

Abstract
Here we demonstrate a novel application of ‘surface enhanced resonance Raman scattering nanoparticles’ (SERRS NPs) for imaging breast cancer (BC) lung metastases with much higher precision than currently feasible. The BC lung metastasis mouse model was established by intravenous injection of LM2 cells. SERRS NPs conjugated with ALT-836, an anti-tissue factor (TF) monoclonal antibody, were administered to these mice intravenously and subjected to Raman imaging to visualize the expression of TF both in vivo and ex vivo. Raman imaging indicated marked uptake of αTF-SERRS NPs by the lung metastases (n=4) compared to isotype (n=4) and blocking controls (n=3). Conversely, little uptake of αTF-SERRS NPs was observed in the lungs of healthy mice. Successful detection and delineation of pulmonary micrometastatic lesions as small as 200 μm, corroborated by histology, immunohistochemistry, and bioluminescence imaging (BLI) confirmed the suitability of both TF as a target and αTF-SERRS NPs as an effective contrast agent for imaging breast cancer lung metastases. Further advancements of this technique in the form of Raman endoscopes coupled with ultrabright SERRS NPs developed in this work could lead to minimally invasive detection and resection of lung metastases.
Keywords: Tissue factor, breast cancer, pulmonary metastases, Surface enhanced resonance Raman spectroscopy (SERRS), Nanoparticles
Graphical Abstract
Introduction
Breast cancer (BC) is the most common malignant disease in women both in the developed and the developing world 1. Although the primary BC tumor can be identified and treated with existing medical imaging methods, patients still face high morbidity and mortality from recurrence and, more importantly, from metastasis due to the preferential dissemination of cancer cells to distant sites such as the lungs, liver, and bones 2, 3. Management and treatment of metastasis in BC patients would ideally make use of advanced diagnostic tools including molecular imaging techniques. Currently available imaging techniques, such as magnetic resonance imaging (MRI), computed tomography (CT), positron-emission tomography (PET), and ultrasound (US), possess significant limitations with regard to imaging metastases. CT and US lack the sensitivity to identify small metastatic lesions 4–8 and therefore may underestimate the tumor burden. MRI, although more sensitive, cannot detect microscopic tumor infiltrations, and it is an expensive technique. Similarly, PET suffers from limited spatial resolution. Additionally, there is the matter of undesired exposure of the patient to ionizing radiation involved with CT and PET 9, 10. More importantly, these imaging techniques are limited in their ability to provide intraoperative guidance as part of resection of metastatic nodules. Therefore, there exists a need to develop novel imaging modalities free of such limitations and with the ability to detect the full extent of metastatic dissemination. Detection of the precise location and extent of metastatic lesions will allow for personalized treatment planning, image-guided tumor removal, and effective monitoring of therapeutic responses.
With the above goals in mind, we have developed a novel method for imaging BC metastases using an ultrasensitive diagnostic tool: ultra-bright surface enhanced resonance Raman scattering (SERRS) nanoparticles (NPs). The SERRS NPs consist of a gold nanostar core, coated with a Raman reporter dye (resonant with the near infrared interrogation laser (785 nm)), and encapsulated by a biocompatible silica shell 11–13. With the addition of a suitable targeting moiety, the SERRS NPs may be used to target a variety of specific markers. Potential targets include molecules known to be involved in physiological processes such as glucose metabolism 14–16, hypoxia 17–19, and angiogenesis 20–22, or signaling cascades linked to tumor aggressiveness and metastasis. Targeting such markers within the tumor microenvironment may enable not only the detection of metastatic potential but also provide profound information regarding disease progression.
Recent progress in unraveling molecular pathways has established the existence of a strong correlation between the coagulation cascade in the hemostatic system and tumor progression. The coagulation cascade initiated by a transmembrane glycoprotein called Tissue Factor (TF) is reported to be expressed in several tumor cell types, including breast cancer 23. Importantly, increased TF expression has been implicated in cancer cell signaling, tumor cell migration, and decreased apoptosis leading to enhanced prospect of metastasis 24–27. This signifies the potential application of TF as a suitable target for imaging and therapy of BC metastasis. A chimeric anti–human-TF monoclonal antibody, ALT-836 (formerly known as Sunol-cH36), has been found to inhibit the coagulation cascade by blocking the activation of Factor X (FX) via binding to the FX binding site in TF with high affinity 28. Currently, ALT-836 in combination with the chemotherapy drug gemcitabine is undergoing phase I clinical trial for locally advanced or metastatic solid tumors (ClinicalTrials.gov Identifier - NCT01325558). Additionally, ALT-836 is undergoing phase II clinical trial for acute lung injury and acute respiratory distress syndrome (ALI/ARDS) after encouraging results from a phase I clinical trial 29.
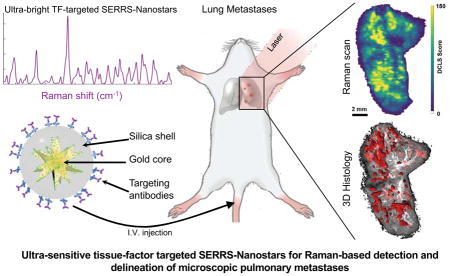
In this study, our goal was to develop a minimally invasive advanced diagnostic probe for imaging breast cancer lung metastasis using TF-targeted SERRS NPs (Figure 1), which in the future could be clinically translated to a novel robot-assisted laparoscopic/endoscopic surgery technique for pulmonary resection.
