

Abstract
AIM
To characterize AXL receptor tyrosine kinase (AXL) expression in relationship to tumor protein P53 (TP53 gene, p53 protein) and its role in tumor invasion and response to therapy.
METHODS
We used 14 cell lines, including 3 isogenic pairs carrying mutant/knockout p53, to gain insight into the relationship between AXL and TP53. These included HCT116, HCT116.p53 mutant, RKO, and RKO.p53-/- lines (all from colon cancers) as well as breast cancer cell lines MCF7 and 1001 (MCF7-p53 mutant clone). HeLa cell line was used as a positive control for epithelial to mesenchymal transition (EMT). AXL expression was determined by Western blotting using rabbit monoclonal antibody clone C89E7. AXL siRNA silencing was performed and followed by collagen invasion assay. Cell viability analysis using the sulforhodamine B assay and the invasion assay were performed after exposure to chemotherapeutic agents (doxorubicin for breast cancer cells; 5FU or irinotecan for colon cancer cells).
RESULTS
We showed that the introduction of p53 mutations or knockout increased expression levels of AXL in isogenic cells compared to the matching p53 wild-type parental cells. Overall, we found a trend for correlation between the potential EMT candidate AXL, p53 alterations, and EMT markers in colorectal and breast cancers. The expression of AXL in RKO cells, a rare colon cancer cell line with inactive Wnt signaling, suggests that the AXL oncogene might provide an alternative genetic pathway for colorectal carcinogenesis in the absence of Wnt signaling activation and TP53 mutation. AXL silencing in the TP53 mutant isogenic cell lines 1001, HCT116.p53 mutant and RKO.P53-/- was > 95% efficient and the silenced cells were less invasive compared to the parental TP53 wild-type cells. AXL silencing showed a subtle trend to restore colon cancer cell sensitivity to 5FU or irinotecan. Importantly, AXL expressing cells developed more invasive potential after exposure to chemotherapy compared to the AXL-silenced cells.
CONCLUSION
AXL is influenced by p53 status and could cause the emergence of aggressive clones after exposure to chemotherapy. These findings could have applications in cancer management.
Keywords: AXL, Breast cancer, Chemotherapy, Colon cancer, Invasion
Core tip: AXL receptor tyrosine kinase (AXL) is emerging as an attractive molecular target in cancer therapy. We showed that it is regulated by TP53 in colon and breast cancer cells, and it contributes to epithelial to mesenchymal transition and response to therapy in these tumors. We also showed that it could be linked to other carcinogenic pathways, such as the Wnt/β-catenin signaling pathway in colorectal cancer. These interactions should be considered carefully when designing AXL based therapy, because AXL could trigger the emergence of aggressive clones after inappropriate therapy.
INTRODUCTION
Colorectal cancer is the third most common cancer in men (746000 cases, 10.0% of the total) and the second most common in women (614000 cases, 9.2% of the total) worldwide. Additionally, the latest estimates indicate that colorectal cancer is the third leading cause of cancer-related deaths in the United States[1]. Mortality is higher (52%) in the less developed regions of the world[1]. Colorectal cancers in regions/ethnicities other than the West, such as the Middle East, are often associated with intriguing clinical characteristics. Examples include a younger age at onset, advanced stage at presentation, and poor prognosis[2]. Studies have identified contrasting molecular features in specific subsets of colorectal cancers including those that occur in the Middle East[3]. These data highlight the need for novel molecular therapeutic targets to enable the design of personalized therapy for treatment of colorectal cancer. The receptor tyrosine kinases are attractive targets in this regard[4,5].
AXL receptor tyrosine kinase (AXL) is a receptor tyrosine kinase in the TAM family (Tyro3, AXL and Mer). It transduces signals from the extracellular matrix into the cytoplasm by binding to its main ligand: the growth factor “growth arrest-specific 6 (Gas6)”[6]. Ligand binding induces dimerization and autophosphorylation of AXL, which then binds and induces tyrosine phosphorylation of PI3-kinase, GRB2, PLCG1, LCK and PTPN11. Other downstream substrate candidates for AXL are CBL, NCK2, SOCS1 and TNS2. Recruitment of GRB2 and PI3-kinase regulatory subunits by AXL leads to the downstream activation of the AKT kinase. Thus, AXL signaling plays a role in a wide array of processes, including cell survival, cell proliferation, migration, invasion and epithelial to mesenchymal transition (EMT)[7]. AXL is considered a proto-oncogene that is overexpressed in lung, breast, ovarian, gastric, pancreatic and prostate cancers. It was first cloned from chronic myelogenous and lymphoblastic leukemia cells[8]. AXL is regulated by tumor suppressor microRNAs, such as miR-34a[9]. Furthermore, AXL is associated with increased metastasis potential[10,11], poor prognosis[6,12,13] and resistance to therapy[14-16] in many cancer types. More recently, TAM receptors were shown to foster immune escape through regulation of PD-L1 in breast cancer cells[17].
Many oncogenic mechanisms mediated by AXL, such as EMT, are known to be controlled by tumor protein P53 (TP53 gene, p53 protein), which often induces apoptosis in invading cancer cells in a hostile micro-environment. TP53 is one of the most commonly mutated/altered genes involved in colorectal carcinogenesis[18,19]. Previous work suggested a relationship between AXL and the p53/miR-34a axis in B-cell chronic lymphocytic leukemia[20]. Therefore, we hypothesized that AXL could be controlled by TP53 in colorectal cancers where a substantial fraction of tumors has mutant/altered TP53. Additionally, many of the downstream mechanisms of AXL remain unclear. Based on our recent data[21], we hypothesized that AXL might activate Wnt signaling and play an important role in development of some rare subsets of colon cancer when Wnt signaling activity is lacking[21].
