

Abstract
The rational and predictable enhancement of protein stability is an important goal in protein design. Most efforts target the folded state, however stability is the free energy difference between the folded and unfolded states thus both are suitable targets. Strategies directed at the unfolded state usually seek to decrease chain entropy by introducing cross-links or by replacing glycines. Cross-linking has led to mixed results. Replacement of glycine with an L-amino acid, while reducing the entropy of the unfolded state, can introduce unfavorable steric interactions in the folded state, since glycine is often found in conformations that require a positive φ angle such as helical C–capping motifs or type I′ and II″ β-turns. L-amino acids are strongly disfavored in these conformations, but D-amino acids are not. However, there are few reported examples and conflicting results have been obtained when glycines are replaced with D-Ala. We critically examine the effect of Gly-to-D-Ala substitutions on protein stability using experimental approaches together with molecular dynamics simulations and free energy calculations. The data, together with a survey of high resolution structures, show that the vast majority of proteins can be stabilized by substitution of C-capping glycines with D-Ala. Sites suitable for substitutions can be identified via sequence alignment with a high degree of success. Steric clashes in the native state due to the new sidechain are rarely observed, but are likely responsible for the destabilizing or null effect observed for the small subset of Gly-to-D-Ala substitutions which are not stabilizing. Changes in backbone solvation play less of a role. Favorable candidates for D-Ala substitution can be identified using a rapid algorithm based on molecular mechanics.
Keywords: Protein Stability, Protein Design, Unfolded State, Protein Folding, Thermodynamic Integration
TOC image
Introduction
A primary goal of protein design is to improve the stability of proteins since marginal stability can lead to loss of function, difficulty in formulating protein based pharmaceuticals, increased aggregation and degradation1–5. Small stable proteins are of interest as alternative scaffolds for presenting sequences in a defined structural context and as alternatives to antibodies for drug delivery, for targeting and as analytical tools6–7. Stabilizing small domains can be a challenge especially if the number of sites which can be targeted is limited by the need to preserve a subset of sites for functional reasons. Stability is dictated by the free energy difference between the unfolded state and the folded state. In order to increase the free energy difference, and thus improve stability, one can stabilize the folded state or destabilize the unfolded state, however the vast majority of approaches to rational design seek to manipulate folded state energetics by exploiting the known three-dimensional structure of the folded state8–12. The unfolded state is a dynamic ensemble, containing transient as well as longer lived elements of structure that can include both native and non-native interactions. The dynamic nature of the unfolded ensemble has made it difficult to target using rational design. Here we describe a general approach to rational protein design that exploits structurally conserved glycine residues and targets both the unfolded ensemble and the native state.
Folded state stabilization usually involves decreasing native state enthalpy, while unfolded state destabilization usually seeks to decrease its entropy. Increasing stability by decreasing the enthalpy of the folded state is more broadly studied, however, implementation of this strategy requires detailed structural information on the folded state9, 11. A decrease in the conformational entropy of unfolded states can be achieved by adding disulfide bonds or substituting glycine with non-glycine amino acids8, 10, 12–17. The former approach also requires tertiary structural information of the folded state, since disulfide bonds can introduce strain into the native state and have strict stereochemical requirements. In theory, the effect of adding a disulfide can be estimated using arguments based on loop entropy; the disulfide introduces a cross link in the chain and thereby reduces the configurational entropy of the unfolded state. However, introduction of a disulfide can stabilize compact conformations in the unfolded state and lead to new unfolded state enthalpic interactions. These effects, together with native state strain, often result in engineered disulfides having only a modest or even unfavorable effect on protein stability10, 18. Complete cyclization of a protein by covalently linking the N and C termini has been employed in an attempt to enhance protein stability, but the same considerations come into play19.
Targeting glycine residues is an attractive alternative strategy since introduction of a sidechain is a simple and effective way to decrease configurational entropy owing to the more restricted allowed region of the Ramachandran plot for an L or D amino acid relative to glycine. The approach should be effective provided that the addition of a sidechain does not lead to steric clashes in the folded state and provided the stereochemical constraints introduced by the sidechain are compatible with the native backbone geometry. The latter point is a significant issue since glycine is often located at sites which require a positive value of the backbone dihedral angle φ20. D-amino acids are the more attractive choice when targeting glycine residues that have positive values of φ, since these conformations are disfavored for L-amino acids, but allowed for D-amino acids21–23. Glycine residue with positive values of φ are commonly found in α-helical C-capping motifs and in type I′ and II″ β-turns, where a left-handed conformation (positive φ)is required22, 24–26. These glycines can often be identified using multiple sequence alignments since they are conserved for structural reasons; helical capping motifs have specific sequence requirements and there are well established sequence rules for type I′ and II″ β-turns21–24, 26–27. Glycines located at C-caps are often solvent exposed, thus any perturbation caused by substituting with a D-amino acid should be minimal since the new side chain is less likely to make steric clashes. This potentially opens the door to rational design in the absence of structural information, however conflicting results have been reported for D-Ala substitutions.