Figure 1.

Schematic diagram of the experimental set up, depicting the stepwise synthesis of ultrabright targeted SERRS-NPs, administration to mice with pulmonary metastases, and Raman imaging.
Experimental Methods
Reagents
ALT-836 was provided by Altor Bioscience Corp (Miramar, FL). Alexa Fluor® 647 C2-maleimide, Alexa Fluor® 647 NHS were purchased from Life Technologies (Eugene, OR). Dialysis cassettes (MWCO 3.5 kDa; 15 ml Slide-A-Lyzer® G2) were purchased from Thermo-Scientific (Rockford, IL). D-Luciferin (Gold Biotechnology, Olivette, MO, USA). All other reagents, unless otherwise mentioned, were purchased from Sigma-Aldrich (St. Louis, MO).
Synthesis of SERRS NPs and antibody conjugation
SERRS NPs were synthesized according to a previously described procedure with appropriate modifications (Figure 1). Briefly, gold nanostars were synthesized by rapidly adding 10 ml of 20 mM HAuCl4 to 990 ml of 60 mM L-Ascorbic acid at 4°C under constant stirring. The nanostars were collected by centrifugation (20 min, 3220g, 4°C) and dialyzed (MWCO 3.5 kDa) against deionized water. The dialyzed gold nanostar dispersion (10.8 ml; 2.0 nM) was mixed with 117 ml absolute ethanol, 1.8 ml 28% (v/v) ammonium hydroxide, 4.5 ml 99.999% tetraethyl orthosilicate (TEOS), and 180 μL (25 mM) resonant Raman reporter (IR780 perchlorate) in N, N-dimethylformamide and allowed to react for 35 minutes to produce silicated SERRS-NPs. The SERRS-NPs were washed and resuspended in ethanol. To enable antibody conjugation, sulfhydryl groups were introduced on the silica surface by heating the SERRS-NPs (3 nM concentration) for 2 hours at 72°C in 850 μl absolute ethanol, 100 μl of (3-Mercaptopropyl) trimethoxy-silane (MPTMS), and 50 μl deionized water. The surface-modified SERRS-NPs were thoroughly washed with ethanol and water and redispersed in 10 mM PBS buffer (pH 7.4).
For antibody conjugation ALT-836 or isotype IgG (1 mg/ml) in 500 μL of PBS were reacted with 10-fold molar excess of the heterobifunctional linker, poly (ethylene glycol) (N-hydroxy-succinimide 5-pentanoate) ether N′-(3-maleimidopropionyl) amino ethane (NHS-PEG4k-Mal) for 30 minutes at RT. The excess crosslinker was removed using Amicon® Ultra-0.5 Centrifugal Filters (MWCO 100 kDa) purchased from Merck Millipore (Tullagreen, Ireland). The purified NHS-PEG4k-Mal–conjugated antibodies were added to sulfhydryl-modified SERRS-NPs in PBS and reacted for 30 minutes at RT. The SERRS-NP conjugated with ALT836 and isotype IgG (non-targeted control) were washed by centrifugation and gentle sonication to remove unconjugated antibodies and redispersed in PBS for intravenous (i.v). injection.
For flow cytometry, equimolar concentration of NHS-PEG4k-Mal conjugated antibodies and Alexa Fluor® 647 C2-maleimide were reacted with sulfhydryl-modified SERRS-NPs in PBS using the previously mentioned procedure to get αTF-SERRS-NP-A647 and IgG-SERRS-NP-A647. For positive and negative control, the antibodies were reacted with molar excess of Alexa Fluor® 647 NHS for 30 minutes at RT and purified using Amicon® Ultra-0.5 Centrifugal filters to get ALT-836-A647 and control IgG-A647.
Physicochemical characterization SERRS NPs
For characterization by electron microscopy, aqueous drops of the SERRS NPs were deposited on carbon film-coated copper grids (300 Mesh, Electron Microscopy Sciences) and air-dried. Images were typically acquired at magnification factors ranging from 25,000× to 500,000× using a JEOL 1200EX (JEOL USA, Inc.) transmission electron microscope operating at 80 kV. The concentration of the SERRS NPs was determined by nanoparticle tracking analysis (NTA; Nano Sight NS500; Malvern Instruments Inc.; Westborough, MA). The size distribution and zeta potential of the SERRS NPs were measured by dynamic light scattering (DLS) (Zetasizer Nano, Malvern, Worcestershire, UK). Raman signal intensity of varying concentrations of SERRS nanoparticle dispersions were measured in a 384 well plate using an inVia Raman microscopy system (Renishaw, Hoffman Estates, IL) at 7.5 mW laser power and 1 s acquisition time, using a 5× objective. BCA protein assay (Pierce™ BCA Protein Assay Kit) was used to quantify the antibody load of the NPs after conjugation.