Therefore, we sought to determine how AXL expression levels are affected by TP53 in colorectal and breast cancers and to address its role in response to chemotherapy.
MATERIALS AND METHODS
Cell lines and cell culture
A total of 14 cell lines were used. The breast cancer cell lines were MCF7, 1001 (a Tumor Necrosis Factor (TNF)-resistant/TP53 mutant clone from MCF7 parental cell line), CAL-51, MDA-MB-231, MDA-MB-361, ZR-75-1, T47D and BT-549. Colon cancer cell lines were HCT116, HCT116.p53 mutant, RKO, RKO.P53-/- and SW480. A HeLa cell line was used as a positive control for EMT. The abovementioned cell lines were cultured in their corresponding growth media (Table 1), maintained at 37 °C in a humidified 5% CO2 incubator. The cells were split when their confluency reached 80%-90% according to supplier instructions. Sources and maintenance conditions of the cell lines are detailed in Table 1. The origin and p53 status of the cells were detailed on the commercial supplier data sheet, the ATCC data sheets/website, the Cancer Cell Line Encyclopedia (CCLE), and/or the supplied references in Table 2.
Table 1.
Cell lines sources and maintenance
| Cell line | Source | Media | FBS % | Additives |
| MCF7 | France1 | DMEM | 10% | 1% P/S |
| 1001 | France | DMEM | 10% | 1% P/S |
| ZR-75-1 | Helsinki2 | RPMI 1640 | 10% | 1% P/S + Sodium pyruvate (1 mmol/L) |
| RKO | Horizon3 | RPMI 1640 | 10% | 1% P/S + Sodium pyruvate (1 mmol/L) |
| RKO.p53-/- | Horizon | RPMI 1640 | 10% | 1% P/S + Sodium pyruvate (1 mmol/L) |
| HCT116 | Horizon | RPMI 1640 | 10% | 1% P/S + Sodium pyruvate (1 mmol/L) |
| HCT116.p53 | Horizon | DMEM | 10% | 1% P/S + 50% F12 |
| HeLa | France | DMEM | 10% | 1% P/S |
| MDA-MB-361 | France | DMEM | 10% | 1% P/S |
| MDA-MB-231 | France/ATCC | DMEM | 10% | 1% P/S + 50% F12 + insulin (10 mg/mL in 25 mmol/L HEPES) + hydrocortisone (0.5 μg/mL) |
| CAL-51 | Helsinki | DMEM | 20% | 1% P/S |
| T47D | France | DMEM | 10% | 1% P/S + 50% F12 + EGF (20 ng/mL) |
| BT-549 | Helsinki | RPMI 1640 | 10% | 1% P/S + Sodium pyruvate (1 mmol/L) + insulin (10 mg/mL in 25 mmol/L HEPES) |
| SW480 | Helsinki | DMEM | 10% | 1% P/S + 50% F12 + insulin (10 mg/mL in 25 mmol/L HEPES) + hydrocortisone (0.5 μg/mL) + EGF (20 ng/μL) |
France: Kind Gift from Professor Salem Chouaib, Institute Gustave Roussy, France;
Helsinki: Kind gift from Professor Paivi peltomaki; Department of Medical Genetics, Helsinki University, Finland;
Horizon Discovery, Cambridge, United Kingdom.
Table 2.
Cell lines origin and p53 status
| Cell lines | Cell line description1 | p53 status1 |
| Breast cancer cell lines | ||
| MCF7 | Adenocarcinoma of the mammary gland, derived from 69-year-old female, cells were obtained from metastatic site; pleural effusion | Wild-type[34] |
| 1001 | 1001 is derived from its parental MCF7 (MCF7/R-A1; which are cells exposed to increasing dose of recombinant TNF, transfected by p55 TNF receptor cDNA, Mutation in R280K)[35] | TNF resistant associated with loss of p53 function[35] |
| CAL51 | Adenocarcinoma isolated from a malignant pleural effusion of a 44-year-old female with metastatic breast cancer, normal karyotype with genetic stability[36] | Wild-type[37] |
| ZR-75-1 | Ductal carcinoma of the mammary gland, derived from 63-year-old female, cells were obtained from metastatic site: ascites | Wild-type |
| MDA-MB-361 | Adenocarcinoma of the mammary gland, derived from 40-year-old female, cells were obtained from metastatic site; Brain | Wild-type |
| T47D | ductal carcinoma of the mammary gland, derived from 54-year-old female, cells were obtained from metastatic site; pleural effusion | Heterozygous mutant. |
| MDA-MB-231 | Adenocarcinoma of the mammary gland, derived from 51-year-old female, cells were obtained from metastatic site; pleural effusion. | Homozygous mutant |
| BT549 | Ductal carcinoma of the mammary gland, derived from 72-year-old female, cells were obtained from mammary gland. | Homozygous mutant |
| Colon Cancer Cell lines | ||
| RKO | poorly differentiated colon carcinoma cell line developed by Michael Brattain | Wild-type. |
| RKO-p53 | RKO parental cell line double negative mutant for p53 | Homozygous knock out2 |
| HCT 116 | Colorectal carcinoma of Adult Male derived from primary tumor site | Wild-type |
| HCT 116.p53 | HCT116 parental cell line with hemizygous p53 mutation and knock out of the homologous p53 | R284w/-2 |
| SW480 | Dukes' type B, colorectal adenocarcinoma, derived from 50-year-old male, cells obtained from primary tumor site | Homozygous mutant |
| Positive control for EMT | ||
| HeLa | Cervical adenocarcinoma of 31-year-old female derived from primary tumor site | Wild-Type but HPV inactivated[34] |
The cell line origin and the p53 status were from ATCC data sheets/website, the Cancer Cell Line Encyclopedia or the indicated references;
As per the source/supplier: Horizon Discovery, Cambridge, United Kingdom.
