction of miRNA::RNA heteroduplexes. miRBShunter was tested and experimentally validated on the in-house Ago2-dataset and on an Ago2-PAR-CLIP dataset in human stem cells. Overall, we provide guidelines to choose a suitable peak detection program and a new method for miRNA-target identification.
INTRODUCTION
MicroRNAs (miRNAs) are small non-coding RNAs of ∼22 nucleotides (nt) that together with Argonaute proteins, including Ago2, form the miRNA-Induced Silencing Complex (miRISC) to inhibit the expression of target mRNAs by either repressing the translation or promoting the degradation (1,2). miRNAs target mRNAs by using a seed sequence of 6–8 nt in their 5΄ end (from 2 to 7 nt position of miRNA sequence) to bind to either the 3΄UTR or the coding sequence (CDS) of mRNAs (2). This mode of binding defines the so-called canonical miRNA-binding site (2). However, it has been experimentally found that about 60% of miRNA binding activity is non-canonical, which involves other portions of miRNA sequence outside the seed or with seed-like motifs including mismatches or bulges (3,4).
Despite the biological importance of the miRNAs, little is known about the specificity of miRNA binding sites on mRNAs (5). Different bioinformatics programs, such as miRANDA, TargetScan and PITA, use algorithms to predict canonical miRNA-binding sites based on different features, which include sequence complementary matches between miRNA seed and 3΄UTR, conservation of the target sequence across species, low free-energy of the predicted duplex and degrees of complementary matches beyond the seed sequence. Another method, called MIRZA, calculate the strength of the miRNA–mRNA heteroduplexes found in Ago2 CLIP-seq datasets by using 27 different energy parameters (6). MIRZA uses these parameters to predict the frequencies with which miRISCs bind to mRNA fragments in the mRNA pool (6). Although some experimentally validated miRNA-target mRNAs were successfully identified by using the support of these programs in combination with mRNA profile analysis, this approach can mislead to high rates of false positive and false negative targets (7,8). Moreover, these programs cannot predict non-canonical miRNA binding sites. On top of that, these programs hardly give a genome-wide point of view of how miRNA pathway can globally impacts on gene expression programs in cells. Indeed, one miRNA can potentially interact with a large number of target mRNAs through canonical and non-canonical activities (2). Conversely, one mRNA can be targeted by different miRNAs with additive or synergistic effects. Therefore, these are the reasons why, nowadays, more and more laboratories prefer to use genome-wide experimental approaches to define the totality of the miRNA targetome. The experimental approach would provide a better description of the underlying functional networks of miRNAs in cells or tissues and a more precise description of the features for miRNA binding sites.
CLIP-seq is a relatively new experimental technique to study the specificity of the binding activity for RNA-Binding Proteins (RBP) (9). This technique provides a comprehensive and genome-wide map of the direct RNA binding sites for RBP. The application of CLIP-seq to Ago2 has been used to identify the miRNA-binding sites (10–12). Three main variants of CLIP-seq have been developed so far, which chronologically include: (i) the HIgh-Throughput Sequencing of RNA isolated by CLIP (HITS-CLIP) (9), (ii) the high-throughput sequencing of RNA isolated from PhotoActivable-Ribonucleoside-enhanced-CLIP (PAR-CLIP) (11); and (iii) the individual-nucleotide resolution CLIP (iCLIP) (13). Briefly, all methods involve UV-cross-linking to bind RNA to proteins, partial RNA digestion, immunoprecipitation of the protein of interest, sodium dodecyl sulphate-polyacrylamide gel electrophoresis to get rid of the aspecific proteins, RNA extraction, reverse transcription and high-throughput sequencing. Although the PAR-CLIP and iCLIP provide higher resolution, the HITS-CLIP method is more largely used because of its wide adaptability, relatively easy sample preparation and the straightforward bioinformatics analysis (14). For this reason, here we focused on HITS-CLIP data analysis.
In this study we have developed a new method to identify miRNA-binding sites from CLIP-seq datasets, called miRBShunter. This method allows a comprehensive unbiased identification of both canonical and non-canonical miRNA-binding sites defined by the de novo identification of enriched motifs from Ago2 CLIP-seq peaks and the calculation of a miRNA::RNA heteroduplex score. The search for enriched de novo motifs permits the identification of miRNA binding sites, without restricting the analysis on the seed region, as other methods propose (15–18). Since the quality of the detected peaks is very important, to ensure a reliable output of the pipeline, first we have performed a comprehensive, quantitative and qualitative comparative evaluation of four different publicly available programs for HITS-CLIP peak detection, including CIMS, PIPE-CLIP, Piranha and Pyicoclip on four published Ago2 HITS-CLIP datasets (10,19–20) and on an unpublished in-house Ago2 HITS-CLIP dataset generated from P19 stem cells. By tuning the programs in default parameters, we found that Pyicoclip outperformed the other programs in terms of sensitivity, positional accuracy, agreement with TargetScan, specificity, as well as for consistency in finding the same results on different databases from the same tissue. Based on the benchmarking, we secondly validated the proposed method by running Pyicoclip-detected peaks of the in-house Ago2 HITS-CLIP dataset on a new developed pipeline, called miRBShunter. We found 40 potential canonical and 75 non-canonical miRNA binding sites with either a miRNA seed-like motif or outside the seed. Among them, we experimentally validated six new miRNA-binding sites (three canonicals and three non-canonicals) out of nine by gene reporter assay. To demonstrate that the method works independently on the CLIP-seq technique used, we tested it on an Ago2 PAR-CLIP dataset from human stem cells (21). miRBShunter pipeline found 215 canonical and 611 non-canonical miRNA binding sites and we have experimentally validated 5 out of 7 (3 canonicals and 2 non-canonicals), confirming the validity of our method. Therefore, the proposed method can be usefully used for detection of both canonical and non-canonical miRNA binding sites. Finally, we have developed a user-friendly tool with a graphic interface to use miRBShunter available at https://github.com/TrabucchiLab/miRBShunter.
MATERIALS AND METHODS
Publicly HITS-CLIP-seq datasets
Raw sequencing reads for all datasets except for the in-house one were downloaded by GEO (Leung et al. (20): GSE25310, Karginov et al. (19): GSE4404) and by the website: http://ago.rockefeller.edu/rawdata.php for Chi et al. (10) datasets. Pre-processing and alignment for all datasets were done following the protocol in Moore et al. (14). Briefly, reads were mapped on either the mouse (mm10) or the human (hg19) genome using the program Novoalign (http://www.novocraft.com/products/novoalign/). In Supplementary Table S1 is reported the number of mapped reads for each replicate of each dataset. The reproducibility of the replicates was assessed by performing principal component analysis using the R package ‘htSeqTools’ (R package version 1.14.0) (Supplementary Figure S1). Enriched peaks for the Ago2 PAR-CLIP dataset of Lipchina et al. (21) were downloaded by StarBase web server (22).