Cell line and cell culture
MDA231-LM2 (LM2 for short) is a lung metastatic subline derived from the MDA-MB-231 breast cancer cell line 30. For bioluminescence imaging, this cell line consists of a triple-fusion protein reporter construct encoding firefly luciferase along with herpes simplex virus thymidine kinase 1 and green fluorescent protein (GFP). These cells were grown in high-glucose DMEM medium with 10% fetal bovine serum and incubated at 37°C with 5% CO2. Cells were used for in vitro and in vivo experiments when they reached ~80% confluence.
Flow cytometry
The immunoreactivity of ALT-836-A647 and TF-SERRS NP-A647 to MDA-231 LM2 cells were evaluated by fluorescence flow cytometry analysis. Briefly, cells were harvested from culture and suspended in ice cold PBS with 0.5% bovine serum albumin (BSA) and 0.1% sodium azide at a concentration of 5×106 cells/ml. The cells were incubated with ALT-836-A647 (0.5 μg/ml) and TF-SERRS NP-A647 for 1hr at RT. For control studies, the cells were incubated with isotype IgG-A647 (5 μg/ml) and isotype IgG-SERRS NP-A647. For blocking studies, before incubation with the ALT-836-A647 and TF-SERRS NP-A647, cells were initially incubated with unconjugated ALT-836 (500 μg/ml) for 1hr at RT and washed three times with cold PBS. Afterwards, the cells were washed four times with cold PBS and analyzed using a BD LSR II flow cytometer, which is equipped with 488 and 647 nm lasers (Becton–Dickinson, San Jose, CA, USA). The data analysis was done using FlowJo analysis software (Tree Star, Inc., Ashland, OR, USA).
Animal model
All animal studies were conducted under a protocol (#06-07-011) approved by the Institutional Animal Care and Use Committees of Memorial Sloan Kettering Cancer Center. The breast cancer experimental lung metastasis model was established by i.v. injection of 0.5×105 LM2 cells in 150 μL of phosphate-buffered saline (PBS) into (4–5) weeks old female athymic nu/nu mice (Envigo, Indianapolis, IN) via tail vein. The tumor progression was monitored by noninvasive bioluminescence imaging (BLI) carried out using an IVIS Spectrum system (Caliper Life Sciences) after intraperitoneal (i.p.) injection of D-luciferin (150 mg/kg of mouse body weight) on day 0, day 1, day 3, day 7 and continued in subsequent weeks.
Raman imaging
When the BLI signal from the thoracic area of mice was substantially strong (typically 4 weeks after injection), the metastatic mice were administered SERRS NPs either functionalized with ALT-836 (αTF-SERRS-NPs) or isotype IgG (IgG-SERRS-NPs) at a concentration of 3.5 nM in 150 μL of PBS (10 mM, pH 7.4) via tail vein injection. For blocking studies, a cohort of four metastatic mice were injected with 2 mg of ALT-836 Ab 2 hours prior to administration of αTF-SERRS-NPs to evaluate in vivo TF specificity. Similarly, a group of five healthy mice (8–9 weeks) were intravenously administered αTF-SERRS-NPs and used as control. After an 18–24 hour circulation period, mice were injected with 0.5 mg luciferin per mouse, and euthanized by carbon dioxide asphyxiation. Depending on the experimental setting, BLI imaging was performed either on the whole mouse or on dissected lungs placed on glass slides for direct BLI to Raman correlation. Subsequently, either in situ Raman images of lungs in the exposed thoracic region or ex vivo Raman scans of resected tissues (lungs, liver, and spleen) were obtained. All Raman scans and measurements were performed with an inVia Raman microscope (Renishaw, Hoffman Estates, IL) equipped with a 785 nm diode laser (300 mW cm−2) and a 1 in. charge-coupled device detector with a spectral resolution of 1.07 cm−1. Raman spectra were acquired through a 5× objective (Leica, Buffalo Grove, IL), where laser output at the objective was measured to be 100 mW cm−2 using a hand-held laser power meter (Edmund Optics, Inc., Barrington, NJ). The focal plane was selected by focusing on the region of interest with a parfocal bright-field camera. The Raman scans were collected in “streamline mode” and have a resolution of 14 μm in the x-direction and 150–300 μm in the y-direction. A typical Raman scan took between 60 and 120 min, depending on the resolution and area scanned.
Data analysis
Data analysis of the spectral images was performed in MATLAB (R2014b) and PLS Toolbox v.8.0 (Eigenvector Research, Inc., Wenatchee, WA). For the DCLS model reference spectra were collected from pure solutions of SERRS-NPs. The reference spectra were first subjected to baseline subtraction using a Whittaker filter with λ = 200 cm−1, then an L∞-norm (normalization by max), followed by a Savitzky–Golay filter (second degree polynomial fit, first-order derivative, width = 15 steps). The experimental imaging data were subjected to the Whittaker and Savitzky–Golay filters (with no normalization) and then assessed by the model. In this way, the scores assigned by the model correspond roughly to physical intensity units as shown in Figure S5.
Biodistribution
Tissue samples of known masses were taken up in Savillex vials having 5 mL of freshly prepared aqua regia (1:3 ratios of concentrated nitric acid and hydrochloric acid) and heated overnight at 60 °C for complete digestion. When the samples fully dissolved in the acidic solution, 40 μl of the digest were taken up in 15 ml centrifuge tubes and the volume made up to 10 ml using deionized water and analyzed at the Brooklyn College Environmental Sciences Analytical Center on a PerkinElmer Elan DRC-e ICP-MS instrument and referenced to a HAuCl4-derived calibration.
