

Abstract
Despite being a simple dehydration reaction, the industrially relevant conversion of lactic acid to acrylic acid is particularly challenging. For the first time, the catalytic cracking of lactide and poly(lactic acid) to acrylic acid under mild conditions is reported with up to 58 % yield. This transformation is catalyzed by strong acids in the presence of bromide or chloride salts and proceeds through simple SN2 and elimination reactions.
Keywords: acrylic acid, biobased economy, lactic acid, renewable resources, sustainable chemistry
Environmental concerns and the impending scarcity of oil resources result in increasing attention for the concept of a biobased economy. To replace oil‐based processes, energyefficient and economically competitive processes using biobased feedstocks have to be developed. Lactic acid (LA) is already an important platform chemical obtained by fermentation. Stimulated by technological improvement and the diversification of fermentation feedstocks, and driven by the increasing demand for the biodegradable polymer poly(lactic acid) (PLA), its production capacity is expected to rise in the coming years, accompanied by a drop in price.1 Besides PLA, value‐added commodities derived from LA such as alkyl lactates (solvents) are already produced industrially. The cyclic diester lactide is produced industrially from oligo(lactic acid): it is used as a monomer for the production of PLA. LA can also be transformed into propylene glycol, pyruvic acid, acetaldehyde, 2,3‐pentanedione, or acrylic acid (AA).2 AA is an important feedstock for the production of various polymers applied in, for example, plastics and elastomers as well as paints.3 Petroleum‐based AA is relatively expensive owing to the relatively high cost of the propene feedstock, the energy‐demanding two‐step process, and purification.4 A feasible process for the conversion of LA to AA is highly desired; however, this simple dehydration reaction is surprisingly challenging.
The dehydration of alcohols is typically catalyzed by strong acids.5 Because LA bears a carboxylic acid function, the acid‐catalyzed dehydration of LA under mild and concentrated conditions does not lead to acrylic acid but instead results in linear oligomers and a small amount of lactide.6 Under harsher conditions, a range of products is formed, such as acetaldehyde, propionic acid, CO, and CO2.7 The use of dehydration catalysts based on rhenium or molybdenum complexes has been reported to lead to traces of AA together with polymers,8 whereas in the presence of a reductant mainly propionic acid is obtained.9
The best systems reported so far for the direct dehydration of LA to AA are based on heterogeneous phosphate or sulfate catalysts in the gas phase (Scheme 1 a).10 The highest reported yields are in the range of 80 %.11 The main drawbacks of these processes are their high temperature (300–400 °C), the relatively high dilution required to avoid the formation of non‐volatile oligomers of LA, and poisoning of the catalysts. Alternatively, LA can be converted to AA in supercritical water without the need of added catalysts. The polymerization of LA is suppressed in this system. However, the temperature and pressure required are very high (1000 bar) and AA yields are low (44 % selectivity at 23 % conversion).7b, 12 Another reported two‐step process relies on the use of acetic acid: 2‐acetoxypropionic acid is first produced by the esterification of LA and acetic acid catalyzed by strong acids,13 or by the reaction of lactide with acetic acid catalyzed by a nickel catalyst at 250 °C.4 Pyrolysis of 2‐acetoxypropionic should lead to AA and acetic acid (Scheme 1 b). However, recycling of the acetic acid reagent/coproduct is mandatory to achieve an economically viable route,4, 11, 13, 14 impeding the development of an industrial process.
Scheme 1.
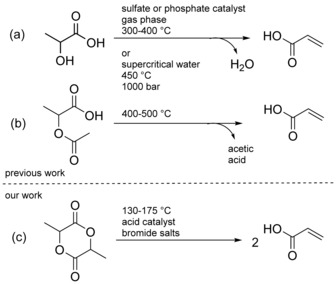
(a) Direct dehydration of lactic acid. (b) Pyrolysis of 2‐acetoxypropionic acid. (c) Catalytic cracking of lactide (our work).