A list of most expressed Ago2-associated miRNAs in each dataset is shown in Supplementary Table S2. For the in-house dataset, we used the web-tool omiRas (23) to map the reads against mouse miRNA database and to normalize toward the reads count.
Main characteristics of publicly peak detection programs for HITS-CLIP analysis
Briefly, diverse strategies have been employed by different programs to identify peaks. In particular, ‘PIPE-CLIP’ (24) and ‘Pyicoclip’ (25) cluster together reads based on positional overlap, while ‘Piranha’ (26), ‘MiCLIP’ (27) and ‘HITS-CLIP data analysis’ (http://qbrc.swmed.edu/softwares.html) bin on genomic portions by fixed size. On the contrary, CIMS focuses on the mere identification of cross-linked sites by looking for clusters containing mutated reads (28). All these programs follow a statistical procedure to discriminate enriched clusters over the background (29). Briefly, PIPE-CLIP and Piranha employ the zero-truncated negative binomial likelihoods to model the distribution of the reads in peaks. These programs also use additional information to refine the peak detection, such as identification of cross-linked-dependent mutations for PIPE-CLIP or the use of covariates provided by the user for Piranha, including cross-linked-dependent mutations and transcript abundance. On the other hand, Pyicoclip performs a background estimation implementing the modified false discovery rate (FDR) procedure proposed by Yeo et al. (30). Particularly, it calculates the background frequency after randomly placing the same number of extended reads within the region and iterating the process many times. MiCLIP and HITS-CLIP data analysis employ Hidden Markow Models (HMM) to model the reads distribution in peaks. Finally, CIMS assesses statistical significance employing a permutation based model. Unfortunately, none of the programs employing HMM could be included in this analysis, since ‘MiClip’ is no longer available in the R CRAN repository (27) and ‘HITS-CLIP analysis’ is implemented in Matlab (http://qbrc.swmed.edu/softwares.html) that is not freely available. To our knowledge, there are two more programs that could be used to analyze HITS-CLIP data, namely ClipSeqTools (31) and GraphProt (32). Because ClipSeqTools is a complete pipeline for HITS-CLIP analysis more than a peak detection program, while GraphProt is a program for modeling the binding of RBPs, they were not considered for the present study.
Program implementation
All mappings and peak detection programs were run on linux workstation with two 2.6 GHz Intel Core Ubuntu machine equipped with 4 × 32 GB of RAM. All programs were installed and run locally on a workstation.
Peak detection parameters and peak list creation
All programs were run with default or suggested parameters, as follows:
- PIPE-CLIP: -m 1 –c 0 –l 20 –r 1 –M 0.001 –C 0.001 –s mm10/hg19
- Piranha: -b 20 –p 0.001-d zeroTruncatedNegativeBinomial
- Pyicoclip: –P-value 0.001 –region *.bed
- CIMS: parameters as in Moore et al. (14)
Because Pyicoclip requires the user to provide a BED file with the genome regions in which to look for peaks, we took from Homer database the genomic coordinates of the following categories: 3΄UTR (UnTranslated Region), 5΄UTR, CDS (protein CDS), introns, promoters, TTS (Transcription Termination Site), non-coding RNAs and intergenic sequences (33). Pyicoclip was run with each BED file separately on each dataset. To avoid any possible duplicates, after filtering (FDR < 0.001 and number of reads > 10) all peaks were put together and only one peak was retained in case of duplicates.
Because in CIMS the peak length is inferred by the user and automatically centered in the mutation points, we selected 40 nt of peak length as suggested by the developers. To avoid overlapping peaks, a custom python script was used to merge overlapping peaks into one single peak.
For all lists of peaks detected by the four programs, we filtered them by applying a threshold of FDR < 0.001 and reads count > 10.
Comparative evaluation analysis of peak detection programs
All comparative analyses were done using custom python and R scripts. Genomic location of peaks and peak sequence extraction were performed using Homer software (33). miRNA sequences were downloaded from miRBase (34) while high-confidence predicted miRNA-binding sites were downloaded from the newest TargetScan version (http://www.targetscan.org/ Release 7) (35). Intersection of peaks identified with different programs was calculated with the program BEDtools.
Motif analysis
For de novo motif discovery we ran the findMotifs.pl tool from Homer software (33) on the peak sequences identified by the 4 peak detection programs on each dataset, with the following parameters: -norevopp –len 7 –bits –noknown –mcheck ‘motif_file’. The last parameter is needed to compare the motifs found enriched in the peak sequences with the miRNA sequences. As background we used 3 times the scrambled sequences of the peak sequences (homer default option). To build the ‘motif_file’ we used the program ‘seq2profile.pl’ from Homer software with the reverse complement sequences of the most expressed miRNAs in each dataset.
To select the most confident motifs, we set up a combined score (CS), similar to the one used in (36), with the following formula:
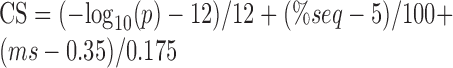 |
Where p is the P-value associated to the motif by Homer software, %seq is the percentage of the peak sequences in which the motif is present, ms is the match score of the motif with the given miRNA by Homer software. For an easier interpretation of the CS, we decided to subtract from each term of the equation a threshold quantity, in order to obtain a negative score when the three parameters are below such thresholds. Hence, a negative contribution to the CS is given by motif with P-value bigger than 1*10−12, present in <5% of the sequences or with a match score smaller than 0.35. The first two thresholds were suggested by Homer software to select most reliable motifs, while the third one is the threshold previously used to identify significant matches (36). Finally, we divided each term of the equation by ad hoc quantities, namely –log10 of the P-value threshold for the first term, the maximum percentage value for the second term and half of the score match threshold for the last term. Although this makes each term of the equation comparable to each other, the P-value score weights more than the other two scores.
To find peaks bearing the motifs selected with a CS ≥ 1 we used the FIMO program (37) with 0.0002 as highly stringent P-value, requiring not to search for motifs in the reverse complement of the provided sequences (–norc). Frequency matrices relative to the motifs in Homer software format were converted in MEME format using a custom python script.
miRNA-target RNA heteroduplex structure prediction
Duplex structure prediction between miRNAs and cognate RNA sequences was carried out with RNAduplex tool from Vienna package (38) using ‘RNAduplex –noLP < sequences.fa > output’, in the file sequences.fa there are the sequences of the miRNAs and their targets.
All duplexes without pairing in the motif were discarded.
Assessment of the duplex score
To sort the miRNA::mRNA heteroduplexes we set up the duplex score (DS) that summarize all the parameters considered in one score, as follows:
 |
This score is based on the following parameters:
MFE is the free energy of the most stable structure given by RNAduplex tool;
N_paired_nt is the number of paired nucleotides in the predicted heteroduplex;
N_paired_nt_motif is the number of paired nucleotides in the motif found;
N_paired_nt_seed is the number of paired nucleotides in the seed region;
N_bulges_seed is the number of bulges in the heteroduplex in the seed.
