

The limb-girdle muscular dystrophies (LGMD) comprise a large group of genetic disorders that lead to shoulder and pelvic girdle muscle weakness. Although these disorders are grouped together based on phenotypic presentation, there is extensive genetic variability among them. The dysfunctional protein within each disorder may be found at any level of the muscle fiber structure. Mutations to proteins within the extracellular matrix, including collagen VI and laminin α2 proteins, can disrupt the normal basal lamina architecture and lead to muscle weakness in a limb-girdle distribution.1 We present an interesting case with a novel mutation in the laminin α2 (LAMA2) gene, which has not been reported previously; the patient has a mixed clinical phenotype without contractures.
CASE REPORT
A 6-year-old boy who had recently emigrated from Southeast Asia presented with progressive muscle weakness since birth. His mother reported decreased fetal movements during pregnancy when compared to her first child. He was born at full term with very low muscle tone and had feeding difficulties as a neonate. He had marked motor developmental delay and first walked at the age of 3 years. His language, social, and cognitive development were normal. Of note, there was consanguinity between his parents, as they were first cousins. On physical examination, he had profound shoulder and pelvic girdle muscle weakness (3/5 muscle power on Medical Research Council scale), with relative sparing of the distal muscles. There was also significant proximal muscle atrophy and hypotonia. Contractures were not present, and the deep tendon reflexes were diminished. A prior workup at an outside hospital revealed consistently high creatine phosphokinase (CK) levels (range 1,100 to 1,200 IU/L) since 1 year of age. Chromosome karyotype was normal. Cardiac workup was normal. Pulmonary function tests revealed a restrictive pattern. A left vastus lateralis muscle biopsy was performed that was suggestive of chronic muscular dystrophy (figure 1). A congenital muscular dystrophy gene sequencing panel identified 2 homozygous nucleotide variants of unknown significance in the collagen VI α1 (COL6A1) and laminin α2 (LAMA2) genes: c.1823-8G>A (ivs29-8G>A) and c.713C>A (p.aA238D), respectively. Using current modeling software (polyphen-2 and SIFT), the COL6A1 nucleotide change was suggested to be benign, as it had previously been reported in the general population (communication with Emory Genetics Laboratory, Decatur, Georgia). Since a limb-girdle pattern of weakness has previously been described with both merosin deficiency and collagen VI deficiency, immunofluorescence staining was performed (figure 2). Partial deficiency of laminin α2 (merosin) was observed using 3 different antimerosin antibodies (figure 2, A–F), while normal staining was observed for collagen VI (figure 2, G and H). Brain MRI revealed diffusely abnormal cerebral white matter (figure 3).
Figure 1. Hematoxylin & eosin photomicrographs from the patient's muscle biopsy.

(A, B) These images show wide variation in fiber size (atrophy and hypertrophy), moderately severe endomysial fibrosis and fatty replacement, and a mild degree of ongoing myonecrosis/regeneration (upper left corner of B). The size bar in A is 100 μm for A and B.
Figure 2. Immunofluorescence of muscle biopsy.

(A–H) Anti-laminin α2 immunofluorescence images are from a normal control (A, C, E) and the patient (B, D, F). Partial deficiency of merosin is demonstrated with 3 different antibodies. The 80 kd antibody (clone 5H2; Chemicon, Temecula, CA), the 300 kd antibody (clone Mer3/22B2; Leica, Newcastle, UK), and the 4H8 antibody (Millipore, Billerica, MA) detect different epitopes of laminin α2. G and H are dual label immunofluorescence images of anti-collagen VI (green) and antiperlecan (red). Nuclei in G and H are stained with DAPI (blue). The size bar in A is 100 μm for A–H.
Figure 3. Brain MRI and brain magnetic resonance spectroscopy.