Total protein extraction and Western blot analysis
Cell lysates were made using RIPA buffer (150 mmol/L NaCl; 1% NP-40; 0.5% deoxycholic acid; 0.1% SDS; 50 mmol/L Tris-HCl pH 7.6 and 1 × protease/phosphatase inhibitors (Cell Signaling Technology). Protein concentration was quantified using the BCA protein assay (Thermo Fisher Scientific). Approximately 30 μg of total protein was subjected to SDS-PAGE (7.5% resolving gel, 4% stacking gel). The gel, with the separated proteins, was transferred to a PVDF membrane (Bio-Rad) using a Trans-Blot® TurboTM Blotting system (Bio-Rad) according to the standard transfer protocol. The membrane was blocked for 1 h at room temperature using 5% non-fat dry milk (Sigma-Aldrich) in 1 × TBST (TBS + 1% tween), followed by incubation with the primary antibody overnight at 4 °C on a shaker. The primary antibodies were against AXL (clone C89E7, rabbit monoclonal antibody, Cell Signaling), beta-actin (clone 13E5, rabbit, rabbit monoclonal antibody, Cell Signaling) and E-Cadherin (clone 24E10, rabbit monoclonal antibody, Cell Signaling). All primary antibodies were used at a 1:1000 dilution. Next, the membrane was washed with 1 × TBST 3 times for 10 min each. Then, it was incubated with horseradish peroxidase labeled secondary antibody for 1 h at room temperature (anti-rabbit IgG HRP-linked antibody at dilution of 1:2000, Cell Signaling). After washing with 1 × TBST for 10 min 3 times, the membranes were incubated with HRP labeled substrate (Pierce™ ECL Western Blotting Substrate) for 1 min, exposed to film (Kodak Cl-XPosure TM Film; catalog No: 34090; Company: Thermo Scientific), fixed and developed. Pictures were then evaluated and scanned.
Gene silencing using siRNA
We used siRNA for AXL protein (siRNA ID: s1847, Life Technologies, United States). We transfected the 1001 cell line, according to the Life-Technologies protocol, using reverse transfection of the MCF7 cell line. The protocol is available at https://www.lifetechnologies.com/content/dam/LifeTech/migration/en/filelibrary/pdf/protocols.par.23973.file.dat/human-breast-cancer.pdf.
Invasion assay
For the invasion assay, we used QCMTM High Sensitivity Non-cross-linked Collagen Invasion Assay kits, available from Millipore, and followed the supplied manufacturer protocol. Briefly, the assay was performed using a modified chamber with filter inserts (pore size 8 μm) coated with matrigel in 24-well dishes. Approximately 0.5 million cells were prepared in serum-free media (RPMI1640). Two hundred and fifty μL of the cell suspension was added into the inserts (top chamber) and 500 μL of 15% FBS-containing media was added to the bottom chamber. After a 48-h incubation, cells remaining in the top chamber were removed, and 400 μL of cell stain was applied to the invasion chamber insert for 15 min. After several washes with water, the inserts were dried, viewed under the microscope and photographed. Inserts were then transferred into 200 μL of extraction buffer and allowed to incubate for 15 min at room temperature. The dye mixture was then assessed by a plate reader at a wavelength of 630 nm.
Doubling times and sulforhodamine assay
Population doubling times were calculated according to standard protocols from the American Type Culture Collection (ATCC) available at https://www.atcc.org/en/Guides/Guides.aspx. The IC50 concentration of drugs used in the experiments were prepared by serial dilution in DMEM medium prior to performing the experiment. The IC50 concentrations were determined from published literature[22-24]. In all of the experiments, the highest dimethyl sulfoxide (DMSO) concentration did not exceed 1% for drugs dissolved in DMSO. The cytotoxicity of the drugs was determined using the sulforhodamine B (SRB) method[25]. Cells were seeded in 96-well plates at a concentration of 104 cells/well. After 24 h, cells were incubated with drug-free medium, medium containing DMSO (1%), or medium containing the drug at varying concentrations (0.1-100 μmol/mL) in a final volume of 200 μL/well. Triplicate wells were prepared for each concentration. Cells were incubated for 48 h at 37 °C in a humidified atmosphere of 5% CO2. Cells were fixed for 1 h at 4 °C by adding 50 μL of 50% trichloroacetic acid (TCA) to the culture medium in each well. Cells were then rinsed with water several times, dried and stained with 0.4% SRB for 30 min. Next, cells were washed several times with 1% acetic acid to remove unbound stain. After drying, the dye was solubilized with 10 mmol/L Tris base. The optical density was measured at 564 nm with an ELISA microplate reader. The absorbance of cells incubated with only DMSO was subtracted from the absorbance reading of each well (to account for the DMSO effect). Fractions of viable cells were calculated by dividing the reading at the respective concentration by the control (untreated cells). Survival curves were constructed by plotting the fraction of viable cells against the concentrations used.
Statistical analysis
When appropriate, Fisher’s exact probability test, χ2 test, or the student t-test was used to evaluate differences between groups. Person correlation analysis was performed to test relationships between variables. Analyses were performed using MS Excel and/or VassarStats Web-based statistical program, found at http://faculty.vassar.edu/lowry/VassarStats.html. All reported P-values were two-tailed and P values < 0.05 were considered significant. The “ImageJ” program (http://imagej.nih.gov/ij/) and MS Excel were used to quantify the Western blot band intensity.