Herein we report the direct formation of AA from lactide (Scheme 1 c): a yield of 58 % AA was obtained from lactide after 10 h at 175 °C. Importantly, using this procedure AA can be produced directly from PLA or oligo‐LA, offering a new route to the recycling of PLA yielding the high‐valued product AA (patent pending).
Our initial aim was to develop catalysts for the carbonylation of LA to succinic acid, for which we investigated the Lapidus system reported for the carbonylation of cyclohexanol.15 This catalytic system comprises a palladium source, a strong acid, and the bromide‐based ionic liquid tetrabutylammonium bromide (TBAB). By analogy with the described mechanism for related reactions,16 we expected the formation of 2‐bromopropionic acid (2BrPA) as the first step of the mechanism. The conversion of LA to 2BrPA in the presence of hydrobromic acid is well known.17 Unexpectedly, a control experiment at 130 °C in the absence of the palladium catalyst showed 24 % conversion of LA and the formation of AA in approximately 4 % selectivity with a large amount of LA oligomers and 2BrPA in 24 % selectivity, (Table S1, entry 1 in the Supporting Information). In the absence of strong acids, no conversion was observed, and if 1 equivalent of water was added the conversion dropped (Table S1, entries 2 and 3).
Because the water formed by the dehydration of LA appeared to be detrimental for the formation of AA, we decided to use lactide as the substrate in this reaction. The main products found in the reaction mixtures of our investigations are shown in Scheme 2.
Scheme 2.
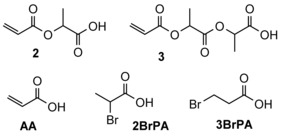
Species observed in the reaction mixtures.
When lactide was heated in TBAB in the presence of p‐toluene sulfonic acid (HOTs), AA was obtained in 2 % selectivity, and compound 2, the ester of AA and LA, in 14 % selectivity at 93 % conversion (Table 1, entry 1). Subsequently, we tried to optimize the reaction for the selective formation of AA. The effect of the quantity of HOTS on the outcome of the transformation was investigated (Table S2). Not surprisingly, an increasing amount of lactide was hydrolyzed to LA when an increasing amount of HOTs was used because the HOTs used for this study was the commercial monohydrate. Furthermore, 3‐bromopropionic acid (3BrPA), the addition product of HBr to AA, was also detected in the reaction mixture.
Table 1.
Rearrangement of lactide, oligo(lactic acid), and poly(lactic acid) forming acrylic acid (AA) and the ring‐opened intermediate 2.[a]
| Entry | Substrate (1 mmol) | Acid [mmol] | Bromide source [mmol] | T [°C] | Conv. [%] | AA [mmol] | 2 [mmol] |
|---|---|---|---|---|---|---|---|
| 1 | lactide | HOTS (0.5) | TBAB (3.1) | 130 | 93 | 0.04 | 0.13 |
| 2 | lactide | HOMs (0.5) | TBAB (3.1) | 130 | 63 | 0.06 | 0.27 |
| 3 | lactide | HOMs (2.4) | TBAB (15) | 130 | 93 | 0.10 | 0.42 |
| 4 | lactide | HOMs (0.83) | TBAB (5) | 150 | >99 | 0.32 | 0.44 |
| 5 | lactide | HOMs (0.83) | PPh4Br[b] (5) | 150 | >99 | 0.64 | 0.44 |
| 6 | lactide | 2BrPA (0.5) | PPh4Br[b] (5) | 150 | >99 | 0.62 | 0.35 |
| 7 | lactide | 3BrPA (0.5) | PPh4Br[b] (5) | 150 | >99 | 0.70 | 0.35 |
| 8 | oligo‐LA[c] | HOMs (0.83) | PPh4Br[b] (5) | 150 | >99 | 0.46 | 0.26 |
| 9 | PLA[c] | HOMs (0.83) | PPh4Br[b] (5) | 150 | >99 | 0.48 | 0.26 |
[a] Full analytical details and mass balance are given in the Supporting Information. For ease of comparison, the quantities of material in this table have been scaled to the use of 1 mmol lactide. Reaction conditions: 130 °C, 16 h. [b] Sulfolane was used as a solvent (1 g sulfolane per g PPh4Br). [c] Amount of PLA and oligo‐LA corresponding to 1 mmol lactide.