The effect of Gly-to-D-Ala substitutions has been reported for four different proteins: the N-terminal domain of the ribosomal protein L9 (NTL9), the C-terminal Ubiquitin associated domain of HHR23A (UBA), the mini-protein construct TC5b (Trp-cage) and human erythrocytic ubiquitin (ubiquitin)12, 17, 28. D-amino acids have also been used to stabilize small β-hairpin peptides25. The limited experimental measurements reveal several apparent contradictions: To first order, the entropic stabilization caused by Gly-to-D-Ala substitution is expected to be system independent, but not all proteins are stabilized by Gly-to-D-Ala substitutions and a significant range of ΔΔG° values have been reported for those that are. The stability of NTL9 and UBA are increased by a favorable 1.87 kcal/mol and 0.6 kcal/mol respectively when a C-capping Gly was replaced with D-Ala12. Note, in this manuscript, we report ΔG° values of unfolding, thus positive values of ΔΔG° indicate stabilization. The stability of Trp-cage was improved by 0.9 kcal/mol when G10 was substituted by D-Ala17. However, a G35D-Ala substitution at a helical C-capping position in ubiquitin was slightly destabilizing at pH=2.528. The lack of an effect was conjectured to be due to unfavorable contributions from backbone desolvation, caused by the introduction of a sidechain, that offset the decreased entropy of the unfolded state28.
The limited data set indicates that replacement of glycines with positive φ-angles by D-Ala can be stabilizing, but it also leads to important questions: will the trend of an increase in stability be preserved if larger data sets are examined? What causes the range of values of ΔΔG°? Why does the replacement lead to no effect in ubiquitin? Can the energetic effects of a D-Ala substitution be quantitatively predicted? From a practical perspective, the key issues are whether or not it is possible to reliably and robustly predict, a priori which Gly to D-Ala replacements will be stabilizing, and by how much. This is critical since D-amino-acids must currently be introduced via solid phase synthesis or via chemical ligation methods.
In this study, we use a combined experimental and computational approach to systematically examine the consequences of replacing C-capping glycines with D-amino acids and develop a rapid algorithm for predicting when such substitutions will be stabilizing. Gly-to-D-Ala substitutions at the C-caps of α-helices in four additional proteins were examined, doubling the number of reported examples: the engrailed homeodomain (EH), the GA albumin-binding module (GA), the peripheral subunit-binding domain (PSBD) and the chicken villin subdomain (HP35)29–32. These proteins are all α-folds and each contains a glycine C-capping residue with a positive φ angle (Figure 1). EH, GA and PSBD were randomly chosen and D-Ala replacements were found to be stabilizing. The small helical protein HP35 was predicted to be destabilized by Gly-to-D-Ala substitutions based on molecular modelling and serves as a negative control. Computational modelling successfully reproduced the experimental stability changes and indicates that intra-molecular van der Waals interactions in the folded state are the reason for the wide range of ΔΔG° caused by Gly-to-D-Ala substitutions. Screening a database of representative high-resolution X-ray structures shows that 95% of C-capping Gly-to-D-Ala substitutions are predicted to be stabilizing and 80% of all substitutions are predicted to enhance stability by more than 1 kT. This work shows that Gly-to-D-Ala substitutions at C-caps of α-helices, under the guidance of molecular modelling, is a general strategy for rational protein design. This works reveals the rules for stabilizing proteins via D-Ala substitutions. This “mirror image” approach to protein design is widely applicable and sites suitable for substitution can be rapidly predicted.
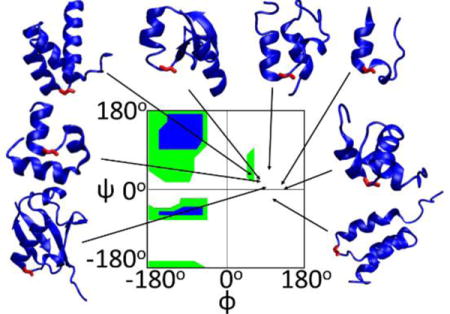
Figure 1.