Histology
Freshly excised tissues (liver, spleen and lungs) from both healthy and metastatic mice were fixed in 4% paraformaldehyde overnight at RT and processed for paraffin embedding using a Leica ASP6025 tissue processor (Leica Biosystems). Tissue sections of 5 μm thickness were stained for immunohistochemistry on a Leica Bond RX (Leica Biosystems) with 0.5 μg/ml CD31 rat anti-mouse monoclonal Ab (Dianova cat#DIA-310) or 1 μg/ml CD142 rabbit anti-human TF polyclonal Ab (Thermofisher cat#PA5-27278) for 1 hour on Protocol F. The sections were pre-treated with Leica Bond ER2 Buffer (Leica Biosystems) for 20 min at 100 °C before each staining. After staining, the sections were dehydrated and mounted with Permount for digital scanning with Pannoramic Confocal (3dHistech) using a 40× water immersion objective.
For three dimensional (3D) histologic reconstruction of metastatic lungs, every 6th section was selected and stained with H&E. Serially sectioned H&E slides (n=40) were digitally scanned using Pannoramic SCAN (3DHistech, Hungary) with a 20× objective. Alignment of these sections was performed using Voloom software (Micro Dimensions, München, Germany) and exported in TIFF format. Segmentation of tumor region from the entire tissue region was performed using a color deconvolution algorithm in FIJI/ImageJ (NIH). A pseudocolor image was then generated using the tumor region and total tissue region as separate channels in FIJI. The aligned resultant stack from FIJI was visualized in Imaris (Bitplane, Zurich, Switzerland), where 3D surfaces were created of the tumor region and compared with image of metastatic lungs obtained by BLI and Raman imaging.
Results and Discussion
Synthesis and in vitro characterization of SERRS NPs
SERRS NPs were synthesized and conjugated with two types of antibodies (ALT-836 or isotype IgG) according to previously established procedures 12, 13. The resulting SERRS NPs (αTF-SERRS-NPs and IgG-SERRS-NPs) were subjected to multiple independent physicochemical and biological characterizations in vitro (Figure 2). Throughout the experiments, the concentration of the SERRS-NPs was determined by nanoparticle tracking analysis (NTA; Nano Sight NS500; Malvern Instruments Inc.; Westborough, MA). The transmission electron micrographs (TEM) (Figure 2a), acquired at magnification factors ranging from 25,000× to 500,000× using a JEOL 1200EX (JEOL USA, Inc.) revealed the average diameter of gold nanostar core and final spherical SERRS NPs (including the silica shell) to be 70±24 nm and 122±22 nm respectively. The hydrodynamic diameter of the nanoparticles (Figure 2b) as measured by dynamic light scattering (DLS) was 79, 141, and 164 nm with polydispersity index of 0.32, 0.17, and 0.20 for the gold nanostar cores, SERRS NPs, and antibody-conjugated SERRS NPs respectively. The difference in size (~5–20 nm) as observed by TEM and DLS was expected, due to the presence of a hydration layer on the SERRS NPs surface 31. The zeta potential (ζ) of −37.3±1.2 mV, −45.9±1.4 mV and −16.6±0.7 mV for the above respective nanoparticles indicates successful silication and antibody conjugation. Furthermore, the final slightly negative surface charge of the antibody-conjugated SERRS NPs is within the desired range to be effectively delivered to metastatic lesions upon intravenous administration 32. The average concentration of the antibody conjugated to SERRS NPs as determined by bicinchoninic acid assay (BCA assay) was found to be 60 μg/ml/nM of NPs, equaling on average 400 antibodies per SERRS NP. The Raman spectrum (Figure 2c) of a 100 pM SERRS NP suspension acquired with parameters used for in vivo imaging showed the characteristic peaks confirming the presence of the Raman reporter molecule (IR 780 dye). The limit of detection (Figure 2d & Figure S1) of SERRS NPs in solution, similarly obtained using conditions amenable to in vivo imaging, was 10 fM, two orders of magnitude lower than previously reported non-resonant SERS NPs 33. The absorbance spectrum of the SERRS NPs was obtained by UV-vis spectroscopy (Figure S2) and showed their maximum absorbance to be at 740 nm, with high absorbance in the wavelength of the Raman interrogation laser (785 nm). The SERRS NPs were found to be very photostable upon constant illumination with the interrogation laser in tissue lysates (Figure S3), retaining strong signal intensities for at least 15 minutes. These SERRS NPs have already been reported to be highly stable in biological solutions such as mouse serum, with no visible aggregation even after incubation for 72 hours 11, 13. Additionally, they have also been found to be nontoxic after extensive cytotoxicity studies 34.
Figure 2. In vitro characterization of SERRS NPs.