We arbitrarily decided to give the same weight to each feature in the formula, thus we divided each term of the equation to the maximum value that can be reached, to have a maximum contribution of one for each term.
miRBShunter pipeline
We collected all programs and scripts used to develop our method in an easy-to-use computational pipeline. The pipeline is written in python language (version 2.7) and it is freely available at https://github.com/TrabucchiLab/miRBShunter. We also provide a user-friendly interface for non-expert users.
Additional ‘Materials and Methods’ section are provided in the Supplementary Data.
RESULTS
Before testing our hypothesis method, we needed to make sure that we have access to good quality Ago2 peaks. Thus, we performed a benchmarking of different publicly available peak detection programs on four illustrative published Ago2 HITS-CLIP datasets (10,19–20) and one in-house unpublished dataset. In the Supplementary Table S3 we provide the main features of the datasets used in this study. To perform the analysis, we used the following peak detection programs: CIMS, PIPE-CLIP, Piranha and Pyicoclip (Table 1).
Table 1. Main features of the peak detection programs used in this analysis.
| CIMS | PIPE-CLIP | Piranha | Pyicoclip | |
|---|---|---|---|---|
| Technology | HITS-CLIP | HITS-CLIP, PAR-CLIP, iCLIP | HITS-CLIP, PAR-CLIP, iCLIP, RIP-seq | HITS-CLIP, PAR-CLIP, iCLIP, RIP-seq, CHIP-seq |
| Website | http://zhanglab.c2b2.columbia.edu/index.php/CIMS_Documentation | https://github.com/QBRC/PIPE-CLIP | http://smithlabresearch.org/software/piranha/ | https://bitbucket.org /regulatorygenomicsupf /pyicoteo |
| Input file format | Bed | Bam | Bed | Eland/sam/bed/bam |
| Preprocessing | Yes | Yes | No | Yes |
| Resolution | Single nucleotide | Cluster of reads | Genome binning | Predefined regions |
| Statistical model | Background estimation | Zero-Truncated Negative Binomial | Many | Background estimation |
| Mutation | Yes | Yes | Yes | No |
| FDR | Permutation | Benjamini - Hochberg | Benjamini - Hochberg | Modified FDR |
| Peak length assessment | User defined | Automatic | Automatic | Automatic |
| Annotation | No | Yes | No | No |
| Control data | No | Yes | Yes | No |
| Implementation | Perl | Python/Galaxy | Executable binary | Python |
Briefly, each replicate from each dataset was processed with the same pipeline (see ‘Materials and Methods’ section for details). After mapping and collapsing identical reads, replicates belonging to the same dataset were merged. Since each program has its own statistical model to calculate the significance of the identified peaks, for the comparative analysis we arbitrarily decided to select peaks with FDR < 0.001 and number of reads > 10 to make the data across programs comparable among each other. Finally, the peak detection programs were run with the default tuning or the recommended parameters. While some programs have tunable parameters, we forgo parameter optimization, which might have improved the results for some datasets, as this task may be beyond the ken of most users.
Comparative analysis of the main characteristics of the identified peaks
For each dataset, the number of peaks identified was different (Figure 1A). CIMS always detected much less peaks compared to the other programs, since it identifies peaks by looking for reads having cross-linked-dependent mutations. Indeed, these mutations are rare and reads containing them represent a small portion of the totality of reads (39,40). On the other hand, the number of peaks identified by Piranha and Pyicoclip varied to the different datasets: from few hundreds to several thousands, reflecting a higher capability to adapt to the data than the other two programs. These differences between programs and datasets are likely caused by different statistical and mathematical models that lead to different results depending on the level of noise/background of the experiments.
Figure 1.

General features of the peaks identified by the four indicated programs for the indicated Ago2 HITS-CLIP datasets. (A) Total number of peaks identified. (B) Boxplots of the peak lengths found. (C) Genomic location of the identified peaks. Color code is indicated. 3΄UTR (3΄ untranslated region), 5΄UTR (5΄ untranslated region), CDS (protein coding sequence), pseudo (pseudogene), rRNA (ribosomal RNA), TTS (terminal transcription site).
In Figure 1B, we reported the length of the identified peaks as an important parameter that influences the peak accuracy toward the miRNA seed-match binding sites. For PIPE-CLIP and Pyicoclip, we found a broader range of peak length, which median is between 40 and 80 nt. For Piranha, the length of the peaks is determined by the binning size, which was set up at 20 nt by default. For this reason, the peak length average found for Piranha was 20 nt and other lengths are multiple of 20. For CIMS all peak measured 40 nt long, as the peak length was defined by us as suggested by the developers of the program. Therefore, this analysis showed how the difference in the strategy employed by the different programs to cluster the reads influences the peak length.
Then, we inspected the genomic location of the identified peaks. As shown in Figure 1C, many Ago2-peaks mapped in genomic regions other than the 3΄UTR and the CDS, as it was already reported in several publications (12,36,41–43). The variability of the percentage of the peaks mapped in different regions is higher across the datasets than across the programs. For example, although the replicates D and E and the replicates A, B and C of both Chi et al. (10) datasets originate from the same biological material but processed with two different antibodies, in the replicates D and E all four programs found about 40% of peaks mapped in the 3΄UTR, while in the replicates A, B and C only about 25% of peaks. Surprisingly, virtually no peaks in 3΄UTR were detected by all programs for the Leung et al. (20) dataset, while around 20% of peaks mapped in 3΄UTR for the in-house dataset, which was performed in the same cell type and same antibody of Leung et al. (20). These results indicate how different experimental conditions can have a dramatic impact on the data analysis output.
In addition, we performed a pairwise comparison of the genomic positions of the identified peaks by the different programs for each dataset (Figure 2A). Overall, the set of shared peaks by all the programs was rather poor, which was assessed around 50% in average. As expected, CIMS, which detected much less peaks, found more shared peaks with the other programs, indicating that they represent a subset of peaks identified by others with less stringent default parameters. In a few number of cases, CIMS has even reached an identity higher than 80% with other programs, including for Chi et al. (10) dataset D and E and for Leung et al. (20) dataset analyzed by Pyicoclip and for Karginov et al. (19) analyzed by Piranha. Similarly, Pyicoclip and Piranha reached a good agreement in many datasets. Conversely, PIPE-CLIP showed the least degree of shared peaks with other programs. Moreover, we calculated the intersection of peaks found by the four programs for each dataset (Figure 2B). The dataset D and E of Chi et al. (10) showed the highest number of shared peaks (46 peaks with length >20 nt), while the Leung et al. (20) dataset showed the lowest one. Therefore, these findings suggest that the detection of different peaks is likely due to different mathematical models employed by the programs.
Figure 2.