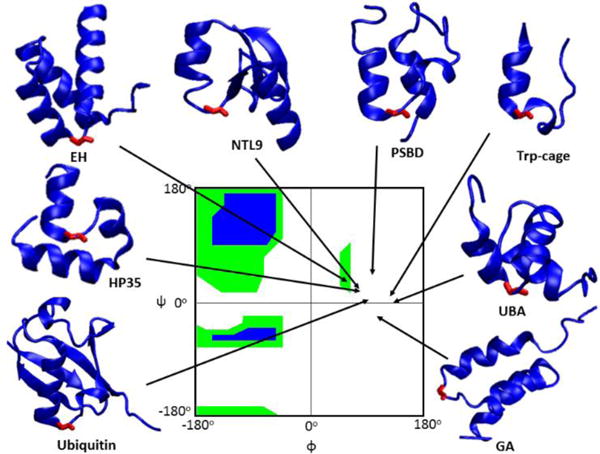
Ribbon representation of the proteins studied with the C-capping Gly colored red. φ/ψ angles of the C-capping glycines are indicated by arrows. The Ramachandran plot is colored green for broadly allowed and blue for most favored regions for L-amino acids, which is adopted from Ramaplot in VMD36. The Ramachandran plot for a D-amino acid is the mirror image about the central point (φ = 0° and ψ = 0°) of the plot shown above.
Results
Proteins are usually stabilized by Gly-to-D-Ala substitution
Published results on a limited set of proteins indicate a range of effects for Gly-to-D-Ala substitution at C-capping sites. However, the number of systems tested to date is too small to draw general conclusions. In order to gain better insight into the consequences of Gly-to-D-Ala substitutions at C-capping sites, Gly-to-D-Ala substitutions were examined in another four proteins (EH, GA, PSBD and HP35). All of these domains have been shown to fold reversibly in a 2-state fashion29, 33–35. Like NTL9, UBA and Ubiquitin, these proteins all have a C-capping glycine that is solvent exposed as judged by standard accessible surface area algorithms (Figure 1). The φ/ψ angles and the solvent accessibility of all of the glycine sites studied are provided in the supporting information (Table S1). Thermal and denaturant induced unfolding curves of EH, GA, HP35, PSBD display sigmoidal transitions and all can be fit by standard methods to extract unfolding free energies (Table 1, Figures S1 and S2). The stability of EH G39D-Ala, GA G16D-Ala and PSBD G15D-Ala are 0.64 kcal/mol, 0.81 kcal/mol and 1.25 kcal/mol higher than the respective wild-type. HP35 G11D-Ala is 0.38 kcal/mol less stable than wild-type HP35, but HP35 was intentionally selected as a negative control using the computational approach described below. The experimental measurements on these four additional proteins, especially the inclusion of an additional example (HP35) in which D-Ala substitution is destabilizing, provide a more robust test set for the computational studies described in the next several paragraphs.
Table 1.
Thermodynamic properties of EH, GA, HP35, PSBD and their D-Ala variants.
| Protein | ΔG° of unfolding at 25°C (kcal/mol) | m (kcal/mol M−1) | Tm (°C) | ΔH° (Tm) (kcal/mol) |
|---|---|---|---|---|
| EH | 1.91 ± 0.03(1) | 0.61 ± 0.01 | 55.6 ± 0.18 | 32.5 ± 0.74 |
| EH G39D-Ala | 2.55 ± 0.13(1) | 0.66 ± 0.03 | 60.7 ± 0.38 | 33.1 ± 1.39 |
| GA | 4.71 ± 0.16(2) | 1.00 ± 0.03 | ND | ND |
| GA G16D-Ala | 5.52 ± 0.19(2) | 1.02 ± 0.04 | ND | ND |
| PSBD | 2.75 ± 0.07(1) | 0.67 ± 0.01 | 52.5 ± 0.14 | 29.6 ± 0.51 |
| PSBD G15D-Ala | 4.00 ± 0.34(1) | 0.73 ± 0.07 | 61.3 ± 0.23 | 31.9 ± 0.84 |
| HP35 | 2.47 ± 0.12(1) | 0.38 ± 0.03 | 76.1 ± 1.78 | 23.8 ± 0.57 |
| HP35 G11D-Ala | 2.08 ± 0.13(1) | 0.45 ± 0.03 | 61.2 ± 1.34 | 21.7± 1.33 |
Determined by urea denaturation;
Determined by GdnHCl denaturation;
ND: Not determined. Uncertainties represent the standard error of the fit.