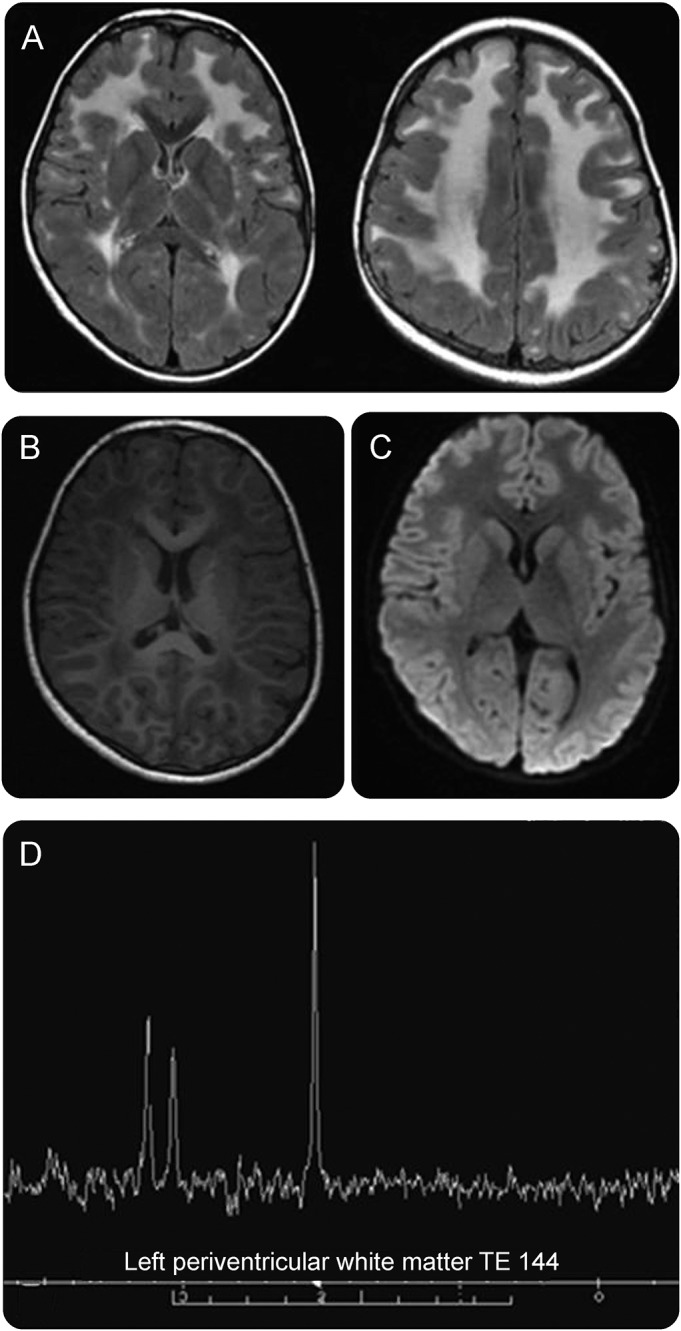
(A–C) Brain MRI. Axial T2 fluid-attenuated inversion-recovery MRI shows white matter hyperintensities in the frontal-parietal regions of the brain with sparing of the corpus callosum, internal capsule, and basal ganglia (A). Subcortical U-fibers and external capsule are also involved. Axial T1 reformatted images show white matter hypointensities in the frontal-parietal regions (B). Diffusion-weighted images show no acute lesions (C). (D) Brain magnetic resonance spectroscopy. Periventricular white matter shows slight elevation in NAA peak with normal choline and creatine peaks.
DISCUSSION
LGMD are a diverse group of disorders that can present with a variety of genotype–phenotype combinations. A single genotypic variation may manifest with a number of different phenotypes and a single phenotype may result from many different genetic abnormalities. This may be due to complex interactions between environmental and genetic factors. Due to this high variability, it is difficult to classify and devise a consistently successful algorithmic approach towards the diagnosis of patients suspected to have LGMD. Conventional diagnostic steps including muscle enzyme levels, electrophysiologic studies, and muscle biopsies may not provide a definitive answer in many cases. Even specialized centers may not be able to identify the pathology in up to 25% of cases. Therefore, utilizing gene sequencing early on during the diagnostic work up may be a more rational approach.2
In our case, novel variants were found in both the LAMA2 and COL6A1 genes, and initial interpretation suggested that either could explain the patient's clinical symptoms of limb-girdle muscle weakness. Abnormal white matter by MRI and partial deficiency of laminin α2 (merosin) by immunofluorescence staining of a muscle biopsy indicated that the LAMA2 variant was very likely to be pathologic. In contrast, muscle biopsy dual label immunofluorescence staining showed normal expression of collagen VI. Furthermore, the COL6A1 variant was later determined to be benign because it is found in more than 1% of the asymptomatic general population (communication with Emory Genetics Laboratory). Here, we review the clinical presentations of collagen VI deficiency and merosin deficiency and how they relate to our patient.
The collagen VI disorders result from mutations in the COL6A1, COL6A2, or COL6A3 genes; they have a broad spectrum of clinical phenotypes. Bethlem myopathy is on the mild end of the spectrum, while Ullrich congenital muscular dystrophy (UCMD) is the most severe form.3 While contractures are prominent in both Bethlem myopathy and UCMD, a limb-girdle distribution of muscle involvement without contractures has been reported in families with COL6A1 and COL6A2 gene mutations.4 On muscle biopsy, these patients had normal collagen VI immunostaining, and instead, had a significant reduction of laminin β1 immunostaining. It was proposed that the mutation in the collagen VI protein caused a disruption of the basal lamina and protein interactions, which then led to abnormalities in the laminin β1 protein. It is possible that the nucleotide change in our patient's COL6A1 gene has also affected the basal lamina and protein interactions; however, the prevalence of our patient's homozygous mutation within the general population makes this conclusion less likely.
Merosin deficiency can present with different patterns of muscle weakness depending on the type and location of mutation, which is similar to collagen VI deficiency. The LAMA2 gene encodes for the α2 subunit of the laminin-2 protein (merosin) and is located on the long arm of chromosome 6 (6q22-q23). There are 3 subunits that comprise the merosin protein: α2, β1, and γ1. Each subunit is encoded by a different gene, and together, the complex is found in the basement membrane of skeletal muscle.5 The merosin protein is important in maintaining the extracellular matrix structure, and mutations in LAMA2 can lead to partial or complete absence of the merosin protein. Two classical forms of merosin deficiency have been described: early-onset and late-onset. The severe, early-onset form is also known as congenital muscular dystrophy (CMD; also known as MDC1A) and presents within the first 6 months of life with severe hypotonia. These patients often have homozygous mutations that lead to the absence of the laminin α2 subunit and complete loss of merosin from muscle and other tissues.6 In contrast, the late-onset form of merosin deficiency has a milder course and typically presents in late childhood or adulthood with a limb-girdle pattern of muscle weakness. Usually there are compound heterozygous mutations in the late-onset form that lead to a partial deficiency of the laminin α2 subunit and reduced merosin immunostaining in muscle. In both the early- and late-onset forms, contractures are classically present, creatinine kinase levels are elevated, and there are T2 hyperintensities seen within brain white matter on MRI.6,7 The above described findings in our patient's MRI are consistent with the typical magnetic resonance brain findings seen in CMD.8,9 It has been proposed that the abnormal T2 white matter signals in CMD may represent increased water content in the white matter rather than a defect in myelination.8 However, an underlying mechanism for this white matter abnormality has not been established.
Our patient is unique in that he has features of both the early- and late-onset forms of merosin deficiency. He has a homozygous LAMA2 mutation and presented with hypotonia at birth, which is consistent with the early-onset form of merosin deficiency. He did not, however, develop contractures or have continued feeding difficulties. Later in childhood, he developed a limb-girdle pattern of weakness and demonstrated a weak immunohistochemical staining pattern for merosin on his muscle biopsy, which is more traditionally seen with the late-onset form of merosin deficiency. One reported case had a similar presentation of hypotonia at birth and became ambulatory at 2 years of age without substantial respiratory or feeding problems; however, contractures were present.6
We present a novel LAMA2 mutation with a clinical presentation of partial merosin deficiency that does not fit into a single classic phenotype. The lack of contractures on examination has rarely been reported and contributes to the uniqueness of this patient. It has been proposed that patients with LAMA2 mutations manifest a spectrum of clinical phenotypes rather than 2 distinct forms.10,11 Our patient supports this proposal, as his clinical course was not typical for either the early- or late-onset form of merosin deficiency.
The absence of contractures with a limb-girdle muscle pattern of weakness should not dissuade physicians from considering the diagnosis of merosin deficiency, as many different phenotypes exist. This further emphasizes the importance of genetic testing early in the diagnostic workup for these patients. It is possible that reaching a diagnosis may not have many clinical implications at this point in time. Nevertheless, it may provide worthwhile information regarding a need to screen other organ systems and direction for counseling, prognostication, and future research.
AUTHOR CONTRIBUTIONS
Marissa Dean: drafting and revising the manuscript, study concept and design. Salman Rashid: revising the manuscript, analysis and interpretation of data. William Kupsky: analysis and interpretation of data. Steven A. Moore: revising the manuscript, analysis and interpretation of data. Huiyuan Jiang: analysis and interpretation of data.
STUDY FUNDING
Steven A. Moore is supported by U54-NS053672 that funds the Iowa, Paul D. Wellstone Muscular Dystrophy Cooperative Research Center.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Laval SH, Bushby KMD. Limb-girdle muscular dystrophies: from genetics to molecular pathology. Neuropathol Appl Neurobiol 2004;30:91–105. [DOI] [PubMed] [Google Scholar]
- 2.Swaiman KF, Ashwal S, Ferriero DM, Schor NF. Swaiman's Pediatric Neurology. 5th ed. Philadelphia: Saunders; 2012. [Google Scholar]
- 3.Lampe AK, Flanigan KM, Bushby KM, Hicks D. Collagen type VI-related disorders [updated 2012 Aug 9]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews [Internet]. Seattle: University of Washington; 2004:1993–2016. [Google Scholar]
- 4.Scacheri PC, Gillanders EM, Subramony SH, et al. Novel mutations in collagen VI genes: expansion of the Bethlem myopathy phenotype. Neurology 2002;58:593–602. [DOI] [PubMed] [Google Scholar]
- 5.Tzu J. Bridging structure with function: structural, regulatory, and developmental role of laminins. Int J Biochem Cell Biol 2008;40:199–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Løkken NN. Muscle & Nerve: LAMA2-related Myopathy: Frequency Among Congenital and Limb-Girdle Muscular Dystrophies. New York: John Wiley & Sons; 2015;52:547. [DOI] [PubMed] [Google Scholar]
- 7.Gavassini BF, Carboni N, Nielsen JE, et al. Clinical and molecular characterization of limb-girdle muscular dystrophy due to LAMA2 mutations. Muscle Nerve 2011;44:703–709. [DOI] [PubMed] [Google Scholar]
- 8.Caro PA, Scavina M, Hoffman E, Pegoraro E, Marks HG. MR imaging findings in children with merosin-deficient congenital muscular dystrophy. AJNR Am J Neuroradiol 1999;20:324–326. [PMC free article] [PubMed] [Google Scholar]
- 9.Geranmayeh F, Clement E, Feng LH, et al. Genotype-phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromuscul Disord 2010;20:241–250. [DOI] [PubMed] [Google Scholar]
- 10.Beytía Mde L. Molecular and Cellular Probes: High Creatine Kinase Levels and White Matter Changes: Clinical and Genetic Spectrum of Congenital Muscular Dystrophies with Laminin Alpha-2 Deficiency. Philadelphia: Elsevier; 2014;28:118. [DOI] [PubMed] [Google Scholar]
- 11.He YY. Congenital muscular dystrophy with primary partial laminin alpha2 chain deficiency: molecular study. Neurology 2001;57:1319. [DOI] [PubMed] [Google Scholar]