To completely eliminate the adverse effect of water in subsequent reactions, anhydrous methanesulfonic acid was used (HOMs, Table S3). Under these water‐free conditions no LA was formed. The selectivity towards the unsaturated compounds AA and 2 was highest at 0.5 equivalents HOMs relative to lactide (entry 2 in Table 1 and entry 3 in table S3: 5 % selectivity towards AA and 43 % towards 2 at 63 % conversion). The use of larger amounts of HOMs resulted in an increase in conversion but did not increase the selectivity towards AA and 2. An increase in the bromide/lactide ratio resulted in increased conversion up to 93 % (when 15 equiv TBAB was used) but had little or no effect on the selectivity: 40–45 % 2 and 5 % AA were formed for Br/lactide ratios from 3 to 15 (Table S4 and Table 1, entry 3).
We then investigated whether TBAB might be replaced by other sources of bromide ions; the use of tetraphenylphosphonium bromide (PPh4Br), 1‐ethyl‐3‐methylimidazolium bromide, 1‐butylpyridinium bromide, or tetrabutylphosphonium bromide as bromide sources afforded similar or lower selectivities and conversions (Table S5). Contrary to the other bromide sources, PPh4Br is not an ionic liquid under the reaction conditions (melting point: 295 °C), and in the reactions with this salt, sulfolane was added as a solvent. In the sulfolane/PPh4Br mixture the use of other (anhydrous) strong acids such as trifluoromethanesulfonic acid or meta‐phosphoric acid afforded similar results as with HOMs. Weak acids such as acetic acid or oxalic acid were inefficient because neither bromo‐substituted nor unsaturated species were formed (Table S6).
Higher selectivity towards AA and 2 was obtained when the reaction temperature was raised to 150 or 175 °C with TBAB or PPh4Br as bromide sources (Table S7 and Table 1, entries 4 and 5). Unfortunately, when the reaction was run at 175 °C for 16 h, the mass balance decreased to 60 % (for both bromide sources), and when the reaction was run at 200 °C, the mass balance dropped to 30 % in TBAB or a mere 2 % in PPh4Br. A tentative explanation for the loss of mass balance may be the formation of polyacrylates under these harsh conditions. Furthermore, TBAB partially decomposes at these higher temperatures, as indicated by the observation of butyl bromide in the reaction mixture. The PPh4Br/sulfolane system is more suitable for the use under harsher reaction conditions because of the higher thermal stability of these species.
The rearrangement of lactide was monitored over 10 h at 175 °C in the HOMs/PPh4Br/sulfolane reaction medium (Figure 1 and Table S8). Under these conditions, lactide quickly disappeared from the reaction mixture: 97 % was consumed within 2 h. The selectivity towards 2 reached a maximum after 2 h (50 % selectivity at 94 % conversion) and then decreased, whereas the selectivity towards 2BrPA increased to 14 % in the first hour and then slowly decreased to reach 0 % after 10 h. The amount of 3BrPA reached a maximum at 4 h with 7 % selectivity and then remained stable until the end. The selectivity towards AA increased continuously over the 10 h reaction time to reach 58 %. In the first few hours of the reaction, the overall mass balance was initially quite good, but after 6 h it decreased significantly.
Figure 1.
