

Abstract
Background
Acute haemorrhagic leukoencephalitis (AHLE) is a rare and rapidly fatal disease of unknown aetiology. There is a paucity of literature on the presentation and management of this rare disease.
Case Description
We report the case of a 33-year-old female presenting with headache and left-sided apraxia. Imaging revealed a right-sided white matter lesion with extensive cytotoxic oedema. Pathology was suggestive of AHLE. She underwent an open excisional biopsy and was treated with high-dose corticosteroids. Three months since symptom onset she remains clinically well with resolving apraxia and radiological appearance.
Conclusion
This case may represent a milder spectrum of AHLE, which responded favourably to corticosteroids.
Keywords: Acute haemorrhagic leukoencephalitis, Hurst disease, Weston-Hurst syndrome, Acute haemorrhagic encephalomyelitis
Introduction
Acute haemorrhagic leukoencephalitis (AHLE) is a rare, usually fulminant demyelinating disease occurring predominantly in young adults. It is considered by some to be a severe form of the more common acute disseminated encephalomyelitis. First described by Hurst in 1941, and also known as Hurst disease, it is characterised by progressive multifocal white matter inflammation, haemorrhage, and necrosis, often diagnosed at autopsy [1, 2]. It typically presents as a monophasic illness with headache, followed by rapid progression to confusion, somnolence and eventually coma and death, frequently preceded by a nonspecific viral illness [3]. The mortality reported in the literature is as high as 70% [4, 5, 6]. The aetiology of the disease is unknown. There are no known prognostic factors to determine the severity of the illness. Herein, we describe a young female with unilateral disease and a more indolent course with a good functional outcome. We postulate that this atypical presentation and clinical course may represent a milder spectrum of the disease.
Case Presentation
A previously healthy 33-year-old female presented with headache and left-sided apraxia.
She had no significant comorbidities, and her only regular medication was the oral contraceptive pill. She had 2 uncomplicated pregnancies. Her children were aged 2 and 4 years, and both were well with no recent illnesses. She was a lifelong nonsmoker, did not drink alcohol regularly, and had no history of recreational drug use. She had no significant family history.
The patient presented with acute onset of right retro-orbital headache following a shower, with associated nausea and photophobia. The day following onset, she developed left-hand clumsiness and presented to her general practitioner, who referred her to the emergency department. She self-administered 1,200 mg of aspirin prior to presentation. She reported no recent infective symptoms, no new medications or recent immunisation, and no unwell contacts or recent travels.
Initial examination findings were left-sided apraxia, a subtle left pronator drift, and mild facial droop. Her higher functions were intact, and she otherwise had no neurological deficit. She was afebrile and showed no signs of meningeal irritation. Fundus examination was not performed.
Brain magnetic resonance imaging (MRI) showed a right frontoparietal lesion with extensive white matter oedema and mass effect. It was hypointense on T1, T2, and fluid attenuation inversion recovery sequences, and demonstrated restricted diffusion and blooming artefact. Susceptibility-weighted imaging revealed multiple petechial haemorrhages throughout the whole temporoparietal region. There was central homogeneous enhancement after contrast (Fig. 1).
Fig. 1.
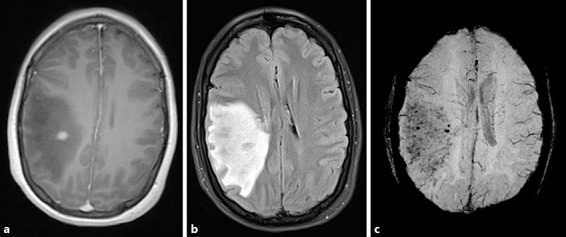
Brain magnetic resonance imaging on day 1 after presentation. a T1 + gadolinium showing a discrete area of contrast enhancement. b Fluid attenuation inversion recovery consistent with significant cerebral oedema. c Susceptibility-weighted imaging demonstrates multiple petechial haemorrhages.
The initial impression from the presentation and imaging was a primary neoplasm. CT of the chest, abdomen, and pelvis did not show any evidence of malignancy. She was commenced on dexamethasone with some improvement in headache but not apraxia. Initial needle biopsy was negative for tumour. It showed scattered reactive astrocytes outnumbered by sheets of foamy macrophages associated with small areas of perivascular necrosis. There was no convincing evidence of vasculitis, infection, or tumour infiltrate.