(a) Transmission electron micrographs showing SERRS NPs, which have an average diameter of 70±24 nm for gold nanostar cores and 122±22 nm for final silicated NPs. (b) Dynamic light scattering (DLS) showing hydrodynamic diameter of 79, 141, and 164 nm with polydispersity index of 0.32, 0.17, and 0.20 and zeta potential (ζ) of −37.3±1.2 mV, −45.9±1.4 mV and −16.6±0.7 mV for gold cores, SERRS-NPs, and antibody conjugated SERRS-NPs, respectively. (c) SERRS spectrum showing characteristic peak at 960 cm−1 confirming the presence of Raman reporter molecule (IR 780 dye) in αTF-SERRS-NPs. (d) Limit of detection of SERRS NPs in solution was approx. 10 fM using conditions amenable to in vivo imaging. (e) Flow cytometry indicating no difference in TF binding affinity or specificity between αTF-A647 and αTF-SERRS-NPs-A647 in comparison to isotype and blocking controls.
The targeting capability of αTF-SERRS-NPs was evaluated in vitro by flow cytometry performed with MDA231-LM2 cells (LM2 for short; a lung metastatic subline derived from the MDA-MB-231 breast cancer cell line). To enable flow cytometry detection, both αTF-SERRS-NPs and IgG-SERRS-NPs were subjected to additional conjugation with Alexa Fluor® 647 C2-maleimide to produce αTF-SERRS-NPs-A647 and IgG-SERRS-NPs-A647. Similarly, for positive and negative controls, the antibodies were directly conjugated to Alexa Fluor® 647 via (N-hydroxy-succinimide) NHS chemistry to yield ALT-836-A647 and IgG-A647. For blocking studies, before incubation with αTF-SERRS-NPs-A647, cells were initially incubated with unconjugated ALT-836 (500 μg/ml) for 1 hr at RT. The flow cytometry results (Figure 2e & Figure S4) indicated increased mean fluorescence intensity (>1000 fold higher than the untreated cells and negative control) of LM2 cells incubated with αTF-SERRS-NPs-A647. This increased fluorescence intensity was comparable to the result obtained from a positive control (LM2 cells incubated with ALT-836-A647). In comparison, cells incubated with αTF-SERRS-NPs-A647 after “blocking” or with IgG-SERRS-NPs-A647 displayed little or no change in fluorescence intensity. The above result demonstrates that αTF-SERRS-NPs-A647 specifically bind to TF on the LM2 cells. Absence of fluorescence signal from LM2 cells incubated with IgG-SERRS-NPs-A647 and IgG-A647 suggests minimal non-specific binding of the covalently functionalized SERRS NPs in cell culture. The TF specificity of αTF-SERRS-NPs-A647, as evidenced by the above in vitro results warranted further in vivo investigation to evaluate its possible application in detecting and imaging BC metastasis.
In vivo Raman imaging and biodistribution studies
The in vivo targeted Raman imaging capability of αTF-SERRS-NPs-A647 was evaluated in a breast cancer experimental lung metastasis mouse model, established by intravenous (i.v.) injection of 0.5 × 105 LM2 cells in 150 μL PBS into 4–5 week old female athymic nu/nu mice via tail vein according to protocol (#06-07-011) approved by the Institutional Animal Care and Use Committee of Memorial Sloan Kettering Cancer Center. The extent of metastasis (Figure 3a) in the murine lung was monitored noninvasively by serial bioluminescence imaging (BLI) after intraperitoneal injection of D-luciferin (150 mg/kg of mouse body weight) on day 0, day 1, day 3, day 7 and subsequently on a weekly basis. It is to be noted that BLI signal was only visible around the thoracic area of these mice as reported by prior literature on LM2 cells which have been selectively sub-populated with genes promoting lung metastatogenicity 30. When this BLI signal was substantially strong (typically 4 weeks after injection) without any adverse symptoms such as body weight loss, or difficulty in breathing, the metastatic mice were administered either αTF-SERRS-NPs or IgG-SERRS-NPs at a concentration of 3.5 nM in 150 μL of PBS (10 mM, pH 7.4) via tail vein injection. For blocking studies, metastatic mice (n=3) were injected with 2 mg of ALT-836 two hours prior to administration of αTF-SERRS-NPs to evaluate in vivo TF specificity. Similarly, a group of five healthy mice were intravenously administered αTF-SERRS-NPs and used as healthy controls. After 18–24h circulation period, mice were injected with 5 mg luciferin per mouse and subjected to BLI. Subsequently, mice were euthanized by carbon dioxide asphyxiation and subjected to in situ Raman imaging of the lungs in the exposed thoracic region and ex vivo Raman imaging of resected tissues (lungs, liver, and spleen) using an InVia (Renishaw) Raman microscope equipped with a 785 nm diode laser (300 mW cm−2) and a 1-inch charge-coupled device detector with a spectral resolution of 1.07 cm−1. The Raman maps visualizing the presence of the Raman signature of the SERRS NPs were derived via a quantitative direct classical least-squares (DCLS) model where the DCLS scores roughly correspond to the physical Raman counts detected (Figure S5). As can be seen from the in vivo and ex vivo Raman maps (Figure 3b), there was low background uptake of αTF-SERRS-NPs in the lungs of healthy mice, with DCLS scores typically around 50 and consistently below 100. Similarly, low background signal was also obtained with regard to the uptake of IgG-SERRS-NPs in the lungs of metastatic mice. In comparison, enhanced uptake of αTF-SERRS-NPs was clearly visible in the lungs of metastatic mice, where the DCLS model produced scores in the range of 150, featuring loci with intensities as high as 250. The intensity of Raman signal was significantly higher in the case of lungs of metastatic mice administered αTF-SERRS-NPs compared to the isotype control and healthy control. High intensity SERRS signal was observed in the liver and spleen of all animals. Since there was no BLI signal seen in any organ other than lungs of the metastatic mice, and because mouse liver and spleen are not known to express human TF, the presence of this signal was largely unrelated to TF expression and attributed to the known sequestration of nanoparticles by the reticuloendothelial system (RES).