Positional overlap of the identified peaks by the indicated programs on the indicated Ago2 HITS-CLIP datasets. (A) Pairwise comparison of shared peaks. Each panel shows the percentage of total peaks from one program (column) that shared with another program (row). Color code is indicated. (B) Number of peaks in the intersection of the peaks found by the four programs applied on each dataset.
Altogether, this analysis indicated that both experimental and bioinformatics conditions need optimization to obtain more consistent data output across both programs and dataset. These results prompted us to perform a benchmark analysis of the peak detection programs.
Sensitivity and rank accuracy toward canonical miRNA binding sites
Next, we asked whether the programs that detected more peaks gained in sensitivity to identify more peaks containing miRNA-binding sites. To address this specific task, we calculated the number of canonical miRNA seed-match occurrences for the top expressed Ago2-loaded miRNAs in each dataset (Supplementary Table S2). As shown in Figure 3A, the additive number of perfect miRNA seed-matches was calculated within intervals of 10 peaks sorted by reads count. The performances of the programs are remarkably different: PIPE-CLIP overall outperformed the other methods showing a higher rate of canonical seed-match discovery in the top ranked peaks. Pyicoclip performances are close to PIPE-CLIP and often the two curves of discovery rate are superimposed especially for the top ranked peaks, while Piranha and CIMS performed worse.
Figure 3.

Sensitivity and rank accuracy of the indicated datasets analyzed by the indicated programs. (A) Additive occurrence of canonical miRNA seed-matches as a function of increasing ranked peaks examined by intervals of 10 peaks sorted by reads count. (B) Frequency of canonical miRNA seed-matches per peak examined in increasing 50 peak intervals (50, 100 peaks, etc.) sorted by reads count.
In addition, programs should provide accurate means for ranking peaks according to some confidence metrics. Therefore, to assess peak ranking accuracy, we calculated the rate of occurrence for miRNA canonical seed-matches in each peak within intervals of 50 peaks sorted by read count (Figure 3B). For PIPE-CLIP and Pyicoclip, the percentage of peaks containing canonical miRNA binding sites decays with decreasing peak rank, indicating that rank by reads count generally discriminates well between high confidence and lower confidence peaks. Piranha and CIMS showed again very poor performances, indicating the absence of different levels of confidence peaks.
Overall, both PIPE-CLIP and Pyicoclip programs show a better sensitivity, which is quite similar over most of the peak lists, and a better peak rank in recovering canonical miRNA-binding sites. To rule out the possibility that PIPE-CLIP and Pyicoclip have wide peak lengths on the top ranked peaks, which may increase the chances to get more seed-match sequences, we calculated the correlation between peak length and reads count. As shown in Supplementary Table S4, only PIPE-CLIP shows a very good correlation, indicating that the good performances in sensitivity of this program are due to the wider peak lengths of the top ranked peaks.
Spatial resolution
A further important feature to take into account is the degree of precision to facilitate the location of the binding site. In fact, the length of identified peaks can dramatically determine the accuracy of de novo motif identification and, in case of miRNAs, the identification of non-canonical binding sites, since wider peaks imply the addition of noisy sequences that can overshadow the miRNA-binding signature. To evaluate the spatial resolution of the different peak detection programs, we calculated the following two parameters, (i) the distance between the canonical seed-matches for the top expressed miRNAs in each dataset and the peak centers, and (ii) the distance between the canonical seed-matches and the cross-linked-dependent mutations. Programs with a higher degree of spatial resolution are expected to have peaks centered on both the seed-matches and the cross-linked-dependent mutations. In Figure 4A and Supplementary Figure S2A the distance distributions between the seed-matches and the peak centers are shown. Piranha outperforms the other programs for all datasets, except for the A, B and C replicates dataset from Chi et al. (10). Pyicoclip also showed rather narrow distributions, despite the wide distribution of peak lengths (Figure 1B). CIMS worked very well just for two datasets, even if the peak length is set up at 40 nt, while PIPE-CLIP performed the worse.
Figure 4.

Spatial resolution of canonical binding sites identified in peaks by the indicated programs for the indicated datasets. (A) Boxplot (upper panel) or frequency (lower panel) of the distance distribution between seed-matches and peak centers. Distances in frequency plots are represented from −40 to 40 nt for a better visualization. (B) Boxplot (upper panel) or frequency (lower panel) of the distance distribution between seed-matches and the most frequent mutations in the peaks. Distances in frequency plots are represented from −40 to 40 nt for a better visualization.
Similar data were obtained by measuring the distance distributions between the seed-matches and cross-linked-dependent mutations (Figure 4B and Supplementary Figure S2B), showing good and consistent performances only for Piranha and Pyicoclip. Interestingly, we found that the cross-linked-dependent mutation was shifted to the left side compared to the peak center, suggesting that Ago2 cross-links to the target mRNA immediately upstream the seed-match sequence.
Overall, for all programs, the positional accuracy of the canonical miRNA binding site was upper for the D and E replicates dataset from Chi et al. (10) than for the other datasets. On the other hand, for almost all datasets the positional accuracy of the peaks identified by Piranha and Pyicoclip was upper that for the other programs. Therefore, this analysis indicates that the spatial resolution of Ago2 binding is not influenced by the peak length.
Agreement toward high confidence miRNA-binding sites predicted by Targetscan
Next, we assessed the specificity of the considered peak detection programs by calculating the level of agreement with TargetScan (35). TargetScan predicts miRNA-binding sites by searching for the presence of perfect match between 3΄UTR sequences and the seed region of miRNAs (44). We calculated the agreement with TargetScan in each dataset by calculating the percentage of peaks containing TargetScan-predicted binding sites for the top expressed Ago2-loaded miRNAs over the total number of identified peaks by each programs (Figure 5). Importantly, because TargetScan only predicts miRNA binding sites in the 3΄UTR, this analysis was restricted to peaks located in this region.
Figure 5.

Agreement with TargetScan-predicted miRNA binding sites and the 3΄UTR peaks found by the indicated programs on the indicated datasets. (A) Percentage of the peaks containing TargetScan-predicted binding sites for each highly expressed-Ago2-loaded miRNA in each dataset calculated over the totality of the 3΄UTR peak number detected by the indicated programs. (B) Sum of the percentages of the peaks containing TargetScan-predicted binding sites for each dataset calculated over the totality of the 3΄UTR peak number detected by the indicated programs.
Overall, Pyicoclip showed the highest percentage of binding sites in common with the TargetScan prediction for three out of five datasets, while PIPE-CLIP for the other two. The agreement between TargetScan and Piranha peaks is very low, under 10%, except for the dataset of Chi et al. replicates A, B and C. Agreement between CIMS and TargetScan is also very poor, except for the dataset of Chi et al. replicates D and E. Finally, Leung et al. dataset (20) showed a very poor percentage of common binding sites with Targetscan, which never overcame 2% considering the four programs. A possible source of disagreement can be that TargetScan only predicts perfect seed-pairing located in 3΄UTR, while the majority of miRNA-binding sites can be expected to be non-canonicals (12,36,41).