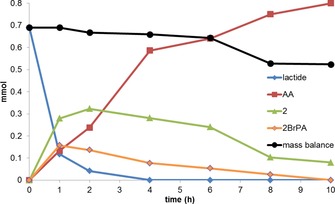
Rearrangement of lactide to acrylic acid at 175 °C as a function of time. Reaction conditions: lactide (0.69 mmol), HOMs (0.83 equiv), PPh4Br (5 equiv), sulfolane. Lines are only given as a guide to the eye.
The chloride salt PPh4Cl can also be used for the reaction, although the selectivity towards rearrangement products is lower than when PPh4Br is used. Whereas AA and 2 were obtained in 32 and 44 % selectivity at full conversion in PPh4Br, under otherwise identical conditions AA and 2 were obtained in 11 and 29 % selectivity at 86 % conversion in PPh4Cl (see Table S9). A significant amount of 2‐chloropropionic acid (2ClPA) was formed in the reaction (26 % selectivity), whereas 2BrPA was formed with only 7 % selectivity under otherwise identical conditions.
We hypothesized that bromopropionic acid itself can also be used as a source of acid because it can eliminate HBr to afford AA.18 The rearrangement of lactide also occurred in the absence of a strong acid but with 2BrPA added at the beginning of the reaction (Table S10, entries 1–3): when lactide (0.69 mmol) and 2BrPA (0.35 mmol) were stirred in BrPPh4/ sulfolane for 16 h at 150 °C, the resulting reaction mixture was found to contain AA (0.54 mmol), 2 (0.31 mmol), and 2BrPA (0.10 mmol). The compound 3BrPA could also be used (Table S10, entries 4 and 5): a reaction of lactide (0.69 mmol) and 3BrPA (0.35 mmol) in BrPPh4/sulfolane for 16 h at 150 °C resulted in a mixture containing AA (0.62 mmol), 2 (0.31 mmol), and 3BrPA (0.18 mmol).
Finally, oligo‐LA and PLA were submitted to the PPh4Br/ sulfolane/HOMs reaction medium at 150 °C for 16 h. Starting from oligo‐LA (100 mg), AA (23 mg) and 2 (26 mg) were obtained. Additional signals in the acrylate region of the NMR spectrum were attributed to the tri‐ester 3 (comprising two LA units and one acrylate unit, see Scheme 2 and Figure S2), which was obtained in 11 mg yield (Table S11, entry 1). When PLA (100 mg) was used as a starting material, AA (24 mg), 2 (26 mg) and 3 (16 mg) were obtained (Table 1, entry 9; Table S11, entry 2). Besides pure commercial PLA, to show that this transformation can be applied for the recycling of PLA, we also used a piece of plastic cutlery (consisting of 67 % PLA and 33 % inert filler) from the university canteen. The results were virtually identical to those obtained when bulk PLA was used (Table S11, entry 3).
Mechanistic considerations
The various intermediates found in the reaction mixtures helped us to devise a mechanism for the transformation of lactide to AA, as shown in Scheme 3. The cyclic lactide ester is first opened by a proton and a bromide ion to give 2‐(2’‐bromopropanoyloxy)propanoic acid (1) (first nucleophilic substitution). Compound 1 then either eliminates HBr to afford 2‐(acryloyloxy)propanoic acid (2) (first elimination) or may react with a second equivalent of HBr to yield two equivalents of 2BrPA. Compound 2 can also be hydrobrominated to afford AA and 2BrPA (second nucleophilic substitution). Finally, 2BrPA eliminates HBr to yield AA (second elimination reaction). The presence of small amounts of 3BrPA in the reaction mixture could be the result of the reversible addition of HBr to AA.
Scheme 3.
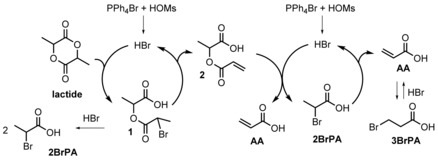
Proposed mechanism for the catalytic cracking of lactide by strong acid and bromide salts.