Five of the six proteins which were randomly chosen without computational guidance exhibit enhanced stability when a C-capping Gly is replaced by D-Ala, suggesting that Gly-to-D-Ala substitutions at C-capping sites are likely to improve protein stability. Left unanswered are the questions why there is a significant range of ΔΔG° values and why are HP35 and ubiquitin destabilized?
Gly-to-D-Ala substitutions can modulate ΔΔG° via other interactions in addition to entropic stabilization
Recent computational work reported that Gly-to-L-Ala substitution entropically destabilizes the unfolded state by −TΔS = 0.3 kcal/mol when the unfolded states are modeled as tri and pentapeptides37, while earlier work provide estimates ranging from 0.05 to 0.72 kcal/mol38–41. The wide range of experimental unfolding free energy changes (0.39 kcal/mol destabilizing to 1.87 kcal/mol stabilizing) argues that interactions beyond entropic destabilization of the unfolded state play an important role in determining the change. A range of effects could counteract or supplement the entropic stabilization of replacing a C-capping Gly. Introduction of a sidechain at a C-capping Gly site can lead to increased desolvation of the polypeptide backbone, a process which is energetically unfavorable28. All else being equal, desolvation in the native state will destabilize a protein. However, desolvation of the backbone in the folded state is likely compensated by desolvation of the backbone in the unfolded state. Moreover, the desolvation penalty may also be compensated by new favorable intramolecular interactions such as buried hydrogen bonds or favorable van der Waals interactions. Desolvation of the backbone is thus unlikely to be the sole reason for the wide range of experimental ΔΔG° values. On the other hand, unfavorable van der Waals interactions, such as steric clashes between D-Ala and other residues in the folded state can offset the decrease of entropy in the unfolded states. These new folded state interactions will usually be alleviated upon unfolding and are less likely to perturb the unfolded state. We hypothesized that a significant contribution to the difference in ΔΔG° values reflects differences in van der Waals interactions between the C-capping Gly/D-Ala and the rest of the protein in the folded state.
In order to test our hypothesis, molecular dynamics simulations (MD) of wild-type proteins and their D-Ala variants were conducted using the Amber ff14SB force field. MD simulations were also conducted for simplified unfolded state models to account for local unfolded state effects. Per-residue energy decomposition provided an estimate of the intramolecular van der Waals energy (Evdw) contributed by C-capping Gly/D-Ala to the total potential energy of the protein. New unfavorable intramolecular van der Waals interactions in the folded state caused by the D-Alasidechain lead to a negative value of ΔΔEvdw, while new favorable intramolecular van der Waals interactions in the folded state lead to a positive ΔΔEvdw value. A good correlation between ΔΔEvdw and ΔΔG° is expected if the variation in ΔΔG° values is determined by whether or not the D-Ala residue generates new contacts, and how strong these interactions are. ΔΔEvdw can be calculated from snapshots derived from the MD simulations, while the contribution of backbone desolvation to ΔΔG° can be studied by counting the number of water molecules that are blocked from interacting with the peptide backbone at the C-capping site in the folded and unfolded states using snapshots from the MD simulations. The difference provides an estimate of the net desolvation effect. It is important to validate the models used for these analyses and the applicability of the force field employed with more rigorous methods. Consequently, we first tested if our MD simulations were sufficiently converged and our force field accurate enough to reproduce the experimental data using thermodynamic integration (TI) free energy calculations.
Thermodynamic integration validates more approximate computational models and provides further insight into C-capping energetics
The model used for the unfolded states are tetrapeptides with neutral capping groups and the length of the MD simulations can only reach a time scale that is much smaller than the experimental time scale. A recently parametrized force field was chosen in this study, but, like all force fields, is still an approximate description of molecules42. Therefore, we tested our models by asking if we can reproduce the experimental values of ΔΔG° using TI. 34 λ windows were simulated for 12 ns each. TI is computationally expensive and reaching complete ergodic convergence in each λ window is unlikely, thus two different starting structures of each protein were used for two independent TI calculations in order to evaluate precision. For each protein, one of the starting structures was the PDB structure, while the other one was the last frame of a 50 ns MD simulation. (supplemental information).