We calculated the degree of overlap of the peaks containing the miRNA-binding sites predicted by TargetScan found by the different programs for the same dataset. As shown in Supplementary Figure S3A, we found that Pyicoclip and PIPECLIP overall share the highest degree of overlap, while Piranha showed a modest overlap with Pyicoclip, but very poor with PIPE-CLIP.
In addition, to evaluate the data reproducibility between two datasets originated from the same biological material, we compared the number of binding sites predicted by TargetScan in peaks shared by the two datasets of Chi et al. (Supplementary Figure S3B). These two datasets only differ by the anti-Ago2 antibody used to perform the immunoprecipitation. We found that 75% of peaks identified in the 3΄UTR by Pyicoclip containing miRNA-binding sites predicted by TargetScan for the replicates A, B and C were also identified in the replicates D and E. However, because in the replicates D and E we found much more peaks containing miRNA-binding sites predicted by TargetScan, this dataset only shows about 15% of overlap with the replicates A, B and C. As shown in Supplementary Table S5, similar percentages were found for each miRNA, except for miR-410/344de/344b-1-3p, miR-125a-5p/125b-5p/351/670/4319 and miR-193/193b/193a-3p. Therefore, these data indicate that, although Pyicoclip once again better performed, different experimental conditions (i.e. different antibody used for immunoprecipitation) can change the results of the analysis.
Overall our benchmark analysis of four different peak detection programs showed that Pyicoclip outperformed in term of sensitivity, positional accuracy of the seed-match binding site, agreement with predicted sites by TargetScan.
Identification of miRNA binding sites through de novo motif finding and RNA duplex prediction
Since canonical binding sites do not represent the totality of miRNA-binding sites (12,36,41), we propose a novel strategy to find and characterize potential binding sites. We reasoned that Ago2 peak sequences would potentially show enriched motifs reverse-complementary to any portion of the sequence of the most expressed miRNAs. Based on this assumption, we have developed a computational pipeline, called ‘miRBShunter’ (Supplementary Figure S4). miRBShunter (freely downloadable from https://github.com/TrabucchiLab/miRBShunter) is fed with the coordinates of the Ago2 peaks and the sequence of the most expressed Ago2-loaded miRNAs. miRBShunter extracts peak sequences, annotates on the genome and converts miRNA sequences in Homer format by using Homer software (33). Homer software is also used to identify 7 nt-de novo enriched motifs in Ago2 peaks (33) and to map them onto the reverse-complement sequence of miRNAs (for computational parameters, please see the ‘Materials and Methods’ section). To select only the most confident RNA motifs, we used a CS based on the following parameters calculated by Homer software: (i) –log10(P-value) assigned to the motif; (ii) percentage of target sequences in which the motif matches; and iii) the score of the matching of the motif on the miRNA sequence. Then, we classified as ‘significant’ all the motifs with CS > 0 and as ‘best candidates’ the ones with CS ≥ 1. After selecting the most confident motifs, miRBShunter maps them onto the peak sequences using the tool FIMO (37).
We tested de novo motif finding for the identified Ago2 peaks by the four programs in the five datasets here considered. As showed in Figure 6, Pyicoclip found the highest number of motifs with CS ≥ 1 in two datasets and with CS > 0 in three datasets, indicating that this program finds the highest number of peaks containing potential binding sites for miRNAs. Among the motifs with CS ≥ 1, we have found several canonical miRNA-binding sites, however the majority of the motifs were non-canonicals. Noteworthy, peaks of the dataset of Chi et al. replicates D and E showed the strongest enrichment for canonical miRNA-binding sites, especially for the peaks found by Pyicoclip (Supplementary Table S6).
Figure 6.

Identification of miRNA binding sites through de novo motif searching. Number of predicted miRNA::RNA motif pairs with combined score (CS) bigger than 0 or 1 for each program in each dataset. Color code indicated.
In addition, we tested de novo motif finding by MEME software for the identified Ago2 peaks by the four programs in the five datasets. MEME software found just a small subset of the motifs found by Homer software (CS ≥ 1) and only for the D and E replicates of Chi et al. (10) and Karginov et al. (19) datasets (data not shown), indicating that for our analysis Homer software is more appropriate.
Thereafter, the motifs have been identified, miRBShunter uses RNAduplex program from the Vienna suite (38) to predict RNA heteroduplexes formed by the association between Ago2 peaks and the most expressed Ago2-loaded miRNAs. miRBShunter filters out the miRNA::RNA duplexes that do not bear any previously identified motifs by Homer software or with CS < 1. Finally, the resultant miRNA::RNA duplexes are ranked by a DS based on the following parameters: (i) the free energy of the miRNA::RNA heteroduplex given by RNAduplex tool (38); (ii) the pairing degree between the full length miRNA and the target RNA sequence; (iii) the pairing degree between the miRNA motif and the target RNA sequence; (iv) the pairing degree between the miRNA seed and the target RNA sequence; and (v) the number of bulges in the seed region. Because canonical miRNA-binding sites confer a more potent targeting (35), we arbitrarily introduced the last two parameters to add more weight on the seed region. Therefore, the DS score summarize the biophysical parameters of the miRNA::mRNA duplex structure through the identified motifs. Additional details about the CS and DS are provided in the ‘Materials and Methods’ section. The final output of miRBShunter is a list of potential canonical and non-canonical miRNA-binding sites sorted by decreasing DS. Higher DS indicate high confidence miRNA-binding sites.
Experimental validation of miRBShunter
To validate the miRNA-binding sites found by miRBShunter, we focused on the in-house dataset. Because Pyicoclip outperformed the other programs, the validation was carried out on the results obtained with this program. Briefly, we feed miRBShunter with the peak coordinates in CDS and 3΄UTR as primary substrate of miRNAs and the sequence of the 22 most expressed Ago2-loaded miRNAs in the dataset. We found 115 potential miRNA::mRNA duplexes, including 40 canonicals and 75 non-canonicals (46 showing a seed-like motif whereas 29 outside the miRNA seed) (Figure 7A and Supplementary Table S7).
Figure 7.

Experimental validation of the miRNA binding sites found by our method. (A) Left panel: the motifs identified by miRBShunter for the in-house dataset with CS ≥ 1 are reported with the corresponding miRNA matching and the type of binding site (BS). Right panel: distribution of the different types of miRNA:mRNA duplexes identified by miRBShunter. Color code is indicated. (B) Relative luciferase activity of reporter constructs containing two either wild-type or mutant miRNA binding sites. HEK-293T cells were transfected with either the indicated mimic miRNA or control. The data were normalized using Renilla activity. miRNA::RNA heteroduplex structure expected to result from base pairing of the indicated miRNAs with the indicated binding sites are shown below the corresponding luciferase activity. A-U and G-C base pairs are represented by solid lines; G-U wobble base pairs are represented by dotted lines. Underlined the motif found by Homer software and matching with the portion of 7 nt of miRNA sequence in bold. Data are presented as mean and s.d. (n = 3). Student's t-test: *P < 0.05; **P < 0.01;; ***P < 0.001.