RESULTS
Upregulation of AXL results from introducing TP53 mutation/knockout
Introduction of TP53 mutations upregulated AXL expression in the HCT116.p53 mutant cells compared to its parental HCT116 TP53 wild-type cell line. The same was true for RKO.P53-/- compared to its parental RKO p53 wild-type cell line (Figure 1A and B). It should be noted that the parental RKO cell line already expressed AXL protein, but knocking out p53 further increased the level of expression, as detected by band intensity quantification. In breast cancer, the 1001 cell line showed high AXL expression in comparison to its parental MCF7 cell line, which has wild-type TP53 functionality (Figure 1C and D). The 1001 cell line originated from a TP53-mutant clone developed from the MCF7 cell line.
Figure 1.
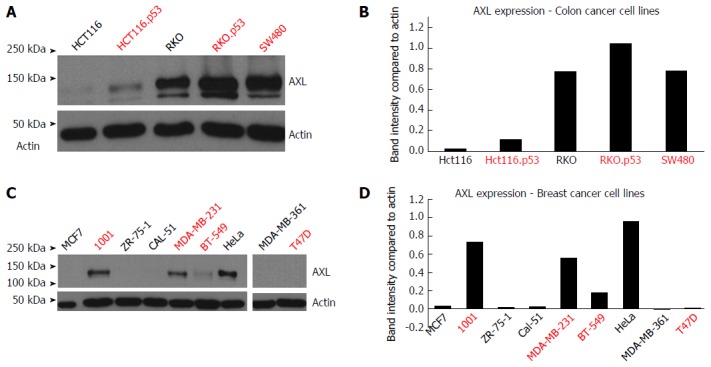
Western blot analysis of AXL protein levels in cancer cell lines. A: Colon cancer cell lines HCT116, HCT116.p53, RKO, RKO.p53-/- and SW480. A band of 140 kDa was observed in AXL positive samples. Actin was used as a loading control; B: This graph shows the quantification of band intensity in comparison to Actin using the “ImageJ” program; value analysis was done using MS Excel. Red font indicates p53 mutation; C: Breast cancer cell line MCF7, MCF7-TP53 mutant clone 1001, ZR-75-1, CAL-51, MDA-MB-231, BT-549, MDA-MB-361, and T47D. HeLa was used as positive control for EMT. Actin was used as a loading control; D: This graph shows the quantification of band intensity in comparison to Actin using the “ImageJ” program and value analysis was done using MS Excel. Red font indicates p53 mutation.
AXL upregulation is correlated to p53 alterations in colorectal and breast cancer cells
As shown in Table 3, there was association between the p53 mutation/knockdown and increased AXL expression in both colon cancer (with the exception of RKO) and breast cancer cell lines (with the exception of T47D).
Table 3.
Summary results of proteins investigated
| Cell lines | p53 status | AXL | E-Cadherin |
| Positive control | |||
| HeLa | HPV inactivated | Highest | Negative |
| Colon | |||
| HCT116 | Wild-Type | Negative | High |
| RKO | Wild-Type | High | Negative |
| HCT116.p53 | Mutant | High | Lowest |
| RKO.p53-/- | Mutant | Highest | Negative |
| SW480 | Mutant | Highest | High |
| Breast | |||
| MCF7 | Wild-Type | Negative | Low |
| MDA-MB-361 | Wild-Type | Negative | Highest |
| ZR-75-1 | Wild-Type | Negative | Highest |
| CAL-51 | Wild-Type | Negative | Low |
| 1001 | Mutant | Highest | Negative |
| MDA-MB-231 | Mutant | High | Negative |
| T47D | Mutant | Negative | Highest |
| BT-549 | Mutant | Lowest | Negative |
AXL is negatively associated with E-Cadherin in both colorectal and breast cancer
Table 3 shows there was also a negative association between increased AXL expression in both colon cancer (with the exception of SW480) and breast cancer cell lines and E-cadherin.
Microarray analysis of AXL, p53, and Wnt signaling
Colorectal cancer cell lines: We extended our analysis of the relationship between AXL and TP53 to available data sets from our own laboratory (Table 4)[21]. The data, acquired from assessing 8 colon cancer cell lines, contradicted the hypothesis that mutations occurring naturally in TP53 (compared to those introduced artificially by manipulating cell lines) can directly cause AXL upregulation. Two p53 mutant cells (HCA7, KM12) were AXL negative and the RKO (p53 wild-type cell) was strongly positive (Table 4) as confirmed by Western blotting above. This suggested the relationship between TP53 and AXL is not direct and that other molecules or signaling pathways are involved. Since RKO is a unique colon cancer cell line that has an inactive Wnt signaling pathway, we hypothesized that AXL is potentially the oncogenic target involved in colorectal carcinogenesis in the absence of active Wnt signaling. We searched the Cancer Cell Line Encyclopedia (CCLE) collection of 62 colon cancer cell lines to further elucidate this hypothesis. We considered AXL to be positive in the CCLE colon cancer cells when it had an expression value of +0.4 or higher, as guided by our Western blot analysis. There was no significant relation between AXL expression and TP53 mutation. We found that AXL was upregulated in 12/62 (20%) colon cancer cell lines. Of these, only 6/12 (50%) were TP53 mutants. However, 3 (RKO, HS698T, HS255T) of these 6 had no mutations in either the APC or CTNNB1 (β-catenin) gene, suggesting inactive Wnt signaling.
Table 4.
AXL mRNA expression from our microarray of colon cancer cell lines[21]
| RKO | HCA7 | KM12 | LoVo | DLD1 | HCT116 | SW48 | LIM 1215 | |
| P53 mutation | WT | mut | mut | WT | WT | WT | WT | WT |
| AXL expression1 | + | - | - | - | - | - | - | - |
Based on Affymetrix probe 202685_s_at considering a cut off value of 70 to differentiate negative from positive.