An open biopsy was subsequently undertaken. Histology demonstrated abundant foamy macrophages with early white matter cavitation. There were perivascular foci of haemorrhage, oedema, and necrosis with fibrin deposition associated with a mild-to-moderate inflammatory infiltrate. This was composed of lymphocytes with occasional neutrophils and eosinophils. Reactive gemistocytic astrocytes, including frequent forms with fragmented nuclei typical of Creutzfeldt-Peters cells, were seen in viable areas of the biopsy. Special and immunohistochemical stains demonstrated perivascular loss of myelin and phagocytosed fragments of myelin within the foamy macrophages. Axons in the demyelinated areas were variably preserved but at times lost. Special stains and microbiological cultures for organisms were negative (Fig. 2).
Fig. 2.
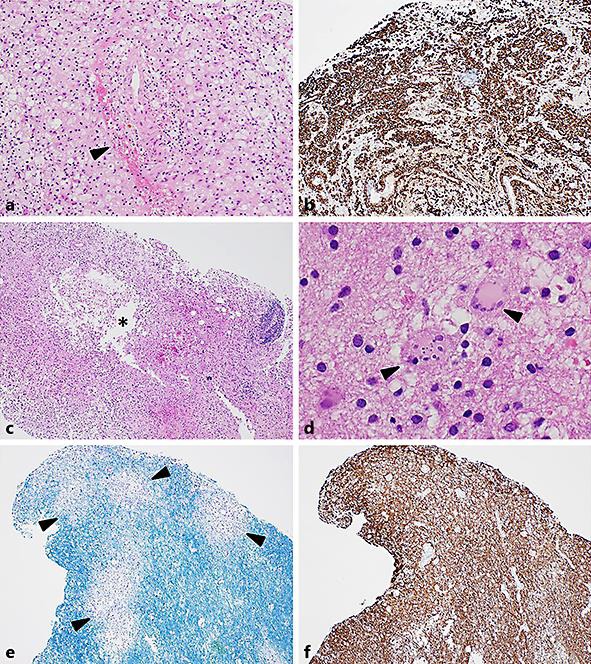
Photomicrographs demonstrating the features of the second biopsy. a Perivascular haemorrhage, haemosiderin, and oedema (black arrowhead). There were plentiful foamy macrophages and a mild lymphocytic inflammatory response (haematoxylin and eosin. Original magnification ×100). b Immunohistochemical staining for the macrophage marker CD163, highlighting foamy macrophages as the dominant cell type (CD163 immunoperoxidase, original magnification ×40). c Evidence of early white matter cavitation (asterisk in white space) with a cystic space lined by foamy macrophages. More marked perivascular inflammation present at the biopsy edge (haematoxylin and eosin. Original magnification ×40). d Adjacent gliosis in surrounding white matter containing plump, reactive gemistocytic astrocytes. Scattered astrocytes had fragmented nuclei typical of Creutzfeldt-Peters cells (black arrowheads; haematoxylin and eosin. Original magnification ×400). e, f Serial sections stained with histochemical stain Luxol Fast Blue(LBF)/Cresyl Violet (CV) and immunohistochemical stain for neurofilament (NF), respectively. There was perivascular loss of blue-staining myelin with an irregular margin (black arrowheads). Neurofilament highlighted variable numbers of preserved axons within the areas of myelin loss. At higher magnification occasional axonal spheroids were demonstrated. There was patchy loss of axons within the areas of cystic degeneration (LFB/CV and NF immunoperoxidase, original magnification ×40).
Based on the perivascular haemorrhage, the necrosis with fibrin deposition and the limited nature of the demyelination with progression to cystic necrosis, a diagnosis of acute haemorrhagic encephalomyelitis was favoured. A progress MRI 2 weeks following her original presentation revealed extensive white matter cystic necrosis, with an increased T2 signal. Thrombosed vessels were visualised throughout the necrotic white matter. The rapid development of cystic necrosis was thought to be highly atypical of a glial tumour and supported a diagnosis of AHLE. An MRI of the whole spine was also performed at this stage to exclude other central nervous system involvement. It was unremarkable.
