

A 13-year-old boy presented to a pediatric clinic with progressive decreased movements. His vision had been decreasing for 8 years, and he had been unable to walk for 3 years.
The patient was born to a healthy nonconsanguineous couple. His mother's pregnancy and his natal history were unremarkable. During infancy his ability to hold his neck and to sit with and without support was delayed. Motor and speech delay were noticed at 2 years, and at 4 years his night vision began to deteriorate, and he had frequent falls with subsequent injuries. An ophthalmologist diagnosed retinitis pigmentosa (RP). At school, his behaviour and academic ability were normal.
At the age of 10, the patient developed spasticity, decreased movements of his lower limbs and dystonic postural abnormalities. The spasticity and dystonia were progressive and disabling enough to restrict him to a wheelchair. His speech deteriorated, and he gradually stopped talking. His parents also observed an intellectual decline. The patient was seen by a number of physicians, none of whom was able to establish a satisfactory diagnosis, until he presented to the pediatric clinic. Funduscopy confirmed the finding of RP, and an MRI showed marked bilateral high-signal intensities surrounding the globus pallidus — the “eye of the tiger” sign that is characteristic of pantothenate kinase-associated neurodegeneration (PKAN) (Fig. 1left; the right image shows an age-matched normal MRI).1
Figure 1.
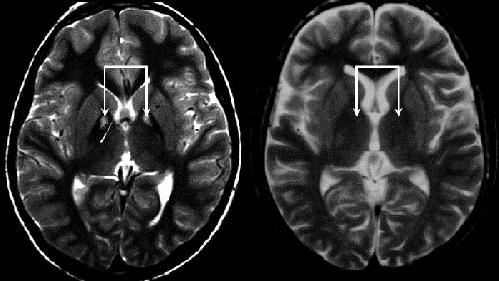
PKAN, formerly known as Hallervorden–Spatz disease, was first described in 1922.2 It is an autosomal recessive neurodegenerative disorder associated with accumulation of iron in the basal ganglia,1 and it has 2 major forms: early onset and late onset.2 Early-onset PKAN is rapidly progressive and characterized by gait impairment, as seen in our patient. Dystonia develops, leading to loss of ambulation and restriction of activities by mid-adolescence. Intellectual impairment, RP and optic atrophy are also associated with the disease.2 Late-onset PKAN presents by 20 years, manifesting a progressive picture of dementia, parkinsonism, dystonia, anarthria, aphonia and incontinence.2 The pathognomonic “eye of the tiger” sign is seen in both forms of the disease. No prevalence studies have yet been done on this disease or its different forms.
PKAN has been mapped to chromosome 20p12.3-p13.3 Mutations in PANK2, the gene encoding the enzyme pantothenate kinase 2, have been demonstrated in the majority of patients with PKAN.1,3 The presence of mutation in PANK2 leads to a genetic diagnosis and makes presymptomatic testing of family members possible, but an MRI can still be considered the “gold standard” in diagnosing early-onset PKAN.1
The differential diagnosis for progressive dystonia in children includes idiopathic generalized dystonia, dopa-responsive dystonia, myoclonic dystonia, metachromatic leukodystrophy, Niemann–Pick disease type C and Lesch–Nyhan syndrome.4
There is no specific treatment. Some patients show residual activity of the enzyme, but the benefit of pantothenate supplementation in ameliorating symptoms has not yet been proven. However, levodopa, anticholinergics and intrathecal baclofen have been shown to improve the patient's quality of life.2
Danish Saleheen Philippe Frossard Mohammed Zeeshan Ozair Mohammad Ali Kazmi Hamza Khalid Department of Biological and Biomedical Sciences Bhojo Khealani Department of Neurology The Aga Khan University Karachi, Pakistan
References
- 1.Hayflick SJ, Westaway SK, Barbara L, Zhou B, Johnson M, Ching K, et al. Genetic, clinical, and radiographic delineation of Hallervorden–Spatz syndrome. N Engl J Med 2003;348:33-40 [DOI] [PubMed]
- 2.Neil Gordon. Pantothenate kinase-associated neurodegeneration (Hallervorden–Spatz syndrome). Eur J Pediatr Neurol 2002;6:243-7. [DOI] [PubMed]
- 3.Zhou B, Westaway SK, Levinson B, Johnson M, Gitshier J, Hayflick SJ. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden–Spatz syndrome. Nat Genet 2001;28:345-9. [DOI] [PubMed]
- 4.Assmann B, Surtees R, Hoffmann GF. Approach to the diagnosis of neurotransmitter diseases exemplified by the differential diagnosis of childhood-onset dystonia. Ann Neurol 2003;54(Suppl 6):S18-24. [DOI] [PubMed]