Breast cancer cell lines: When a threshold of 0.4 or greater was set for identifying AXL positive expression in breast cancer cell lines, we found AXL positive expression in 22/60 (37%), but no significant relationship to TP53 mutation was identified in the CCLE collection.
AXL silencing reduces invasiveness of both colon and breast cancer cells
AXL silencing decreased the invasion of breast cancer cell line 1001, and colorectal cell lines RKO.p53-/- and HCT116.p53 mutant.
AXL predisposes to emergence of invasive clones after chemotherapy
AXL silencing did not show a slight trend to restore breast cancer cells sensitivity to Doxorubicin and colon cancer cell sensitivity to 5FU or irinotecan (Figure 2A and B). Interestingly, analysis of cell invasion after therapy showed that AXL expressing cells developed more invasive potential after exposure to chemotherapy compared to the AXL-silenced cells (Figure 2C and D)
Figure 2.
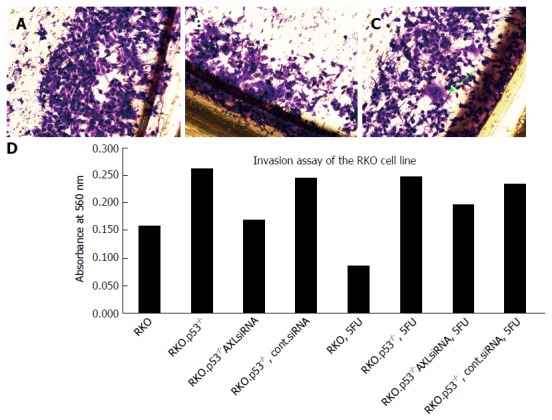
Invasion assays of different RKO clones. A: The base line invasion of RKO.p53-/-, which was not silenced or exposed to treatment, was high, as indicated; B: The same cell line after AXL siRNA silencing shows some decreased invasion; C: RKO.p53-/- treated with siRNA mock control has increased invasiveness and showed some aggressive cells with increased nuclear size, clumped chromatin, increased cytoplasmic extensions (arrow), after exposure to 5FU treatment; D: Quantitative evaluation of different cells after dye elution and spectrophotometric reading at 560 nm.
DISCUSSION
The current work sheds light on the relationship between AXL and TP53 in cancer cells. Our data from isogenic cell lines showed that the introduction of TP53 mutation and/or knockout upregulated AXL expression. As such, this finding indicates AXL is closely regulated by p53 protein. Recent data suggest few alternative mechanisms to explain this effect. Boysen et al[20] showed that the 3’UTR of the AXL gene includes a binding site for miR-34a. The expression of miR-34a is regulated by p53 in B-cell chronic lymphocytic leukemia, ovarian cancer[26] and some other solid tumors[27]. Conversely, miR-34 family members did not correlate with AXL mRNA or protein levels in other tumors, such as renal cell carcinoma[28]. The p53-miR-34a-AXL link in colorectal and breast cancers still needs to be clarified.
Alternatively, mutant TP53 was shown to upregulate AXL in lung cancer cells at both the RNA and protein level[29]. This effect was noted for mutant p53-R175H, R273H, and D281G but was refractory to mutations at positions 22 and 23 in the p53 transactivation domain. Mutant p53 was shown to directly nucleate and induce histone acetylation on the AXL promotor region and knockdown of mutant TP53 was shown to reduce histone acetylation on the AXL promotor. In the presence of mutant TP53, the transcription factors binding capacity to the AXL promotor increased by up to 8-fold. This action was independent of p53-mediated transactivation because the p53/p63 binding site in the AXL promotor was not needed. This work also concluded that AXL partially mediated a change in functional activity of TP53 mutants[29]. It would be interesting to prove this relationship in other tumor lineages, such as colorectal and breast cancers. However, we could not find a consistent association between AXL expression and TP53 mutation status in our microarray data (Table 4) and, more generally, in the CLLE cell lines. This suggests that the relationship between AXL and TP53 could depend on tumor lineage, or more likely, on the carcinogenic mechanisms and molecular subtypes of the individual tumor.
Thus, we were intrigued by the exceptionally high levels of AXL expression in the TP53 wild-type cell line RKO. Because RKO cells have inactive Wnt signaling, a rare phenomenon in colon cancer types, AXL expression might be an alternative oncogenic pathway for colorectal carcinogenesis in the absence of β-catenin/Wnt signaling activation and TP53 mutation[21]. After searching the literature for potential links between AXL and Wnt signaling, we found that AXL was shown to modulate miR-374a and miR548b. These miRNAs play essential roles in gefitinib-induced apoptosis, EMT, tumorigenesis and migration of gefitinib-resistant lung cancer cells by targeting Wnt5a and CCNB1, respectively[15]. MiR-374a was already known to induce canonical Wnt signaling activation in breast cancer cells[30]. More recently, Wang et al[16] showed that AXL stimulation by Gas6 significantly increased β-catenin levels and induced its nuclear translocation, while AXL knockdown caused a decrease in nuclear β-catenin in MCF7 breast cancer cells. Collectively, these data support our argument that AXL potentially acts as an alternative carcinogenic mechanism to exert β-catenin activation in colorectal development, which is one of the very common early changes, in the absence of APC/β-catenin mutations.