Cerebrospinal fluid (CSF) analysis performed 26 days after commencement of corticosteroids showed an elevated IgG index but no oligoclonal bands. There was no pleocytosis, and protein, glucose, and flow cytometry were all normal. Markers of autoimmunity and vasculitis were negative, including negative ANA, ENA, dsDNA, ANCA, and anti-NMO antibodies. Erythrocyte sedimentation rate and serum angiotensin-converting enzyme were normal. HIV testing was not performed. Although visual evoked potentials were considered, they were not performed.
She continued to improve with home-based occupational therapy exercises and oral dexamethasone at a dose of 8 mg twice daily maintained for 4 weeks, followed by a slow wean over 2 months. Serial MRIs showed stable appearance of the right frontoparietal lesion (Fig. 3). At day 90, she remains clinically well with no evidence of progression and resolving neurological deficit.
Fig. 3.
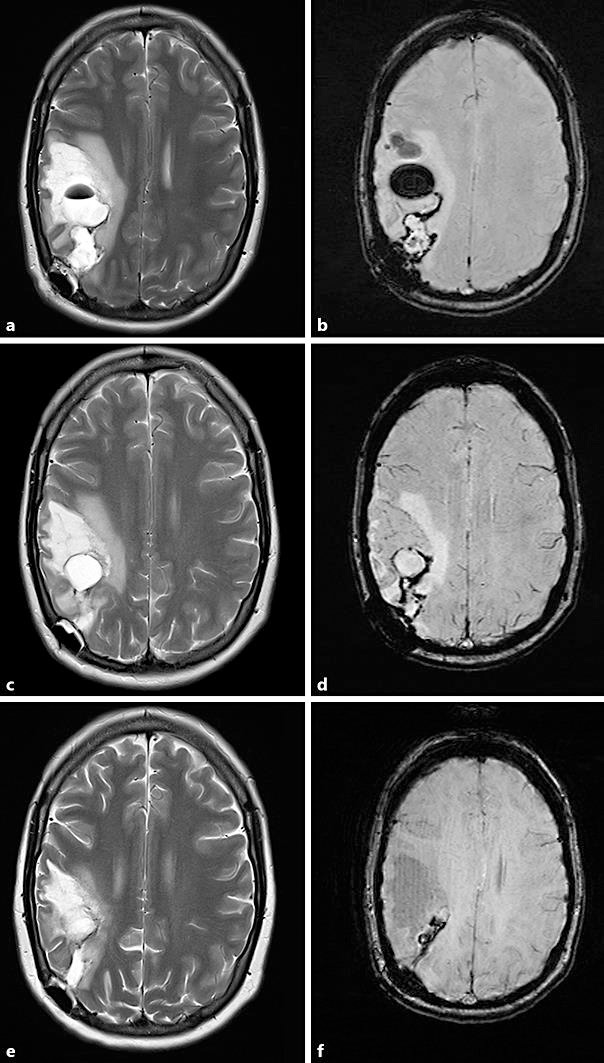
Progress magnetic resonance imaging. a T2. b Susceptibility-weighted imaging. Three weeks after presentation, showing a cystic lesion and ongoing surrounding mass effect, consistent with multiple areas of haemorrhagic necrosis. This may represent the natural progression of the disease, though it is difficult to evaluate how much is secondary to postoperative changes. c T2. d Susceptibility-weighted imaging. Five weeks after presentation, showing improving oedema and resolving haemorrhagic cysts. e T2. f Susceptibility-weighted imaging. Two months after presentation, showing significant reduction in the areas of cystic necrosis.
Discussion
AHLE presents suddenly and rapidly leads to neurological decline, often death [1]. The aetiology is undetermined, and there are no guidelines in treatment. Recent case reports show improved outcomes with a variety of immunomodulatory treatments [4, 7]. However, there are no established prognostic features to determine the severity of the disease. The above case highlights an unusual presentation of AHLE, with a successful outcome following a prolonged course of high-dose corticosteroids. We propose the disease may have a spectrum of severity, and a consideration to possible prognostic factors should be further evaluated.
The aetiology of AHLE is unknown. Cross reactivity between human myelin antigens and viral or bacterial antigens is postulated, with a preceding respiratory illness evident in approximately half of the cases [3, 4, 7, 8]. There are reported cases of presumed cross reactivity with Mycoplasmapneumoniae, Epstein-Barr virus, herpes simplex virus, and influenza A [9, 10]. There was no evidence of recent infection on history or examination in our case; however, serological markers were not tested.