At high temperatures, secondary alkyl halides can be transformed to alkenes through the E1 mechanism involving a carbo‐cationic intermediate. However, in the case of 2BrPA, the involvement of such a carbo‐cationic intermediate is not likely owing to its α‐position with respect to the carboxylic acid group. A strict E2‐type mechanism for this elimination step is also not very probable owing to the extremely acidic reaction medium. An E2C mechanism with bromide ions acting as a weak base is more likely to be operative.19 Alternatively, an intramolecular carboxylic acid‐assisted E1‐type elimination pathway involving a hypothetical oxiranone intermediate has also been proposed.20
The transformation of one equivalent of lactide to two equivalents of AA formally is not a dehydration reaction but rather cracking catalyzed by protons and bromide ions. Molecular HBr is not expected to be present as such in the reaction mixtures: it is likely to be fully dissociated because of its low pK a (−9) compared to the pK a of HOTs or even of protonated acids or esters (pK a in the range of −3 to −2).
Compound 1 remains a hypothetical intermediate because it has not been observed. It is thought to quickly eliminate HBr to afford 2. The reaction of 1 with HBr to yield two equivalents of 2BrPA cannot be ruled out and could contribute to the low concentration of 1. It has been observed that the concentration of 2BrPA and 2 in the reaction mixture increases in the beginning of the reaction and then decreases (Figure 1), which supports their role as reaction intermediates for the conversion of LA to AA. The role of 2BrPA as an intermediate and a source of HBr was further confirmed by an experiment in which no strong acid was added but in which 2BrPA was added at the start of the reaction. Similarly, 3BrPA can also be used as a source of HBr, indicating that the addition of HBr to AA is reversible under our reaction conditions.
Increasing the concentration of sulfonic acid (HOTs⋅H2O/lactide >0.5 or MsOH/lactide >1) in the reaction mixture led to an increase in the formation of 2BrPA but a decrease in the formation of 2 and AA. These observations suggest that the elimination reactions are hindered by high concentrations of protons, which hamper the ability of bromide ions to act as weak bases.
Compared to the bromide ion, chloride is a better nucleophile in aprotic environments (but not so good in protic environments); it is less acidic and an inferior leaving group. As a result, a significant amount of 2‐chloropropionic acid (2ClPA) was formed in the reaction (23 %) with a chloride salt, whereas 6 % 2BrPA was formed under otherwise identical conditions; clearly, the elimination of HCl from 2ClPA is more difficult than the elimination of HBr from 2BrPA. This observation corroborates the proposed mechanism.
To conclude, we disclosed a promising new strategy for the conversion of lactide, oligo(lactic acid) or poly(lactic acid) to acrylic acid (AA). This transformation is catalyzed by strong acids in the presence of bromide salts. Preliminary mechanistic investigations suggest that 2‐(2’‐bromopropanoyloxy)propanoic acid (1), 2‐(acryloyloxy)propanoic acid (2), and 2‐bromopropionic acid are intermediates in the conversion of lactide to AA and that simple SN2 and elimination mechanisms are involved. Selectivities to acrylates up to 64 % were obtained (up to 58 % to AA). Attempts to obtain higher selectivities resulted in lower mass balances, probably owing to polymerization of the acrylates. To circumvent this limitation, continuous removal of AA as it forms seems to be a promising area to be explored because AA has the lowest boiling point of all species involved in this reaction. In that respect, it is foreseen that the catalytic system (bromide salts, acid, and solvent) could be reused after separation of AA by continuous distillation. This new process is strictly different from previously described processes because it is formally a rearrangement and not a dehydration. Contrary to other processes, the reaction temperature is much milder (130–175 °C vs. 300–500 °C), and even oligomers or polymers of lactic acid can be used as substrates. The relatively high cost of the lactide starting material could be a major drawback for the industrial application of our reaction. Thus, a process based on the use of oligomers of lactic acid or mixtures of oligomers and racemized lactide as starting materials will have a higher economic viability. Moreover, poly(lactic acid) is an interesting substrate for recycling to the value‐added product AA.