Our study also concluded that silencing AXL expression in breast and colorectal cell lines decreases their invasiveness. Clearly, this related to the well-established causative role of AXL in induction of EMT[4,6,10,14,16,31]. Because the association between AXL overexpression and EMT markers is currently well-established, we only used E-cadherin expression to confirm this relationship. Overall, we observed a correlation between TP53, AXL and E-cadherin expression (Table 3), but a few exceptions were noted, which may explain the differences in the prognosis observed below. After using siRNA transfection to silence AXL gene expression, we observed a clear reduction in invasiveness of colorectal and breast cancer cell lines, which is consistent with the published literature[10,14,16]. It is important to note that AXL and myeloid zinc finger (MZF-1) were overexpressed in resected colonic cancer compared to normal tissue. MZF-1 increased migration and invasiveness of cancer cells, in part by binding to the promotor region of AXL and enhancing the protein expression. Knockdown of AXL using shRNA decreased migration and invasiveness induced by MZF-1 in RKO cell lines[31]. This finding may explain alternative mechanisms of AXL upregulation and induction of EMT independent of TP53.
Finally, we showed that after exposure to chemotherapy, the AXL expressing cells became more invasive as compared to the AXL-silenced cells. This finding could explain why AXL expression is sometimes associated with poor clinical outcome, often in early stages of colorectal cancer[14]. A recent study showed that AXL overexpression (among other proto-oncogenes) was associated with short overall survival in patients with colorectal cancer and mutations in the tumor suppressor p53[32]. However, after analyzing public datasets we found an inconsistent relationship between AXL and prognostic parameters, such as response to therapy and overall survival in patients with colorectal and breast cancer. Some recent publications[14,33] also hinted at this result; therefore this observation should be considered when designing personalized AXL-based therapy. Our data and analysis suggest that these differences could be explained by the complex AXL signaling pathways activated in different cells, including TP53 and Wnt signaling. These potential factors need to be elucidated for successful use of AXL in tumor therapy.
In conclusion, our data support the role of AXL in EMT and resistance to treatment in colorectal and breast cancer. We also showed that AXL could be regulated by TP53, and AXL could play an oncogenic role in activating the Wnt/β-catenin pathway in colon cancer. The complexity of the interaction of AXL with various signaling pathways should be taken into consideration when designing therapeutic approaches targeting AXL.
COMMENTS
Background
Colorectal and breast cancers remain deadly diseases; hence, there is continuous effort to develop novel molecular therapeutic targets to improve the outcome of these cancers. AXL Receptor Tyrosine Kinase (AXL), a receptor tyrosine kinase, is an attractive candidate because it was shown to play a role in epithelial to mesenchymal transition; thus, it is also involved in cancer invasion and metastasis. The molecular regulations and interactions of AXL must be well understood before it can be used as a target for new therapeutics.
Research frontiers
AXL is an attractive molecule because it was shown to be associated with poor prognosis, resistance to therapy and immune escape in many cancer types. Recent research is focused on its usefulness as a potential therapeutic target.
Innovations and breakthroughs
The authors supply evidence that AXL is regulated by TP53 in cancers of the colon and breast and it contributes to epithelial to mesenchymal transition, cellular invasion and response to therapy in these tumors. AXL could also be a marker for predisposition for emergence of aggressive clones after chemotherapy. This relationship could not be generalized, and we suggest that this could be linked to other carcinogenic pathways, such as Wnt/β-catenin signaling in colorectal cancer. The interaction of AXL with such pathways should be considered carefully when designing AXL based therapy.
Applications
These data are directly applicable to aid in development and design of treatment options for colon and breast cancers based on the AXL gene.
Terminology
Receptor tyrosine kinases (RTKs) are the high-affinity cell surface receptors for many polypeptide growth factors, cytokines, and hormones. RTKs have been shown not only to be key regulators of normal cellular processes but to also play a critical role in the development and progression of many types of cancer. The epithelial to mesenchymal transition (EMT) is a process by which epithelial cells lose their cell polarity and cell-cell adhesion, and gain migratory and invasive properties. EMT is essential for numerous developmental processes. It has been shown to occur in wound healing, and in the initiation of cancer invasion and metastasis.
Peer-review
The proposal has evaluated AXL expression in relationship to p53 status and its role in tumor invasion and response to therapy. The results have shown a directly application in designing therapeutic approaches targeting AXL.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: United Arab Emirates
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B, B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
Institutional review board statement: No patients or patient derived samples were involved in this study.
Conflict-of-interest statement: The authors have no conflicts of interest to declare.
Data sharing statement: All relevant data were presented in the manuscript. Further information is available from the corresponding author.