Histopathologically, the disease is characterised by perivascular microhaemorrhages and necrosis, demyelination accompanied by axonal injury and fibrin exudate. A variable degree of perivascular inflammation is present, typically neutrophilic with some macrophage and lymphocyte infiltration [3, 9]. Massive haemorrhage and oedema leads to death in many cases due to herniation. Ultimately, targets for treatment are immunosuppression and management of cerebral oedema [7].
CSF typically shows pleocytosis with predominant polymorphonuclear cells, in contrast to the lymphocytic predominance in acute disseminated encephalomyelitis [9, 11]. We postulate that the absence of pleocytosis in the CSF of our patient may reflect a milder degree of inflammation, or prior treatment with steroids.
MRI has a critical role in the early diagnosis of AHLE, which typically involves white matter tracts with relative sparing of grey matter, accompanied by haemorrhage on susceptibility-weighted imaging [12, 13, 14]. Classically, the changes are multifocal with predominant involvement of the frontal and parietal lobes, although it may also affect the basal ganglia, brainstem, and spinal cord. In our case, the frontoparietal white matter lesion with multifocal petechial haemorrhages was consistent with previously described cases of this disease. It was unilateral, however. The degree of gadolinium uptake reported in the literature is variable, and homogenous enhancement was clearly present in our patient. Diffusion restriction, which was noted on our patient's MRI, is likely due to cytotoxic oedema. The initial diagnosis was thought to be a primary brain neoplasm; however, the rapid progression to cystic necrosis was strongly suggestive of demyelination with haemorrhage. Less likely differential diagnoses that were considered based on the imaging findings included (a) steroid-treated lymphoma, although the normal flow cytometry and biopsy results made this unlikely, and (b) haemorrhagic encephalitis due to parasite infection; however, she had no peripheral eosinophilia and a more diffuse picture would be expected. Progress imaging was consistent with cystic necrosis, consistent with the available literature [3]. In our case, serial MRI scans provided vital evidence of the oedema, demyelination, necrosis, and haemorrhages that characterise this disease and helped facilitate a timely diagnosis.
There are no published large case series or controlled trials to help guide management of this rare and frequently fatal condition. Management is based largely on the understanding of the underlying pathology, expert opinion, and data from isolated case reports. There are several case reports published reporting favourable outcomes with different modes of immunosuppression, including high-dose corticosteroids, intravenous immunoglobulin, plasmapheresis, and cyclophosphamide [2, 11, 13]. In the case described by Seales and Greer [2], aggressive management of raised intracranial pressure with mannitol, hyperventilation, and phenobarbital was undertaken. In that case, the patient had no lasting neurological deficit. Surgical management of raised intracranial pressure has also been described [6, 7].
Our patient was initially commenced on high-dose dexamethasone when the presumed aetiology was a neoplasm. This was continued once histopathology favoured a diagnosis of AHLE. Prior to definitive diagnosis, she had undergone open excisional biopsy 2 weeks after symptom onset, with resultant debulking of the lesion. Our case differs from most previous reports by her comparatively mild symptoms, which, unlike most described cases, were not followed by rapidly progressive neurological deficit and coma. Consequently, it is not clear whether early immunosuppression or debulking contributed to her improvement or altered her clinical course, or if her disease would have remained indolent without progressive deficit in the absence of treatment.
Previous reports have described a monophasic illness with good prognosis if an individual survives the initial insult [2, 11, 13]. We are not aware of any cases of recurrence of AHLE. There is a paucity of data regarding whether or not immunosuppression has a role in preventing relapse.
Conclusion
In our case, the presentation and progression were inconsistent with previous descriptions of AHLE. However, her biopsy results, progression of imaging findings, and response to immunosuppression were strongly supportive. Possible diagnoses raised by the clinical, radiological, and histological findings in isolation were less likely when these differing modalities were considered in combination. One of the questions raised by this unusual case is whether or not unifocal disease or lack of CSF pleocytosis is predictive of a more indolent clinical course. Given that the patient remained clinically well throughout the course of the illness, the natural history of this patient's disease had she not undergone surgical debulking and immunosuppression is unclear. Whether the presence or absence of a preceding respiratory illness has any prognostic value is also uncertain. Further study is required to better characterise this rare disease and provide further guidance regarding prognosis and treatment.