Experimental Section
Representative experimental procedure (corresponding to entry 5 in Table 1): (S,S)‐lactide (100 mg, 0.69 mmol), methanesulfonic acid (37.6 μL, 0.58 mmol, 0.83 equiv), tetraphenylphosphonium bromide (1.45 g, 3.47 mmol, 5 equiv), and sulfolane (1.5 mL) were introduced in the glass inset of an autoclave equipped with a stirring bar. The autoclave was closed, pressurized with 50 bar N2, and then heated to 150 °C (temperature of the heating mantle) for 16 h under magnetic stirring (400 rpm). Then, the autoclave was placed in an ice bath for 30 min before being vented and opened. 1H NMR spectroscopy of the crude reaction mixture in deuterated DMSO, using the signal of the sulfonic acid as an internal standard, showed the complete consumption of lactide and the formation of acrylic acid (0.44 mmol, 32 % selectivity), 2 (0.31 mmol 44 % selectivity), 2‐bromopropionic acid (0.08 mmol, 6 % selectivity), and 3‐bromopropionic acid (0.09 mmol, 7 % selectivity).
To avoid gaseous HBr leaving the reaction mixture the initial reactions were performed at 50 bar N2 pressure. However, reactions performed at autogenic pressure in closed autoclaves, or at normal pressure in Schlenk glassware led to identical results.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was financially supported by Corbion. Dr. K. van der Voort Maarschalk and Dr. J. Canadell‐Ayats are acknowledged for fruitful discussions. J. A. P. P. van Dijk and J. J. M. van Brussel are thanked for their assistance with the analytical procedures.
F. G. Terrade, J. van Krieken, B. J. V. Verkuijl, E. Bouwman, ChemSusChem 2017, 10, 1904.
References
- 1.Corbion, 2016, http://www.corbion.com/media/press-releases?newsId=2057215.
- 2.
- 2a. Dusselier M., Van Wouwe P., Dewaele A., Makshina E., Sels B. F., Energy Environ. Sci. 2013, 6, 1415–1442; [Google Scholar]
- 2b. Mäki-Arvela P., Simakova I. L., Salmi T., Murzin D. Y., Chem. Rev. 2014, 114, 1909–1971; [DOI] [PubMed] [Google Scholar]
- 2c. Van Wouwe P., Dusselier M., Vanleeuw E., Sels B., ChemSusChem 2016, 9, 907–921. [DOI] [PubMed] [Google Scholar]
- 3. Ohara T., Sato T., Shimizu N., Prescher G., Schwind H., Weiberg O., Marten K., Greim H., Acrylic Acid and Derivatives in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2011. [Google Scholar]
- 4. Fruchey O. S., Malisezewski T. A., Sawyer J. E. (Sga Polymers, Llc, Charleston, WV), WO2013036389 A1, 2013.
- 5. Taber R. L., Champion W. C., J. Chem. Educ. 1967, 44, 620. [Google Scholar]
- 6.
- 6a. Holten C. H., Müller A., Rehbinder D., Lactic acid. properties and chemistry of lactic acid and derivates, VCH, Weinheim, 1971; [Google Scholar]
- 6b. Vu D. T., Kolah A. K., Asthana N. S., Peereboom L., Lira C. T., Miller D. J., Fluid Phase Equilib. 2005, 236, 125–135. [Google Scholar]
- 7.
- 7a. Chuchani G., Martin I., Rotinov A., Dominguez R. M., J. Phys. Org. Chem. 1993, 6, 54–58; [Google Scholar]
- 7b. Mok W. S. L., Antal M. J., Jones M., J. Org. Chem. 1989, 54, 4596–4602. [Google Scholar]
- 8.