Figure 3. Bioluminescence & Raman Imaging of Breast Cancer Lung metastases.

(a) The extent of metastases in murine lungs as indicated by noninvasive serial bioluminescence imaging (BLI) obtained after intraperitoneal (i.p.) injection of D-luciferin, (b) in vivo and ex vivo Raman maps showing SERRS signal in lungs of metastatic mice (n=4) administered with αTF-SERRS NPs as opposed to lungs of metastatic mice administered with isotype control (IgG-SERRS NPs). Negligible SERRS signal from blocking controls (metastatic mice (n=3) injected with 2 mg of ALT-836 two hours prior to administration of αTF-SERRS NPs) and healthy control (healthy mice (n=4) intravenously administered with αTF-SERRS NPs) indicates TF specificity of αTF-SERRS NPs. (c) Biodistribution as determined by ICP-MS performed on organs of interest harvested from both healthy and metastatic mice intravenously injected with αTF-SERRS NPs in (n=3) showed >3-fold higher uptake of gold in metastatic lungs compared to healthy lungs.
Biodistribution analysis in the form of Inductively Coupled Plasma Mass Spectrometry (ICP-MS) for quantitation of gold was performed by the Brooklyn College Environmental Sciences Analytical Center on a PerkinElmer Elan DRC-e ICP-MS instrument on organs of interest harvested from both healthy and metastatic mice (n=3 each) according to a previously published procedure 35. The results show that SERRS NPs were primarily taken up by the organs of the RES, such as the liver and spleen (Figure 3c). The metastatic lungs showed average uptake 3.1 %ID/g of αTF-SERRS-NPs which was >3 fold higher compared to healthy lungs and similar organs of importance such as heart and kidney. The above result is in conformity with the earlier reported data by Blanco et al. 36, pertaining to nanoparticles of similar size, shape, and surface charge with typical low uptake in lungs but rapid clearance from the circulation by Kupffer cells of liver and macrophages of spleen after intravenous administration. Higher specific accumulation of αTF-SERRS-NPs in metastatic lungs suggests that the high intensity Raman signal foci seen in both in vivo and ex vivo Raman maps are likely due to the ability of the αTF-SERRS-NPs to home on TF overexpressed by the metastatic nodules.
2D & 3D Histology
The results from the in vivo and ex vivo Raman maps along with biodistribution studies were further corroborated by histological analysis. Freshly excised lung tissues from both healthy and metastatic mice were processed for paraffin embedding and tissue sections (5 μm thickness) obtained from them were subjected to immunohistochemical (IHC) staining either with 0.5 μg/ml CD31 rat anti-mouse monoclonal or 1 μg/ml CD142 rabbit anti-human TF polyclonal antibody. Adjacent tissue sections were also subjected to H&E staining. As can be seen from Figure 4a, H&E staining clearly revealed the presence of nodules in the lungs of metastatic mice whose locations were completely identical to CD142 staining pattern representing TF expression in these metastatic lesions. As expected, lungs of healthy mice showed neither presence of metastatic lesions nor expression of TF. In addition, IHC staining for mouse CD31 was visible throughout the metastatic lungs except in the areas containing metastatic lesions. Mouse CD31 is expressed by endothelial cells of murine vasculature. The absence of mouse CD31 staining can be explained by the human provenance of these metastatic lesions. The above results confirm the specificity and targeted imaging capability of αTF-SERRS-NPs in pulmonary BC micrometastases.
Figure 4. Immunohistochemical studies.

(a) H&E, anti–mouse-CD31 and anti–human-TF (CD142) staining of healthy and metastatic lungs (Scale bars = 2 mm). (b) Direct comparison of BLI, Raman map of αTF-SERRS-NPs, and 3D H&E in a mouse with breast cancer lung metastases. Note the high degree of correlation between the Raman map, BLI, and 3D histology.
Due to the clinical significance of being able to identify the precise spatial location of these metastatic lesions, it was important to validate the results obtained from ex vivo Raman maps of metastatic lungs by comparing them with their corresponding BLI and H&E images. For this purpose, the mice with lung metastases were subjected to intraperitoneal injection of D-luciferin and were euthanized five minutes later. Subsequently, the metastatic lungs were resected from the mouse, placed on glass slides and subjected to BLI imaging to image the locations of the metastatic nodules. Afterwards, without disturbing the position of the lungs, these glass slides were subjected to Raman imaging and later processed for H&E. As traditional histological analysis relies on very thin (5 μm) slices, it cannot show tumors that may be deeper within the tissue. To remedy this, we performed three-dimensional (3D) histological reconstruction of the metastatic lungs using serially sectioned H&E slides (n=40, every 6th tissue section) (Figure S6). The ex vivo Raman maps could detect the precise locations of tumor lesions and the result was comparable with both BLI and 3D histology (H&E) (Figure 4b). Pulmonary metastases as small as 200 μm were identified and delineated by Raman imaging, with histological confirmation. In addition to the histologically confirmed cancerous lesions, the Raman scans also revealed high-intensity foci as small as 50 μm (Figure 4b). The sensitivity and accuracy obtained by Raman imaging support the suitability of both TF as a target and SERRS NPs as an effective contrast agent for imaging BC lung micrometastases.