Statement of Ethics
Patient consent was obtained.
Disclosure Statement
The authors declare that there are no conflicts of interest to disclose.
Acknowledgements
We would like to thank Dr. Laughlin Dawes for his excellent review of MRI scans and the Imaging Department, Prince of Wales Hospital, Randwick, NSW, Australia.
References
- 1.Hurst EW. Acute hemorrhagic leukoencephalitis: a previously undefined entity. Med J Aust. 1941;2:1–6. [Google Scholar]
- 2.Seales D, Greer M. Acute hemorrhagic leukoencephalitis. A successful recovery. Arch Neurol. 1991;48:1086–1088. doi: 10.1001/archneur.1991.00530220108029. [DOI] [PubMed] [Google Scholar]
- 3.Hart MN, Earle KM. Haemorrhagic and perivenous encephalitis: a clinical-pathological review of 38 cases. J Neurol Neurosurg Psychiatry. 1975;38:585–591. doi: 10.1136/jnnp.38.6.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sotiriou A, Chalkis A, Riga D, Zakynthinos S. Acute hemorrhagic leukoencephalitis: multimodal diagnosis and treatment. Hosp Chron. 2014;9:202–207. [Google Scholar]
- 5.Duggal N, Iftekhar A, Duggal N. Acute hemorrhagic leukoencephalitis associated with autoimmune myopathy. J Vasc Interv Neurol. 2014;7:19–22. [PMC free article] [PubMed] [Google Scholar]
- 6.Leake JA, Billman GF, Nespeca MP, Duthie SE, Dory CE, Meltzer HS, et al. Pediatric acute hemorrhagic leukoencephalitis: report of a surviving patient and review. Clin Infect Dis. 2002;34:699–703. doi: 10.1086/338718. [DOI] [PubMed] [Google Scholar]
- 7.Ryan LJ, Bowman R, Zantek ND, Sherr G, Maxwell R, Clark HB, et al. Use of therapeutic plasma exchange in the management of acute hemorrhagic leukoencephalitis: a case report and review of the literature. Transfusion. 2007;47:981–986. doi: 10.1111/j.1537-2995.2007.01227.x. [DOI] [PubMed] [Google Scholar]
- 8.Yildiz O, Pul R, Raab P, Hartmann C, Skripuletz T, Stangel M. Acute hemorrhagic leukoencephalitis (Weston-Hurst syndrome) in a patient with relapse-remitting multiple sclerosis. J Neuroinflammation. 2015;12:175. doi: 10.1186/s12974-015-0398-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Magun R, Verschoor CP, Bowdish DM, Provias J. Mycoplasma pneumoniae, a trigger for Weston Hurst syndrome. Neurol Neuroimmunol Neuroinflamm. 2016;3:e187. doi: 10.1212/NXI.0000000000000187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeganathan N, Fox M, Schneider J, Gurka D, Bleck T. Acute hemorrhagic leukoencephalopathy associated with influenza A (H1N1) virus. Neurocrit Care. 2013;19:218–221. doi: 10.1007/s12028-013-9880-8. [DOI] [PubMed] [Google Scholar]
- 11.Markus R, Brew BJ, Turner J, Pell M. Successful outcome with aggressive treatment of acute haemorrhagic leukoencephalitis. J Neurol Neurosurg Psychiatry. 1997;63:551. doi: 10.1136/jnnp.63.4.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kao HW, Alexandru D, Kim R, Yanni D, Hasso AN. Value of susceptibility-weighted imaging in acute hemorrhagic leukoencephalitis. J Clin Neurosci. 2012;19:1740–1741. doi: 10.1016/j.jocn.2011.04.034. [DOI] [PubMed] [Google Scholar]
- 13.Lee HY, Chang KH, Kim JH, Na DG, Kwon BJ, Lee KW, et al. Serial MR imaging findings of acute hemorrhagic leukoencephalitis: a case report. AJNR Am J Neuroradiol. 2005;26:1996–1999. [PMC free article] [PubMed] [Google Scholar]
- 14.Mader I, Wolff M, Niemann G, Kuker W. Acute haemorrhagic encephalomyelitis (AHEM): MRI findings. Neuropediatrics. 2004;35:143–146. doi: 10.1055/s-2004-817906. [DOI] [PubMed] [Google Scholar]