Peer-review started: December 28, 2016
First decision: February 10, 2017
Article in press: April 21, 2017
P- Reviewer: Falsafi T, Gazouli M, Santos Riccardi C S- Editor: Qi Y L- Editor: A E- Editor: Zhang FF
References
- 1.Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11 [Internet]. Lyon, France: International Agency for Research on Cancer; 2013. Available from: http://globocan.iarc.fr, 2013. [Google Scholar]
- 2.Veruttipong D, Soliman AS, Gilbert SF, Blachley TS, Hablas A, Ramadan M, Rozek LS, Seifeldin IA. Age distribution, polyps and rectal cancer in the Egyptian population-based cancer registry. World J Gastroenterol. 2012;18:3997–4003. doi: 10.3748/wjg.v18.i30.3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nieminen TT, Shoman S, Eissa S, Peltomäki P, Abdel-Rahman WM. Distinct genetic and epigenetic signatures of colorectal cancers according to ethnic origin. Cancer Epidemiol Biomarkers Prev. 2012;21:202–211. doi: 10.1158/1055-9965.EPI-11-0662. [DOI] [PubMed] [Google Scholar]
- 4.Wu X, Liu X, Koul S, Lee CY, Zhang Z, Halmos B. AXL kinase as a novel target for cancer therapy. Oncotarget. 2014;5:9546–9563. doi: 10.18632/oncotarget.2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kalia M. Biomarkers for personalized oncology: recent advances and future challenges. Metabolism. 2015;64:S16–S21. doi: 10.1016/j.metabol.2014.10.027. [DOI] [PubMed] [Google Scholar]
- 6.Linger RM, Keating AK, Earp HS, Graham DK. TAM receptor tyrosine kinases: biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv Cancer Res. 2008;100:35–83. doi: 10.1016/S0065-230X(08)00002-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Graham DK, DeRyckere D, Davies KD, Earp HS. The TAM family: phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat Rev Cancer. 2014;14:769–785. doi: 10.1038/nrc3847. [DOI] [PubMed] [Google Scholar]
- 8.Liu E, Hjelle B, Bishop JM. Transforming genes in chronic myelogenous leukemia. Proc Natl Acad Sci USA. 1988;85:1952–1956. doi: 10.1073/pnas.85.6.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.El Bezawy R, De Cesare M, Pennati M, Deraco M, Gandellini P, Zuco V, Zaffaroni N. Antitumor activity of miR-34a in peritoneal mesothelioma relies on c-MET and AXL inhibition: persistent activation of ERK and AKT signaling as a possible cytoprotective mechanism. J Hematol Oncol. 2017;10:19. doi: 10.1186/s13045-016-0387-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee HJ, Jeng YM, Chen YL, Chung L, Yuan RH. Gas6/Axl pathway promotes tumor invasion through the transcriptional activation of Slug in hepatocellular carcinoma. Carcinogenesis. 2014;35:769–775. doi: 10.1093/carcin/bgt372. [DOI] [PubMed] [Google Scholar]
- 11.Kirane A, Ludwig KF, Sorrelle N, Haaland G, Sandal T, Ranaweera R, Toombs JE, Wang M, Dineen SP, Micklem D, et al. Warfarin Blocks Gas6-Mediated Axl Activation Required for Pancreatic Cancer Epithelial Plasticity and Metastasis. Cancer Res. 2015;75:3699–3705. doi: 10.1158/0008-5472.CAN-14-2887-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brand TM, Iida M, Stein AP, Corrigan KL, Braverman CM, Coan JP, Pearson HE, Bahrar H, Fowler TL, Bednarz BP, et al. AXL Is a Logical Molecular Target in Head and Neck Squamous Cell Carcinoma. Clin Cancer Res. 2015;21:2601–2612. doi: 10.1158/1078-0432.CCR-14-2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hattori S, Kikuchi E, Kosaka T, Miyazaki Y, Tanaka N, Miyajima A, Mikami S, Oya M. Relationship Between Increased Expression of the Axl/Gas6 Signal Cascade and Prognosis of Patients with Upper Tract Urothelial Carcinoma. Ann Surg Oncol. 2016;23:663–670. doi: 10.1245/s10434-015-4848-x. [DOI] [PubMed] [Google Scholar]
- 14.Dunne PD, McArt DG, Blayney JK, Kalimutho M, Greer S, Wang T, Srivastava S, Ong CW, Arthur K, Loughrey M, et al. AXL is a key regulator of inherent and chemotherapy-induced invasion and predicts a poor clinical outcome in early-stage colon cancer. Clin Cancer Res. 2014;20:164–175. doi: 10.1158/1078-0432.CCR-13-1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Y, Xia H, Zhuang Z, Miao L, Chen X, Cai H. Axl-altered microRNAs regulate tumorigenicity and gefitinib resistance in lung cancer. Cell Death Dis. 2014;5:e1227. doi: 10.1038/cddis.2014.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang C, Jin H, Wang N, Fan S, Wang Y, Zhang Y, Wei L, Tao X, Gu D, Zhao F, et al. Gas6/Axl Axis Contributes to Chemoresistance and Metastasis in Breast Cancer through Akt/GSK-3β/β-catenin Signaling. Theranostics. 2016;6:1205–1219. doi: 10.7150/thno.15083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kasikara C, Kumar S, Kimani S, Tsou WI, Geng K, Davra V, Sriram G, Devoe C, Nguyen KN, Antes A, et al. Phosphatidylserine Sensing by TAM Receptors Regulates AKT-Dependent Chemoresistance and PD-L1 Expression. Mol Cancer Res. 2017 doi: 10.1158/1541-7786.MCR-16-0350. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abdel-Rahman WM, Ollikainen M, Kariola R, Järvinen HJ, Mecklin JP, Nyström-Lahti M, Knuutila S, Peltomäki P. Comprehensive characterization of HNPCC-related colorectal cancers reveals striking molecular features in families with no germline mismatch repair gene mutations. Oncogene. 2005;24:1542–1551. doi: 10.1038/sj.onc.1208387. [DOI] [PubMed] [Google Scholar]