- 8a. Raju S., Moret M.-E., Gebbink R. J. M. K., ACS Catal. 2015, 5, 281–300; [Google Scholar]
- 8b. Dowson G. R. M., Shishkov I. V., Wass D. F., Organometallics 2010, 29, 4001–4003. [Google Scholar]
- 9. Korstanje T. J., Kleijn H., Jastrzebski J. T. B. H., Gebbink R. J. M. K., Green Chem. 2013, 15, 982–988. [Google Scholar]
- 10.
- 10a. Ghantani V. C., Dongare M. K., Umbarkar S. B., RSC Adv. 2014, 4, 33319–33326; [Google Scholar]
- 10b. Hong J. H., Lee J. M., Kim H., Hwang Y. K., Chang J. S., Halligudi S. B., Han Y. H., Appl. Catal. A 2011, 396, 194–200; [Google Scholar]
- 10c. Sun P., Yu D. H., Tang Z. C., Li H., Huang H., Ind. Eng. Chem. Res. 2010, 49, 9082–9087; [Google Scholar]
- 10d. Zhang J. F., Zhao Y. L., Pan M., Feng X. Z., Ji W. J., Au C. T., ACS Catal. 2011, 1, 32–41; [Google Scholar]
- 10e. Lilga M. A., Werpy T. A., Holladay J. E., US2004/0110974 A1, 2004;
- 10f. Werpy T. A., Lilga M. A. (Battelle Memorial Institute, Columbus, OH), WO2002032849 A2, 2002.
- 11. Godlewski J. E., Villalobos J., Collias D. I., Velasquez J. E. (The Procter & Gamble Company, Cincinnati, OH), US20130274513 A1, 2013.
- 12. Aida T. M., Ikarashi A., Saito Y., Watanabe M., R. L. Smith, Jr. , Arai K., J. Supercrit. Fluids 2009, 50, 257–264. [Google Scholar]
- 13. Beerthuis R., Granollers M., Brown D. R., Salavagione H. J., Rothenberg G., Shiju N. R., RSC Adv. 2015, 5, 4103–4108. [Google Scholar]
- 14.
- 14a. Burns R., Jones D. T., Ritchie P. D., J. Chem. Soc. 1935, 400–406; [Google Scholar]
- 14b. Fisher C. H., Ratchford W. P., Smith L. T., Ind. Eng. Chem. 1944, 36, 229–234; [Google Scholar]
- 14c. Fruchey O. S., Malisezewski T. A., Sawyer J. E., US9012686 B2, 2015.
- 15.
- 15a. Eliseev O. L., Bondarenko T. N., Stepin N. N., Lapidus A. L., Mendeleev Commun. 2006, 16, 107–109; [Google Scholar]
- 15b. Lapidus A. L., Eliseev O. L., Solid Fuel Chem. 2010, 44, 197–202. [Google Scholar]
- 16.
- 16a. Seayad A., Jayasree S., Chaudhari R. V., J. Mol. Catal. A 2001, 172, 151–164; [Google Scholar]
- 16b. Seayad A., Jayasree S., Chaudhari R. V., Catal. Lett. 1999, 61, 99–103; [Google Scholar]
- 16c. Jang E. J., Lee K. H., Lee J. S., Kim Y. G., J. Mol. Catal. A 1999, 138, 25–36; [Google Scholar]
- 16d. Jones J. H., Platinum Metals Rev. 2000, 44, 94–105; [Google Scholar]
- 16e. Forster D., Dekleva T. W., J. Chem. Educ. 1986, 63, 204. [Google Scholar]
- 17. Kekulé A., Justus Liebigs Ann. Chem. 1864, 130, 11–31. [Google Scholar]
- 18. Kowski E., Justus Liebigs Ann. Chem. 1905, 342, 124–138. [Google Scholar]
- 19. March J., Advanced Organic Chemistry 4th ed., John Wiley, Chichester, 1992. [Google Scholar]
- 20. Rodriquez C. F., Williams I. H., J. Chem. Soc. Perkin Trans. 2 1997, 953–957. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary