
Table 4.
Select examples of CXCR1/2 inhibitors and their biological properties
| CXCR1/2 Inhibitors | Activities of CXCR1/2 inhibitors |
|---|---|
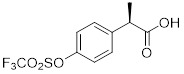
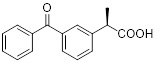 (R)-Ketoprofen |
R)-Ketoprofen inhibited CXCL8-mediated PMN migration (IC50 = 34 nM) and interacts with the TM2 and TM7 region of CXCR1 [398]. |
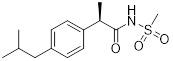 Repertaxin |
Repertaxin inhibited human PMN migration induced by CXCL8 (IC50 = 1 nM) and CXCL1 (IC50 = 400 nM) [392]. Repertaxin inhibited CXCL8-induced migration in CXCR1-transfected (IC50 = 1 nM) and CXCR2-transfected (IC50 = 100 nM) cells [392]. Repertaxin reduced lung neutrophil recruitment and vascular permeability by 50% in LPS-induced acute lung injury model at 15mg/kg [399]. Repertaxin reduced acute inflammation and autonomic dysreflexia in a model of spinal cord injury in rats [400]. Repertaxin inhibited CXCL8-induced PMN adhesion to fibrinogen, CD11b upregulation, and neutrophil activation (granule release) [394]. Reparixin in combination with paclitaxel inhibited brain tumour metastasis due to CSC [293]. Repertaxin inhibited CXCL8-induced T-cell and NK cell migration [394]. Repertaxin reduced granulocyte graft infiltration and serum creatinine post syngeneic transplantation in Lewis rats [401]. Repertaxin inhibited neutrophil recruitment into reperfused livers and reduced myeloperoxidase content in a rat model of liver post-ischaemia reperfusion injury [392]. Repertaxin reduced levels of hypertension-related mediators associated with reduced blood pressure in spontaneous hypertensive rat models [402]. Repertaxin reduced oligodendrocyte apoptosis and neutrophil migration to site of injury post traumatic spinal cord injury in rats [403]. Repertaxin inhibited CXCL8- or CINC-1-induced migration and calcium flux in human or rat neutrophils [395]. Repertaxin reduced neutrophil influx and vascular permeability in a model of mild and severe ischemia/reperfusion (I/R) injury in rats [395]. |
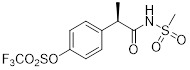 DF2156A |
DF2156A is a noncompetitive allosteric inhibitor that is predicted to be stabilized by a direct ionic bond interaction with Lys99 on CXCR1 and Asp293 on CXCR2 [396]. DF2156A inhibited chemotaxis in CXCR1 and CXCR2 over-expressing transfectants and leukocytes [396]. DF2156A inhibited mice sponge-induced angiogenesis by reducing leukocyte influx and vessel formation [396]. DF2156A inhibited CXCL8-mediated HUVEC proliferation, migration and tube formation [396]. DF2156A decreased PMN influx in a rat model of ischemia and reperfusion injury [396]. DF2156A is a dual inhibitor of CXCR1 and CXCR2. It inhibited human and rat neutrophil migration in response to CXCL1 and CXCL8 [404]. DF2156A has improved in vivo half-life compared to repertaxin [404]. DF2156A decreased cerebral artery PMN infiltrate and improved neurological function in cerebral ischemia/reperfusion rat model [404]. |
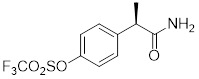 DF2162 |
DF2162 prevented chemotaxis of rat and human neutrophils induced by chemokines acting on CXCR1/2. It is orally bioavailable and inhibited neutrophil influx and production of inflammatory factors in an arthritis rat model [405]. DF2162 reduced neutrophil accumulation in airway, but increased neutrophils in lung parenchyma in a bleomycin-induced pulmonary fibrosis mouse model [406]. |
 Analogue 1  Analogue 2  Analogue 3 |
Analogues 1-3 inhibited CXCL1- and CXCL8-mediated human PMN migration with IC50 < 10 nM [393]. |
 DF2755A |
Allosteric inhibitor, DF2755A, selectively inhibited CXCL8-induced chemotaxis without affecting ligand binding to neutrophils and also reduced inflammatory and post-operative pain in several mouse models. It inhibited both CXCR1 (IC50 = 4.2 nM) and CXCR2 (IC50 = 2.1 nM) [407]. |
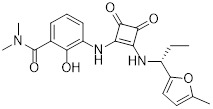 SCH527123 |
SCH527123 inhibited CXCL8 binding to CXCR1 (IC50=36 nM) and CXCR2 (IC50=2.6 nM) and inhibited neutrophil chemotaxis to CXCL8 (IC50=16 nM) and CXCL1 (IC50<1 nM) [408]. Optimization to improve potency of SCH527123 was performed [409-412]. SCH527123 bound to CXCR2 receptors of mice (Kd=0.2 nM), rat (Kd=0.02 nM), and monkey (Kd=0.08 nM) [388]. SCH527123 exhibited less affinity for monkey CXCR1 (5-fold decreased) and was >100 fold less potent in CXCR1-mediated chemotaxis [388]. SCH527123 blocked LPS induced pulmonary neutrophils (ED50=1.2-1.8 mg/kg) in mice and rat [388]. SCH527123 bound to CXCR1 (Kd=3.9 nM) and CXCR2 (Kd=0.049 nM) in CXCR1/2-overexpressing cell lines and inhibited CXCL1 and CXCL8-mediated neutrophil chemotaxis and myeloperoxidase release [389]. SCH527123 reduced sputum neutrophils in patients with severe asthma but had no effect on FEV1, sputum myeloperoxidase, CXCL8 or elastase [390]. SCH527123 reduced ozone induced sputum neutrophil in healthy volunteers. Treatment was safe and well-tolerated (4 day, 50 mg once daily dose) [243]. SCH527123 reduced LPS-induced sputum neutrophil influx (79% inhibition) compared to healthy volunteers [413]. SCH527123 and SCH479833 inhibited colon cancer metastasis, reduced tumor neovascularization, and increased tumor cell apoptosis in mice model [248]. |
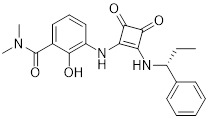 SCH479833 SCH479833 |
SCH527123/SCH479833 inhibited melanoma cell proliferation, chemotaxis, and invasivon in vitro and reduced tumor growth associated with decreased tumor cell proliferation and microvessel density in an in vivo mouse model of melanoma [317]. SCH527123 was well tolerated with neutropenia without severe myelosuppresive effects in phase 1 clinical trial [414]. SCH527123 at 50 mg significantly improved FEV1 in patients with COPD in phase 2 clinical trial although at higher doses led to discontinuation due to decrease in absolute neutrophil count in serum [415]. |
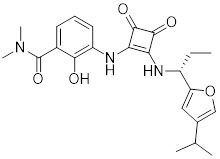 SCH563705 |
SCH563705 exhibited potent inhibitory activities against CXCL8 binding to CXCR2 (Ki = 1 nM) and CXCR1 (Ki = 3 nM) [416]. SCH563705 inhibited CXCL1 and CXCL8 induced human PMN migration (CXCR2: IC50=0.5 nM, CXCR1: IC50=37 nM) [416]. SCH563705 reduced inflammation and bone and cartilage degradation in a mouse model of anti-collagen antibody-induced arthritis [417]. |
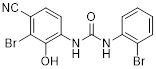 N-(3-bromo-4-cyano-2-hydroxyphenyl)-N-(2-bromophenyl)urea |
N-(3-bromo-4-cyano-2-hydroxyphenyl)-N-(2-bromophenyl)urea is a competitive CXCR2 inhibitor. It inhibited human PMN chemotaxis mediated by CXCL1 (IC50 = 14 nM) and CXCL8 (IC50 = 35 nM) and inhibited CXCL8-mediated neutroprenia in rabbit models [418]. |
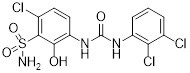 SB332235 |
SB332235 inhibited human CXCL8 binding to rabbit CXCR2 (IC50=40.5 nM) and CXCL8-induced calcium mobilization (IC50 = 7.7 nM). It had lower affinity for CXCR1 (IC50 > 1000 nM) and was less active against CXCR1-mediated calcium mobilization (IC50 = 2200 nM). SB332235 inhibited human CXCL8-induced chemotaxis of rabbit neutrophils (IC50 = 0.75 nM) [26]. SB332235 was optimized to improve pharmacokinetics [419]. SB332235 blocked T-cell entry when rat hippocampus was injected with amyloid β [420]. |
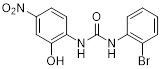 SB225002 |
SB225002 inhibited CXCL8 binding to CXCR2 (IC50 = 22 nM) and binding to CXCR1 with IC50 > 150 fold higher than CXCR2. SB225002 inhibited CXCL1 and CXCL8-induced chemotaxis in rabbit and human neutrophils [381]. SB225002 reduced alveolar neutrophil and exudate macropage influx in mice infected with S. pneumoniae [421]. SB225002 inhibited CXCL8 binding to CXCR2 (IC50 = 9.9 nM) and CXCL1 binding to CXCR2 (IC50 = 87.9 nM) [89]. In a mouse model of hepatic ischemia and reperfusion, post treatment with SB225002 increased hepatocyte proliferation and regeneration, similar to CXCR2-/- mice [422]. Optimization and other analogues of SB225002 were performed and reported [423]. SB225002 exhibited antinociceptive effects in several mouse models of pain (spontaneous nociception) [424]. SB225002 enhanced the activities of agonists for the δ opioid receptor acting in an allosteric fashion [425]. SB225002 reduced tumor progression in a mouse model of pancreatic cancer associated with reduced angiogenesis and improved survival [373]. |
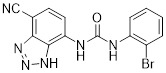 SB265610 |
SB265610 reduced superoxide accumulation and lipid peroxidation in lungs, and preserved alveolar development in hypoxic newborn rats (exposed to 60% oxygen) [426]. SB265610 inhibited BAL neutrophil influx and myeloperoxidase accumulation in the lungs in hypoxia-induced newborn rat lung injury model [427]. SB265610 inhibited rat neutrophil calcium mobilization (IC50=3.7 nM) and chemotaxis (IC50=70 nM) to CINC-1 [427]. SB265610 acts as allosteric, inverse agonist. SB265610 reduced maximal [125I]-CXCL8 binding without affecting its Kd. It also reduced agonist-induced (CXCL1 and CXCL8) CXCR2 activation and basal [35S]-GTPγS binding [428]. |
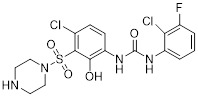 SB656933 |
SB656933 reduced LPS-induced sputum neutrophil influx (52% inhibition) in healthy volunteers [413]. SB656933 inhibited neutrophil CD11b upregulation (IC50=261 nM) and shape change (associated with chemotaxis, IC50=311 nM) in COPD patients [429]. SB656933 inhibited CXCL1-induced CD11b expression on peripheral blood neutrophils (70% inhibition) and reduced sputum neutrophils which correlated with reduced myeloperoxidase concentrations in ozone-induced airway inflammation in healthy patients. SB656933 was safe and well-tolerated at single doses as high as 1100 mg [385]. SB656933 was well-tolerated at 50 mg in cystic fibrosis patients treated for 28 days, and patients had reduced sputum neutrophils (30% reduction) and elastase (26% reduction) compared to baseline. Patients had increased blood levels of fibrinogen, C-reactive protein (CRP), and CXCL8 compared to placebo. No changes in lung function were observed (NCT00903201) [386]. |
| SB455821 (undisclosed structure) | SB455821 inhibited MIP-2-induced neutrophil migration in in vitro (IC50~20 nM) and in in vivo mice models [430]. |
| SB-517785-M (undisclosed structure) | SB-517785-M reduced angiotensin II-induced neutrophils and mononuclear cell recruitment, arteriolar mononuclear leukocyte adhesion, and levels of MIP-1 and RANTES [431]. |
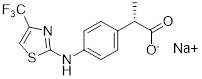
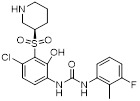 Danirixin (GSK1325756) |
Danirixin, a selective CXCR2 antagonist inhibited CXCL1-induced neutrophil activation (CD11b expression) in healthy adults after repeated daily doses in a dose depedent manner [432]. |
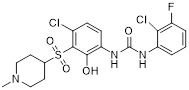 Compound 22 |
A CNS penetrating analog of SB656933, compound 22 inhibited CXCR2 in a Tango assay with IC50 less than 1 nM and showed efficacy in treatment of CNS demyelinating disorders [194]. |
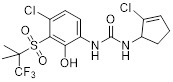 1-Phenyl-3-(cyclopent-2-en-1-yl) urea derivative 2 |
1-Phenyl-3-(cyclopent-2-en-1-yl) urea derivative 2 inhibited CXCR2 in a Tango assay with pIC50 ≥ 9.0 [433]. |
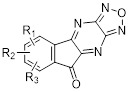 Diazafluorenones |
Diazaflurenones inhibited CXCL8 binding to isolated human neutrophils with IC50 values from 0.05 to 12 μM [434]. |
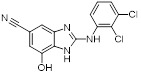 Benzoimidazole analog 2 Benzoimidazole analog 2 |
Benzoimidazole analog 2 inhibited CXCL8 binding to recombinant CXCR2 receptor expressed in CHO-K1 cells with IC50 value of 0.322 μM [435]. |
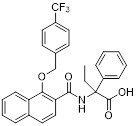 Napthalenecarboxamide 10 |
Napthalenecarboxamide 10 inhibited CXCR2 in calcium fluorescence assay (FLIPR) with an IC50 value of 2.2 μM [436]. |
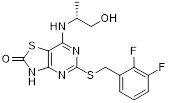 AZD8309 |
AZD8309 reduced leukocyte count (48% inhibition) post nasal LPS challenge in healthy volunteers. No adverse effects were detected after 3 days of dosing [437]. |
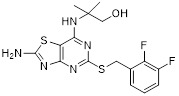 Thiazolopyrimidine 29 Thiazolopyrimidine 29 |
Thiazolopyrimidine 29 inhibited CXCL8 binding to CXCR2 (IC50=14 nM) and calcium mobilization (IC50=40 nM) [438]. |
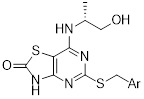 thiazolo[4,5-d]pyrimidine-2(3H)-ones |
Thiazolo[4,5-d]pyrimidine-2(3H)-ones inhibited CXCL8 binding to CXCR2 (IC50=1-60 nM). They showed improved potency and oral bioavailability over thiazolopyrimidines [439]. |
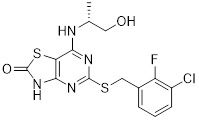 AZ10397767 |
AZ10397767 increased cytotoxicity of geldanamycin and 17-AAG (HSP90 inhibitors) in PC3 but not DU145 prostate cancer cells [253]. AZ10397767 reduced neutrophil infiltration into A549 (NSCLC) spheroids and A549 tumor xenograft models in mice. Treatment did not reduce microvascular density [275]. Optimization studies to improve potency of Thiazolopyrimidine analogs were performed and reported [440] |
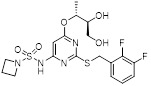 AZD5069 |
AZD5069 inhibited radio labeled CXCL8 binding to human CXCR2 with IC50 = 0.8 nM [441]. Recent clinical data showed that AZD5069 did not adversely affect neutrophil mobilization from bone marrow to peripheral circulation, and thus, did not interfere with normal function of neutrophils in the phagocytosis or the oxidative burst response to bacterial pathogens [442]. AZD5069 significantly reduced sputum neutrophil counts, but failed to improve clinical outcomes in bronchiectasis patients [443]. AZD5069 was well tolerated with no increase in infection rates in phase 2 clinical trial in patients with COPD [444]. |
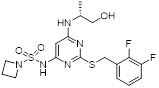 Compound 43 |
Compound 43 inhibited CXCR2 in low picomolar range (pIC50 , 8.4) with lower intrinsic renal clearance and good half life (t1/2, 3.2 h) in mice [445]. |
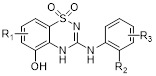 3-Arylamino-2H-1,2,4-benzothiadiazin-5-ol 1,1-dioxides |
3-Arylamino-2H-1,2,4-benzothiadiazin-5-ol 1,1-dioxides inhibited CXCL8 binding to CXCR2 (IC50 = 30 nM) and CXCR1 (IC50 = 3.2µM), and inhibited FLLPR CXCR2 calcium assay (IC50 ~ 300-600 nM) [446]. |
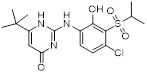 2-Aminopyrimidin-4(1H)-one analog 3e |
2-Aminopyrimidin-4(1H)-one analog 3e, a bioisostere of urea, inhibited CXCR2 (CXCR2-β-arrestin pIC50 = 8.2) [447]. |
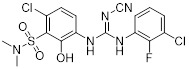 Compound 6i |
Compound 6i inhibited CXCL8 binding to CXCR1 (IC50 = 6.2 µM) and CXCR2 (IC50 = 30 nM) [448]. |
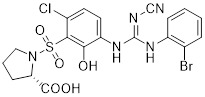 Compound 6j |
Compound 6j was a dual CXCR1 and CXCR2 antagonist and inhibited CXCL8 binding to CXCR1 and CXCR2 with similar IC50 values (~20 nM) [448]. |
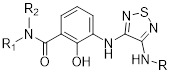 3,4-Diamino-1,2,5-thiadiazoles |
3,4-Diamino-1,2,5-thiadiazoles inhibited CXCL8 binding to CXCR2 (IC50 = 13-126 nM) and CXCR1 (IC50 = 44 nM-10µM) [449, 450]. 3,4-Diamino-1,2,5-thiadiazoles were selective CXCR2 antagonists [449, 450]. |
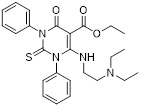 6-amino-4-oxo-1,3-diphenyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbonyl derivative 17 |
6-amino-4-oxo-1,3-diphenyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbonyl derivative 17 inhibited CXCL8-induced PMN migration (IC50 = 0.02 nM) [451]. |
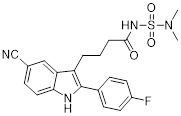 Acylsulfamide 6 |
Carboxylic acid bioisostere acylsulfamide 6 inhibited CXCL8 binding to CXCR2 (IC50 = 50 nM) and CXCL8-induced calcium mobilization (IC50 = 5 nM) [452]. Acylsulfamide 6 inhibited rabbit neutrophil chemotaxis (IC50 = 700 nM) [452]. Acylsulfamide 6 inhibited hyperoxia induced neutrophil (BAL) accumulation (~50% inhibition) in newborn rats at a dose of 10 mg/kg [452]. |
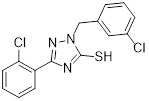 Triazolethiol 45 |
Triazolethiol 45 inhibited CXCL8 binding to CXCR2 (IC50 = 28 nM) and calcium mobilization (IC50 = 48 nM), and exhibited good bioavailability [453]. |
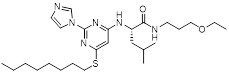 Imidazolylpyrimidine 1 |
Imidazolylpyrimidine 1 blocked CXCL8 binding to CXCR2 with Ki = 60 nM [454]. Imidazolylpyrimidine 1 bound to CXCR2 in transmembrane helices 3, 5, and 6 [455]. |
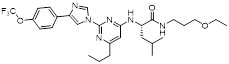 Imidazolylpyrimidine 40 |
Imidazolylpyrimidine 40 blocked CXCL8 binding to CXCR2 with Ki = 25 nM [454] |
| CXCL8(3-73)K11R | Blocked CXCL8 binding to human neutrophils (IC50 = 1.8 pM) and CXCL1 binding with less potency [456]. |
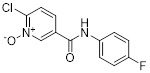 6-chloronicotinamide N-oxide 4a |
6-chloronicotinamide N-oxide 4a inhibited CXCL8-induced human neutrophil chemotaxis (IC50 = 1.3-2.3 μM) [457]. Inhibited CXCL8 binding to CXCR1 and CXCR2 with similar IC50 values (~1µM) [457]. Well-tolerated in mice and stable to rat liver microsomes [457]. |
| CXCL8(3-72)K11R/G31P (G31P) CXCL8 peptide |
G31P was effective at 10 ng/mL in vitro. G31P inhibited both CXCR1- and CXCR2-mediated neutrophil migration and calcium mobilization. G31P blocked neutrophil infiltration in guinea pig model of airway endotoxemia [458]. G31P protected against ischemia and reperfusion injury in rats [203, 204]. G31P had no agonistic or chemotactic activity and antagonized the binding of antibodies to CXCR1/2 [459]. Other analogs and variations of this peptide was also generated [460]. G31P treatment prior to E.coli and LPS challenge in guinea pigs reduced pulmonary neutrophil recruitment (85% inhibition) [461]. G31P treatment significantly inhibited human lung cancer growth and metastasis by blocking the activity of CXCR1 and CXCR2. The treatment also significantly reduced expression of VEGF and NF-κB-p65 as well as phosphorylation of ERK1/2 and AKT [462]. |
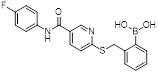 SX-517 |
SX-517 inhibited CXCL1- and CXCL8-stimulated Ca2+ flux in human polymorphonuclear cells (hPMNs) with IC50 values of 38 nM and 36 nM, respectively. It also antagonized CXCL8-stimulated [35S]GTPγS binding (IC50 = 60 nM) in HEK293 cells that stably expressed human recombinant CXCR2 [463]. SX-517 acted as a noncompetitive inhibitor of CXCR2 as it failed to compete with the binding of [125I]-CXCL8 to CXCR2 [463]. |
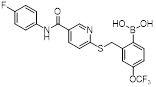 SX-576 |
SX-576 inhibited GRO-α-mediated intracellular calcium release in isolated human PMNs with an IC50 value of 22 nM [464]. SX-576 exhibited significant microsomal stability in rat and monkey liver microsomes with more than 90% still intact after 60 min [464]. |
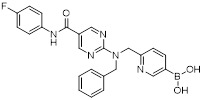 Compound 7 |
Compound 7 inhibited CXCL8-mediated calcium flux in stably CXCR1 or CXCR2 transfected RBL cells with IC50 values of 7 nM and 4 nM, respectively. Compound 7 has improved aqueous solubility, oral bioavailability and plasma stability compared to its analogs [465]. Compound 7 at a dose of 1 mg/kg also significantly reduced neutrophil influx in the BAL fluid of an in vivo rat model [465]. |
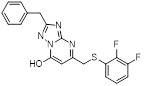 Triazolopyrimidine 14 Triazolopyrimidine 14 |
Triazolopyrimidine 14 inhibited the binding of GRO-α to human recombinant CXCR2 expressed in CHO membranes with an IC50 of 0.33 μM [466]. Triazolopyrimidine 14 showed reasonable lead-like properties with 52% bioavailability (po), 9 h half-life (iv) and low microsomal clearance in rat [466]. |
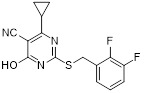 Triazolopyrimidine 20 |
Triazolopyrimidine 20 inhibited the binding of GRO-α and GTPγS to human recombinant CXCR2 expressed in CHO membranes with IC50 values of 0.04 μM and 0.14 μM, respectively [467]. |
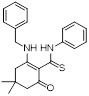 CX4338 |
CX4338 selectively inhibited CXCR2-mediated recruitment of β-arrestin-2 (IC50 = 6.3 μM) and receptor internalization [468]. CX4338 also enhanced CXCR2-mediated MAPK activation [468]. CX4338 inhibited CXCL8-mediated chemotaxis in CXCR2-overexpressing cells at a concentration as low as 1 μM [468]. CX4338 significantly reduced neutrophils in bronchoalveolar lavage in a LPS-induced mouse inflammation model [468]. |
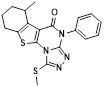 CX797 |
CX797 inhibited IL8-induced chemotaxis in CXCR2-bla U2OS Tango cells [469]. |
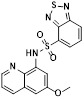 CX25 |
CX25, which was identified using a ligand-based pharmacophore approach, inhibited CXCR2 in a Tango assay (IC50 = 0.36 μM). It also inhibited CXCR4 with an IC50 of 0.59 µM [20]. |
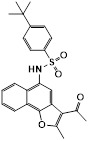 CX4152 |
CX4152 inhibited CXCR2 (IC50 = 7.6 μM) and exhibited selectivity over CXCR4 (IC50=64.7 µM) in the Tango assay [20]. CX4152 induced receptor internalization in a dose- and time-dependent manner. It also down-regulated expression of total CXCR2 at 5 h treatment [20]. CX4152 did not induce rapid calcium mobilization in CXCR2-overexpressing cells (293T-CXCR2-GFP), whereas CX25 induced peak calcium flux within 1 min stimulation [20]. CX4152 inhibited CXCL8-induced chemotaxis in a concentration-dependent manner with an IC50 of 51 μM [20]. CX4152, at a dose of 50 mg/kg, significantly inhibited polymorphonuclear leukocyte migration (about 2-fold decrease) in a murine model of neutrophilic airway inflammation induced with LPS [20]. |
 CX815 |
CX815 inhibited CXCR2 (IC50 = 0.4 µM) and exhibited selectivity over CXCR4 (IC50 > 50 µM) [20]. |