- 19.Puisieux A, Brabletz T, Caramel J. Oncogenic roles of EMT-inducing transcription factors. Nat Cell Biol. 2014;16:488–494. doi: 10.1038/ncb2976. [DOI] [PubMed] [Google Scholar]
- 20.Boysen J, Sinha S, Price-Troska T, Warner SL, Bearss DJ, Viswanatha D, Shanafelt TD, Kay NE, Ghosh AK. The tumor suppressor axis p53/miR-34a regulates Axl expression in B-cell chronic lymphocytic leukemia: implications for therapy in p53-defective CLL patients. Leukemia. 2014;28:451–455. doi: 10.1038/leu.2013.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abdel-Rahman WM, Lotsari-Salomaa JE, Kaur S, Niskakoski A, Knuutila S, Järvinen H, Mecklin JP, Peltomäki P. The Role of Chromosomal Instability and Epigenetics in Colorectal Cancers Lacking β-Catenin/TCF Regulated Transcription. Gastroenterol Res Pract. 2016;2016:6089658. doi: 10.1155/2016/6089658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang W, McLeod HL, Cassidy J. Disulfiram-mediated inhibition of NF-kappaB activity enhances cytotoxicity of 5-fluorouracil in human colorectal cancer cell lines. Int J Cancer. 2003;104:504–511. doi: 10.1002/ijc.10972. [DOI] [PubMed] [Google Scholar]
- 23.Li XX, Zheng HT, Peng JJ, Huang LY, Shi DB, Liang L, Cai SJ. RNA-seq reveals determinants for irinotecan sensitivity/resistance in colorectal cancer cell lines. Int J Clin Exp Pathol. 2014;7:2729–2736. [PMC free article] [PubMed] [Google Scholar]
- 24.Tsou SH, Chen TM, Hsiao HT, Chen YH. A critical dose of doxorubicin is required to alter the gene expression profiles in MCF-7 cells acquiring multidrug resistance. PLoS One. 2015;10:e0116747. doi: 10.1371/journal.pone.0116747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.El-Awady RA, Saleh EM, Ezz M, Elsayed AM. Interaction of celecoxib with different anti-cancer drugs is antagonistic in breast but not in other cancer cells. Toxicol Appl Pharmacol. 2011;255:271–286. doi: 10.1016/j.taap.2011.06.019. [DOI] [PubMed] [Google Scholar]
- 26.Li R, Shi X, Ling F, Wang C, Liu J, Wang W, Li M. MiR-34a suppresses ovarian cancer proliferation and motility by targeting AXL. Tumour Biol. 2015;36:7277–7283. doi: 10.1007/s13277-015-3445-8. [DOI] [PubMed] [Google Scholar]
- 27.Mudduluru G, Ceppi P, Kumarswamy R, Scagliotti GV, Papotti M, Allgayer H. Regulation of Axl receptor tyrosine kinase expression by miR-34a and miR-199a/b in solid cancer. Oncogene. 2011;30:2888–2899. doi: 10.1038/onc.2011.13. [DOI] [PubMed] [Google Scholar]
- 28.Fritz HK, Gustafsson A, Ljungberg B, Ceder Y, Axelson H, Dahlbäck B. The Axl-Regulating Tumor Suppressor miR-34a Is Increased in ccRCC but Does Not Correlate with Axl mRNA or Axl Protein Levels. PLoS One. 2015;10:e0135991. doi: 10.1371/journal.pone.0135991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vaughan CA, Singh S, Windle B, Yeudall WA, Frum R, Grossman SR, Deb SP, Deb S. Gain-of-Function Activity of Mutant p53 in Lung Cancer through Up-Regulation of Receptor Protein Tyrosine Kinase Axl. Genes Cancer. 2012;3:491–502. doi: 10.1177/1947601912462719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cai J, Guan H, Fang L, Yang Y, Zhu X, Yuan J, Wu J, Li M. MicroRNA-374a activates Wnt/β-catenin signaling to promote breast cancer metastasis. J Clin Invest. 2013;123:566–579. doi: 10.1172/JCI65871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mudduluru G, Vajkoczy P, Allgayer H. Myeloid zinc finger 1 induces migration, invasion, and in vivo metastasis through Axl gene expression in solid cancer. Mol Cancer Res. 2010;8:159–169. doi: 10.1158/1541-7786.MCR-09-0326. [DOI] [PubMed] [Google Scholar]
- 32.Zhang SD, McCrudden CM, Yuen HF, Leung KL, Hong WJ, Kwok HF. Association between the expression levels of TAZ, AXL and CTGF and clinicopathological parameters in patients with colon cancer. Oncol Lett. 2016;11:1223–1229. doi: 10.3892/ol.2015.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martinelli E, Martini G, Cardone C, Troiani T, Liguori G, Vitagliano D, Napolitano S, Morgillo F, Rinaldi B, Melillo RM, et al. AXL is an oncotarget in human colorectal cancer. Oncotarget. 2015;6:23281–23296. doi: 10.18632/oncotarget.3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kralj M, Husnjak K, Körbler T, Pavelić J. Endogenous p21WAF1/CIP1 status predicts the response of human tumor cells to wild-type p53 and p21WAF1/CIP1 overexpression. Cancer Gene Ther. 2003;10:457–467. doi: 10.1038/sj.cgt.7700588. [DOI] [PubMed] [Google Scholar]
- 35.Diarra-Mehrpour M, Arrabal S, Jalil A, Pinson X, Gaudin C, Piétu G, Pitaval A, Ripoche H, Eloit M, Dormont D, et al. Prion protein prevents human breast carcinoma cell line from tumor necrosis factor alpha-induced cell death. Cancer Res. 2004;64:719–727. doi: 10.1158/0008-5472.can-03-1735. [DOI] [PubMed] [Google Scholar]
- 36.Gioanni J, Le François D, Zanghellini E, Mazeau C, Ettore F, Lambert JC, Schneider M, Dutrillaux B. Establishment and characterisation of a new tumorigenic cell line with a normal karyotype derived from a human breast adenocarcinoma. Br J Cancer. 1990;62:8–13. doi: 10.1038/bjc.1990.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yuli C, Shao N, Rao R, Aysola P, Reddy V, Oprea-llies G, Lee L, Okoli J, Partridge E, Reddy ES, et al. BRCA1a has antitumor activity in TN breast, ovarian and prostate cancers. Oncogene. 2007;26:6031–6037. doi: 10.1038/sj.onc.1210420. [DOI] [PubMed] [Google Scholar]