Conclusion
We report the development, characterization, and utilization of ultra-bright αTF-SERRS-NPs as an imaging probe for not only detecting but also delineating microscopic pulmonary metastatic lesions. This was possible due to rapid, persistent and TF specific uptake of αTF-SERRS NPs into the metastatic lesions, validated by several in vitro, in vivo and ex vivo experiments. To the best of our knowledge, this is the first example of the successful use of Raman nanoparticles for the imaging of lung metastases. In addition to providing clinical validation of this novel prognostic tool, the present findings shed new light on TF as a possible target for imaging and treatment of BC lung metastasis. Further advancement of this diagnostic probe could result in the development of novel minimally invasive (robotically-assisted) endoscopic systems for resection of lung metastases.
Supplementary Material
Acknowledgments
We thank the MSKCC Electron Microscopy, Molecular Cytology and Flow Cytometry core facilities for technical support. Matthew Brendel assisted with the 3D histology reconstruction. The following funding sources (to M.F.K.) are acknowledged: NIH R01 EB017748 and K08 CA16396; M.F.K. is a Damon Runyon-Rachleff Innovator supported (in part) by the Damon Runyon Cancer Research Foundation (DRR-29-14); Pershing Square Sohn Prize by the Pershing Square Sohn Cancer Research Alliance; MSKCC Center for Molecular Imaging & Nanotechnology (CMINT) Grant; MSKCC Technology Development Grant; Mr. William H. and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research and The Experimental Therapeutics Center of Memorial Sloan Kettering Cancer Center. Acknowledgments are also extended to the grant-funding support provided by the MSKCC NIH Core Grant (P30-CA008748).
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Weigelt B, Peterse JL, van ‘t Veer LJ. Nat Rev Cancer. 2005;5:591–602. doi: 10.1038/nrc1670. [DOI] [PubMed] [Google Scholar]
- 3.Husemann Y, Geigl JB, Schubert F, Musiani P, Meyer M, Burghart E, Forni G, Eils R, Fehm T, Riethmuller G, Klein CA. Cancer Cell. 2008;13:58–68. doi: 10.1016/j.ccr.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 4.Lobrano MB. Oncologist. 2006;11:774–779. doi: 10.1634/theoncologist.11-7-774. [DOI] [PubMed] [Google Scholar]
- 5.Woodard PK, Slone RM, Sagel SS, Fleishman MJ, Gutierrez FR, Reiker GG, Pilgram TK, Jost RG. Radiology. 1998;209:705–709. doi: 10.1148/radiology.209.3.9844662. [DOI] [PubMed] [Google Scholar]
- 6.van den Brekel MW. Eur J Radiol. 2000;33:230–238. doi: 10.1016/s0720-048x(99)00145-x. [DOI] [PubMed] [Google Scholar]
- 7.Kruskal JB, Kane RA. Radiographics. 2006;26:1067–1084. doi: 10.1148/rg.264055120. [DOI] [PubMed] [Google Scholar]
- 8.Mui LW, Pursell LJ, Botwinick IC, Allendorf JD, Chabot JA, Newhouse JH. J Ultrasound Med. 2014;33:47–51. doi: 10.7863/ultra.33.1.47. [DOI] [PubMed] [Google Scholar]
- 9.Fazel R, Krumholz HM, Wang Y, Ross JS, Chen J, Ting HH, Shah ND, Nasir K, Einstein AJ, Nallamothu BK. N Engl J Med. 2009;361:849–857. doi: 10.1056/NEJMoa0901249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang B, Law MW, Khong PL. Radiology. 2009;251:166–174. doi: 10.1148/radiol.2511081300. [DOI] [PubMed] [Google Scholar]
- 11.Andreou C, Neuschmelting V, Tschaharganeh DF, Huang CH, Oseledchyk A, Iacono P, Karabeber H, Colen RR, Mannelli L, Lowe SW, Kircher MF. ACS Nano. 2016;10:5015–5026. doi: 10.1021/acsnano.5b07200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harmsen S, Bedics MA, Wall MA, Huang R, Detty MR, Kircher MF. Nat Commun. 2015;6:6570. doi: 10.1038/ncomms7570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harmsen S, Huang R, Wall MA, Karabeber H, Samii JM, Spaliviero M, White JR, Monette S, O’Connor R, Pitter KL, Sastra SA, Saborowski M, Holland EC, Singer S, Olive KP, Lowe SW, Blasberg RG, Kircher MF. Sci Transl Med. 2015;7:271ra277. doi: 10.1126/scitranslmed.3010633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi JY, Jang HJ, Shim YM, Kim K, Lee KS, Lee KH, Choi Y, Choe YS, Kim BT. J Nucl Med. 2004;45:1843–1850. [PubMed] [Google Scholar]
- 15.Sasaki R, Komaki R, Macapinlac H, Erasmus J, Allen P, Forster K, Putnam JB, Herbst RS, Moran CA, Podoloff DA, Roth JA, Cox JD. J Clin Oncol. 2005;23:1136–1143. doi: 10.1200/JCO.2005.06.129. [DOI] [PubMed] [Google Scholar]
- 16.Shankar LK, Hoffman JM, Bacharach S, Graham MM, Karp J, Lammertsma AA, Larson S, Mankoff DA, Siegel BA, Van den Abbeele A, Yap J, Sullivan D I National Cancer. J Nucl Med. 2006;47:1059–1066. [PubMed] [Google Scholar]
- 17.Rasey JS, Koh WJ, Evans ML, Peterson LM, Lewellen TK, Graham MM, Krohn KA. Int J Radiat Oncol Biol Phys. 1996;36:417–428. doi: 10.1016/s0360-3016(96)00325-2. [DOI] [PubMed] [Google Scholar]
- 18.Rajendran JG, Mankoff DA, O’Sullivan F, Peterson LM, Schwartz DL, Conrad EU, Spence AM, Muzi M, Farwell DG, Krohn KA. Clin Cancer Res. 2004;10:2245–2252. doi: 10.1158/1078-0432.ccr-0688-3. [DOI] [PubMed] [Google Scholar]
- 19.Padhani AR, Krohn KA, Lewis JS, Alber M. Eur Radiol. 2007;17:861–872. doi: 10.1007/s00330-006-0431-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gasparini G, Brooks PC, Biganzoli E, Vermeulen PB, Bonoldi E, Dirix LY, Ranieri G, Miceli R, Cheresh DA. Clin Cancer Res. 1998;4:2625–2634. [PubMed] [Google Scholar]
- 21.Bello L, Francolini M, Marthyn P, Zhang J, Carroll RS, Nikas DC, Strasser JF, Villani R, Cheresh DA, Black PM. Neurosurgery. 2001;49:380–389. doi: 10.1097/00006123-200108000-00022. discussion 390. [DOI] [PubMed] [Google Scholar]
- 22.Beer AJ, Haubner R, Sarbia M, Goebel M, Luderschmidt S, Grosu AL, Schnell O, Niemeyer M, Kessler H, Wester HJ, Weber WA, Schwaiger M. Clin Cancer Res. 2006;12:3942–3949. doi: 10.1158/1078-0432.CCR-06-0266. [DOI] [PubMed] [Google Scholar]
- 23.Callander NS, Varki N, Rao LV. Cancer. 1992;70:1194–1201. doi: 10.1002/1097-0142(19920901)70:5<1194::aid-cncr2820700528>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 24.Bromberg ME, Konigsberg WH, Madison JF, Pawashe A, Garen A. Proc Natl Acad Sci U S A. 1995;92:8205–8209. doi: 10.1073/pnas.92.18.8205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bromberg ME, Sundaram R, Homer RJ, Garen A, Konigsberg WH. Thromb Haemost. 1999;82:88–92. [PubMed] [Google Scholar]
- 26.Jiang X, Bailly MA, Panetti TS, Cappello M, Konigsberg WH, Bromberg ME. J Thromb Haemost. 2004;2:93–101. doi: 10.1111/j.1538-7836.2004.00545.x. [DOI] [PubMed] [Google Scholar]
- 27.Kasthuri RS, Taubman MB, Mackman N. J Clin Oncol. 2009;27:4834–4838. doi: 10.1200/JCO.2009.22.6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiao JA, Kelly AB, Marzec UM, Nieves E, Acevedo J, Burkhardt M, Edwards A, Zhu XY, Chavaillaz PA, Wong A, Wong JL, Egan JO, Taylor D, Rhode PR, Wong HC. Thromb Haemost. 2010;103:224–233. doi: 10.1160/TH09-06-0400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morris PE, Steingrub JS, Huang BY, Tang S, Liu PM, Rhode PR, Wong HC. BMC Pulm Med. 2012;12:5. doi: 10.1186/1471-2466-12-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, Massague J. Nature. 2005;436:518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chakravarty R, Valdovinos HF, Chen F, Lewis CM, Ellison PA, Luo H, Meyerand ME, Nickles RJ, Cai W. Adv Mater. 2014;26:5119–5123. doi: 10.1002/adma.201401372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yuan YY, Mao CQ, Du XJ, Du JZ, Wang F, Wang J. Adv Mater. 2012;24:5476–5480. doi: 10.1002/adma.201202296. [DOI] [PubMed] [Google Scholar]
- 33.Jokerst JV, Cole AJ, Van de Sompel D, Gambhir SS. ACS Nano. 2012;6:10366–10377. doi: 10.1021/nn304347g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thakor AS, Luong R, Paulmurugan R, Lin FI, Kempen P, Zavaleta C, Chu P, Massoud TF, Sinclair R, Gambhir SS. Sci Transl Med. 2011;3:79ra33. doi: 10.1126/scitranslmed.3001963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karabeber H, Huang R, Iacono P, Samii JM, Pitter K, Holland EC, Kircher MF. ACS Nano. 2014;8:9755–9766. doi: 10.1021/nn503948b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blanco E, Shen H, Ferrari M. Nat Biotechnol. 2015;33:941–951. doi: 10.1038/nbt.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.