To assess experimentally the validity of the identified miRNA-binding sites, we performed a gene reporter assay. Briefly, we sorted the list of 115 miRNA-target RNA heteroduplexes by decreasing value of DS and randomly selected 9 miRNA-target RNA heteroduplexes (Supplementary Table S7). Then, we constructed reporter plasmids in which two copies of nine potential binding sites were cloned downstream to a luciferase CDS. Mimic miRNA overexpression in HEK-293T cells significantly repressed luciferase activity of six reporter constructs containing the wild-type binding site for three different miRNAs, but not the mutant reporter constructs (Figure 7B and data not shown). The validated six binding sites are all located in the 3΄UTR and they are composed by three canonical binding sites for miR-302b and three non-canonical binding sites for miR-302b, miR-181b and miR-99a, respectively. The validated non-canonical motifs were seed-like motifs with one-mismatch for both miR-302b and miR-181b, and with the motif outside the seed (position 9–15) for miR-99a. Two validated miR-302b-binding sites are also being predicted by TargetScan.
To prove that both method and the associated tool are adaptable to all CLIP-seq variants, we tested it on an Ago2 PAR-CLIP dataset from human embryonic stem cells (21). We ran the pipeline on the peaks that map in the 3΄UTR and the CDS downloaded from StarBase (22) and considered the 20 most expressed Ago2-loaded miRNAs (21). Briefly, we found 826 miRNA:mRNA target duplexes, 215 canonical and 611 non-canonical miRNA binding sites, which include 357 seed-like and 254 outside the seed binding sites (Supplementary Figure S5A and Table S6). Sixty-six percent of the identified binding sites contained either the canonical or the seed-like binding sites for stem cell specific miR-302 family. Importantly, 22% of the miR-302-binding sites identified by miRBShunter are in common with those identified in the original publication (21), supporting the validity of our approach. By gene reporter assay, we validated five binding sites out of seven (Supplementary Figure S5B and Table S6). As expected, both canonical and non-canonical miRNA binding sites were experimentally validated. Therefore, these data confirm the adaptability of our method to any Ago2 CLIP-seq dataset.
Noteworthy, all the validated miRNA-binding sites in both datasets are top ranked in the DS list of potential heteroduplexes, confirming the validity of our method/approach.
In conclusion, we have demonstrated that miRNA-binding sites containing either seed-like or outside the seed region motifs are bona-fide miRNA-binding sites actively silenced. These data confirm the validity of our method to find in an unbiased fashion both canonical and non-canonical binding sites for miRNAs. Altogether, we can conclude that higher CS and DS are associated with increased chances to identify bona-fide miRNA-binding sites.
DISCUSSION
High-throughput sequencing technologies applied to gene expression control, such as RNA-seq, ChIP-seq and CLIP-seq, provide huge amount of data making the computational analysis a straightforward mean to extract meaningful biological information. In RNA biology, CLIP-seq and in particular HITS-CLIP, is becoming a fundamental experimental approach to understand protein–RNA interactions to map the exact binding sites and study biophysical features. When this technique is applied to the Ago2 protein, HITS-CLIP can assess the miRNA-binding sites and help the researcher to investigate the direct and specific role played by miRNAs in a cell or a tissue type manner. Based on the assumption that highly expressed miRNAs would define a reverse-complementary motif signature(s) from any portion of their sequence on the peaks found by Ago2 CLIP-seq experiments, we propose an original and unbiased method to find and characterize canonical and non-canonical miRNA-binding sites from Ago2 CLIP-seq data.
Since a key step in the CLIP-seq data analysis workflow is peak detection, we have performed a quantitative and qualitative analysis of the performances of four publicly available peak detection programs on four published Ago2 HITS-CLIP datasets (10,19–20) and one in-house unpublished dataset. To our knowledge, this is the first time that a benchmarking of Ago2 CLIP-seq peak detection programs has been performed. The peak detection programs we considered include PIPE-CLIP, Pyicoclip, Piranha and CIMS. Our analysis shows that these programs found a different number of peaks on the same dataset with a poor overlap of peaks demonstrating that different strategies and mathematical models used by these programs lead to quantitative and qualitative differences of the output. The benchmarking was performed on the identification of seed-match occurrences and agreement with TargetScan predicted miRNA-binding sites for the top expressed Ago2-loaded miRNAs in each dataset. Because of the lack of a comprehensive list of bona-fide miRNA binding sites, we could not calculate true/false positive rates. Furthermore, we decided not to use a simulated training set generated in silico, because such a set are unreliable due to challenges in mimicking the form and variability of CLIP-seq peaks (45). Overall, Pyicoclip showed better performances in the whole analysis. Indeed, PIPE-CLIP outperformed only regarding the sensitivity, however, the very high correlation between peak length and read count, indicates that the top ranked peaks have many seed-matches just by chance. While Piranha outperformed concerning the spatial resolution, its performance was poor regarding the sensitivity. This is probably caused by an underestimation of peak lengths due to the binning procedure. CIMS is designed for the identification of the cross-linked-dependent mutations, rather than for the proper peak detection function. As expected, CIMS found much less peaks than the other programs, therefore underestimating them. According to this conclusion, in HITS-CLIP analysis the proportion of reads containing cross-linked-dependent mutations is very low (39,40). Taken together, depending on the biological question, one may want to end up with a stringent list of most confident peaks or a more comprehensive set of peaks, with the risk to include false positives. This balance of stringency and sensitivity can be tuned by changing the parameters of the detection peak programs to meet the needs of the researcher. Alternatively, the researcher can rank the detected peaks according to statistics, such as number of reads, fold of enrichment over the background, or P-value and apply a threshold according to the desired stringency.
To test the reliability of the miRNA-binding sites found by the four programs, we checked whether they are high-confidence binding sites predicted by TargetScan. In agreement with other analyses, Pyicoclip outperformed others showing around 15% of agreement for single miRNAs and around 40–50% considering all miRNAs by datasets. Importantly, significant differences were observed in datasets originating from the same biological material but treated with different experimental conditions. In particular, the two datasets of Chi et al. (10) originated from the mouse neocortex were obtained with two different antibodies to immunoprecipitate Ago2. In fact, we detected many more peaks bearing high-confidence miRNA-binding sites predicted by the TargetScan in the replicates D and E compared to the replicates A, B and C. These discrepancies indicate that experimental conditions have a big impact on the ultimate outcome of the peak detection analysis.
We took advantage of this comparative analysis on the performances of different peak detection programs to test our novel method to identify miRNA-binding sites from CLIP-seq data. Briefly, we hypothesized that enriched motifs on the detected peaks from Ago2 CLIP-seq experiments are defined by the binding activity of any portion of the sequence of highly expressed Ago2-loaded miRNAs. Based on this hypothesis, we set up a de novo motif discovery analysis with Homer software (33) and a CS to select enriched motifs, as putative binding sites for the most expressed miRNAs. We have found that, once again, Pyicoclip found the highest number of motifs that significantly match with the sequence of the top expressed miRNAs for each experiments, demonstrating the validity of both Pyicoclip and our original approach to find miRNA-binding sites.
To further validate our hypothesis, we applied our method to the 3΄UTR- or CDS-located peak set obtained by Pyicoclip from the in-house dataset and obtained a sub-set of 115 miRNA canonical and non-canonical heteroduplexes having a 7 nt-de novo motifs with CS ≥ 1, corresponding to seven motifs matching with eleven miRNAs among the 22 most expressed in P19 cells. In order to rank them, we set up a DS based on different parameters, including biophysical, architectural (presence or absence of mismatches or bulges) and, finally, position of the pairing, based on the assumption that the miRNA seed sequence has much more weight in defining miRNA-binding sites and silencing activity. In concordance with earlier findings (3,4), we found a diversity on miRNA targetome comprising both canonical and non-canonical mRNA binding activity. Moreover, we found that many of the endogenous highly expressed miRNAs exhibited consistent seed-like or outside the seed non-canonical targeting preferences, such as miR-181b and miR-99a, respectively. However, miR-302b mainly acts through canonical binding sites. To prove the adaptability of miRBShunter to any Ago2 CLIP-seq dataset, we successfully ran our pipeline on a publicly available Ago2 PAR-CLIP dataset from human stem cell (21). Importantly, experimental validation of our method was carried out with gene reporter assay for both datasets. In total, we showed that 11 new miRNA::mRNA target interactions are bona-fide miRNA binding sites showing silencing activity. Among them, for the in-house dataset, we validated three canonical binding sites for miR-302b-3p and three non-canonical binding sites, including two seed-like or one outside the seed motif for miR-302b-3p, miR-181b and miR-99a, respectively. Whereas, for the public PAR-CLIP dataset from human stem cells (21), we validated three canonical binding sites for miR-302b-3p and two non-canonical binding sites outside the seed position for miR-106b-5p. These data demonstrate the validity of our method to consider the entire miRNA sequence as potentially active for binding and silencing target mRNAs.
Presently, miRBShunter works for mouse and human miRNA-mRNA pairs, but it can be further extended to other species.
In conclusion, our method illustrates the advantage of considering the de novo identification of enriched motifs from Ago2 CLIP-seq peaks to find miRNA matches for highly expressed Ago2-loaded miRNAs. Alternatively to other methods (15–18), we did not restrict the search of motifs to the miRNA seed sequence, but we considered the unbiased possibility for miRNAs to bind to any portion of the sequence. We believe this method provides a more global aspect of the role played by miRNAs in a determined cell type or tissue system, providing a comprehensive list of canonical and non-canonical miRNA targetome and defining the gene expression program under the control of the miRNA pathway. Furthermore, this method can be usefully adopted when differential CLIP-seq cannot be applied, such as in human biopsies in which knockout or knockdown of specific miRNAs or RBPs is not doable.
Finally, we have developed a new tool, named miRBShunter, that implements the pipeline herein described. It takes as input the coordinates of the enriched peaks and the Ago2-loaded miRNAs expressed in CLIP-seq experiments and finds exact binding sites for these miRNAs in the peak sequences. We believe that miRBShunter is a valuable resource for researchers working on miRNA biology due to its easy applicability and reliability of the results.
ACCESSION NUMBER
All sequencing results for the in-house dataset were submitted to the GEO database under accession number Series GSE85219.
Supplementary Material
ACKNOWLEDGEMENTS
We are indebted to Dr Pascal Barbry for UCA Genomix platform and Dr Kevin Lebrigand for bioinformatics support.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
FRM [DEQ20140329551 to M.T.]; ANR through the ‘Investments for the Future’ [ANR-11-LABX-0028-01 (LABEX SIGNALIFE) to M.T.]; FRM [ING20140129224 to E.R.]. France Genomique to the UCA Genomix platform. Funding for open access charge: Inserm.
Conflict of interest statement. None declared.
REFERENCES
- 1. Ha M., Kim V.N.. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014; 15:509–524. [DOI] [PubMed] [Google Scholar]
- 2. Pasquinelli A.E. MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat. Rev. Genet. 2012; 13:271–282. [DOI] [PubMed] [Google Scholar]
- 3. Helwak A., Kudla G., Dudnakova T., Tollervey D.. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell. 2013; 153:654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chi S.W., Hannon G.J., Darnell R.B.. An alternative mode of microRNA target recognition. Nat. Struct. Mol. Biol. 2012; 19:321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hausser J., Zavolan M.. Identification and consequences of miRNA-target interactions–beyond repression of gene expression. Nat. Rev. Genet. 2014; 15:599–612. [DOI] [PubMed] [Google Scholar]
- 6. Khorshid M., Hausser J., Zavolan M., van Nimwegen E.. A biophysical miRNA-mRNA interaction model infers canonical and noncanonical targets. Nat. Methods. 2013; 10:253–255. [DOI] [PubMed] [Google Scholar]
- 7. Friedman R.C., Farh K.K., Burge C.B., Bartel D.P.. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009; 19:92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu C., Mallick B., Long D., Rennie W.A., Wolenc A., Carmack C.S., Ding Y.. CLIP-based prediction of mammalian microRNA binding sites. Nucleic Acids Res. 2013; 41:e138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Licatalosi D.D., Mele A., Fak J.J., Ule J., Kayikci M., Chi S.W., Clark T.A., Schweitzer A.C., Blume J.E., Wang X. et al. . HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 2008; 456:464–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chi S.W., Zang J.B., Mele A., Darnell R.B.. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009; 460:479–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hafner M., Landthaler M., Burger L., Khorshid M., Hausser J., Berninger P., Rothballer A., Ascano M. Jr, Jungkamp A.C., Munschauer M. et al. . Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010; 141:129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zisoulis D.G., Lovci M.T., Wilbert M.L., Hutt K.R., Liang T.Y., Pasquinelli A.E., Yeo G.W.. Comprehensive discovery of endogenous Argonaute binding sites in Caenorhabditis elegans. Nat. Struct. Mol. Biol. 2010; 17:173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sugimoto Y., Konig J., Hussain S., Zupan B., Curk T., Frye M., Ule J.. Analysis of CLIP and iCLIP methods for nucleotide-resolution studies of protein-RNA interactions. Genome Biol. 2012; 13:R67–R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Moore M.J., Zhang C., Gantman E.C., Mele A., Darnell J.C., Darnell R.B.. Mapping Argonaute and conventional RNA-binding protein interactions with RNA at single-nucleotide resolution using HITS-CLIP and CIMS analysis. Nat. Protoc. 2014; 9:263–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Clark P.M., Loher P., Quann K., Brody J., Londin E.R., Rigoutsos I.. Argonaute CLIP-Seq reveals miRNA targetome diversity across tissue types. Sci. Rep. 2014; 4:5947–5958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Corcoran D.L., Georgiev S., Mukherjee N., Gottwein E., Skalsky R.L., Keene J.D., Ohler U.. PARalyzer: definition of RNA binding sites from PAR-CLIP short-read sequence data. Genome Biol. 2011; 12:R79–R95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Erhard F., Dolken L., Jaskiewicz L., Zimmer R.. PARma: identification of microRNA target sites in AGO-PAR-CLIP data. Genome Biol. 2013; 14:R79–R98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Majoros W.H., Lekprasert P., Mukherjee N., Skalsky R.L., Corcoran D.L., Cullen B.R., Ohler U.. MicroRNA target site identification by integrating sequence and binding information. Nat. Methods. 2013; 10:630–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Karginov F.V., Hannon G.J.. Remodeling of Ago2-mRNA interactions upon cellular stress reflects miRNA complementarity and correlates with altered translation rates. Genes Dev. 2013; 27:1624–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Leung A.K., Young A.G., Bhutkar A., Zheng G.X., Bosson A.D., Nielsen C.B., Sharp P.A.. Genome-wide identification of Ago2 binding sites from mouse embryonic stem cells with and without mature microRNAs. Nat. Struct. Mol. Biol. 2011; 18:237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lipchina I., Elkabetz Y., Hafner M., Sheridan R., Mihailovic A., Tuschl T., Sander C., Studer L., Betel D.. Genome-wide identification of microRNA targets in human ES cells reveals a role for miR-302 in modulating BMP response. Genes Dev. 2011; 25:2173–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li J.H., Liu S., Zhou H., Qu L.H., Yang J.H.. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014; 42:D92–D97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Muller S., Rycak L., Winter P., Kahl G., Koch I., Rotter B.. omiRas: a web server for differential expression analysis of miRNAs derived from small RNA-Seq data. Bioinformatics. 2013; 29:2651–2652. [DOI] [PubMed] [Google Scholar]
- 24. Chen B., Yun J., Kim M.S., Mendell J.T., Xie Y.. PIPE-CLIP: a comprehensive online tool for CLIP-seq data analysis. Genome Biol. 2014; 15:R18–R28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Althammer S., Gonzalez-Vallinas J., Ballare C., Beato M., Eyras E.. Pyicos: a versatile toolkit for the analysis of high-throughput sequencing data. Bioinformatics. 2011; 27:3333–3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Uren P.J., Bahrami-Samani E., Burns S.C., Qiao M., Karginov F.V., Hodges E., Hannon G.J., Sanford J.R., Penalva L.O., Smith A.D.. Site identification in high-throughput RNA-protein interaction data. Bioinformatics. 2012; 28:3013–3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang T., Chen B., Kim M., Xie Y., Xiao G.. A model-based approach to identify binding sites in CLIP-Seq data. PLoS One. 2014; 9:e93248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang C., Darnell R.B.. Mapping in vivo protein-RNA interactions at single-nucleotide resolution from HITS-CLIP data. Nat. Biotechnol. 2011; 29:607–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jothi R., Cuddapah S., Barski A., Cui K., Zhao K.. Genome-wide identification of in vivo protein-DNA binding sites from ChIP-Seq data. Nucleic Acids Res. 2008; 36:5221–5231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yeo G.W., Coufal N.G., Liang T.Y., Peng G.E., Fu X.D., Gage F.H.. An RNA code for the FOX2 splicing regulator revealed by mapping RNA-protein interactions in stem cells. Nat. Struct. Mol. Biol. 2009; 16:130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Maragkakis M., Alexiou P., Nakaya T., Mourelatos Z.. CLIPSeqTools-a novel bioinformatics CLIP-seq analysis suite. RNA. 2015; 22:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Maticzka D., Lange S.J., Costa F., Backofen R.. GraphProt: modeling binding preferences of RNA-binding proteins. Genome Biol. 2014; 15:R17–R35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K.. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010; 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kozomara A., Griffiths-Jones S.. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014; 42:D68–D73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Agarwal V., Bell G.W., Nam J.W., Bartel D.P.. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015; 4:e05005–e05043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Moore M.J., Scheel T.K., Luna J.M., Park C.Y., Fak J.J., Nishiuchi E., Rice C.M., Darnell R.B.. miRNA-target chimeras reveal miRNA 3΄-end pairing as a major determinant of Argonaute target specificity. Nat. Commun. 2015; 6:8864–8881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Grant C.E., Bailey T.L., Noble W.S.. FIMO: scanning for occurrences of a given motif. Bioinformatics. 2011; 27:1017–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lorenz R., Bernhart S.H., Honer Zu Siederdissen C., Tafer H., Flamm C., Stadler P.F., Hofacker I.L.. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011; 6:26–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Licatalosi D.D., Yano M., Fak J.J., Mele A., Grabinski S.E., Zhang C., Darnell R.B.. Ptbp2 represses adult-specific splicing to regulate the generation of neuronal precursors in the embryonic brain. Genes Dev. 2012; 26:1626–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sei E., Wang T., Hunter O.V., Xie Y., Conrad N.K.. HITS-CLIP analysis uncovers a link between the Kaposi's sarcoma-associated herpesvirus ORF57 protein and host pre-mRNA metabolism. PLoS Pathog. 2015; 11:e1004652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Loeb G.B., Khan A.A., Canner D., Hiatt J.B., Shendure J., Darnell R.B., Leslie C.S., Rudensky A.Y.. Transcriptome-wide miR-155 binding map reveals widespread noncanonical microRNA targeting. Mol. Cell. 2012; 48:760–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Taliaferro J.M., Aspden J.L., Bradley T., Marwha D., Blanchette M., Rio D.C.. Two new and distinct roles for Drosophila Argonaute-2 in the nucleus: alternative pre-mRNA splicing and transcriptional repression. Genes Dev. 2013; 27:378–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tan G.S., Garchow B.G., Liu X., Yeung J., Morris J.P.t., Cuellar T.L., McManus M.T., Kiriakidou M.. Expanded RNA-binding activities of mammalian Argonaute 2. Nucleic Acids Res. 2009; 37:7533–7545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lewis B.P., Burge C.B., Bartel D.P.. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005; 120:15–20. [DOI] [PubMed] [Google Scholar]
- 45. Wilbanks E.G., Facciotti M.T.. Evaluation of algorithm performance in ChIP-seq peak detection. PLoS One. 2010; 5:e11471. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.