Similar values of ΔΔG° were obtained for a given protein independent of the starting structure chosen, suggesting that the TI calculation has reached reasonable convergence during the time-scale of the simulations (Figure 2A). The only significant difference between ΔΔG°calc values determined using the different starting structures occurs for EH. We believe the effect is due to the poorly resolved N-terminus of EH in the X-ray structure rather than issues with the computational models implemented here. Residues 1–4 are unresolved and not shown in the crystal structure, while residues 5–7 are resolved, but with low confidence30. We appended the 4 missing residues as an extended peptide to the crystal structure and conducted a MD simulation with restraints on all resolved residues to relax the four appended residues. The last frame of this restrained MD simulation was used as the starting structure for one of the TI calculations for EH (Figure 2A red bar). Following the restrained MD simulation, Gly 39 was changed to D-Ala and unrestrained MD simulation was carried out to fully relax the conformation. The last frame of this simulation was used as the starting structure for the other TI calculation of EH (Figure 2A cyan bar). During the unrestrained MD simulation, residues 1–7 formed contacts with Gly39 or D-Ala39; this was not observed during the restrained MD simulation. The difference in the calculated ΔΔG° of EH may be caused by the difference in the extent of relaxation of the starting structures. Since residues 1–7 in the PDB structures are either unresolved or poorly resolved, the fully relaxed structure is likely a better representation of the structure of EH. The better agreement between ΔΔG°exp and ΔΔG°cal when the fully relaxed structure was used as starting structure is consistent with this hypothesis.
Figure 2.
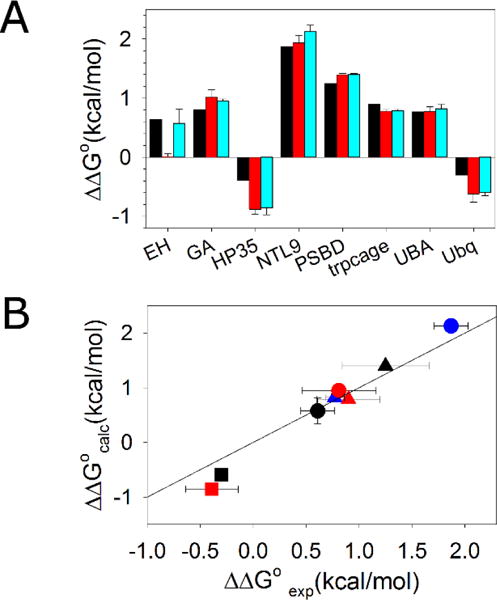
Thermodynamic integration reproduces experimental values of ΔΔG°. (A) Experimental ΔΔG° values are shown in black. Calculated ΔΔG° values using experimental structures as starting structures are shown in red. Calculated ΔΔG° values using the last frames of 50 ns simulations as the starting structures are in cyan. (B) A scatter plot of experimental ΔΔG° and calculated ΔΔG° values using the last frames of a 50 ns simulation as the starting structure. Solid line represents ΔΔG°exp = ΔΔG°cal. EH ●; GA
 ; HP35
; HP35
 ; NTL9
; NTL9
 ; PSBD ▲; Trp-cage
; PSBD ▲; Trp-cage
 ; UBA
; UBA
 ; Ubiquitin ■. The calculated value for the EH domain used in the plot was derived by using the unrestrained MD structure as the starting structure for the TI calculation. Positive ΔΔG° values indicate stabilization.
; Ubiquitin ■. The calculated value for the EH domain used in the plot was derived by using the unrestrained MD structure as the starting structure for the TI calculation. Positive ΔΔG° values indicate stabilization.
A small root-mean-square error of 0.23 kcal/mol is obtained for the complete set of ΔΔG°exp and ΔΔG°cal values calculated using the last frames of 50 ns MD simulations as the starting structures (Figure 2B). This indicates that the simplified unfolded state model, sampling sufficiency and choice of force field provide accurate energetics for these systems. The good agreement also argues that the large span in experimental ΔΔG° values is neither caused by complexity in the unfolded states nor by the different conditions and methods used for the experimental protein stability measurements since a simplified model for the unfolded states and a consistent computational approach were able to reproduce the experimental trends.
The calculated change in van der Waals energy, ΔΔEvdw, is strongly correlated with ΔΔG°, but ΔΔG° does not correlate with predicted desolvation effects
To test our hypothesis that the entropic stabilization is modulated by variation in van der Waals interactions, ΔΔEvdw values were calculated from the MD simulations. There is a strong correlation between ΔΔEvdw and the ΔΔG° values obtained experimentally or computationally with correlation coefficients of 0.89 in both cases (Figure 3). The results strongly support the hypothesis that van der Waals interactions between the D-Ala/Gly site and the rest of the protein play an important role in determining ΔΔG°.
Figure 3.
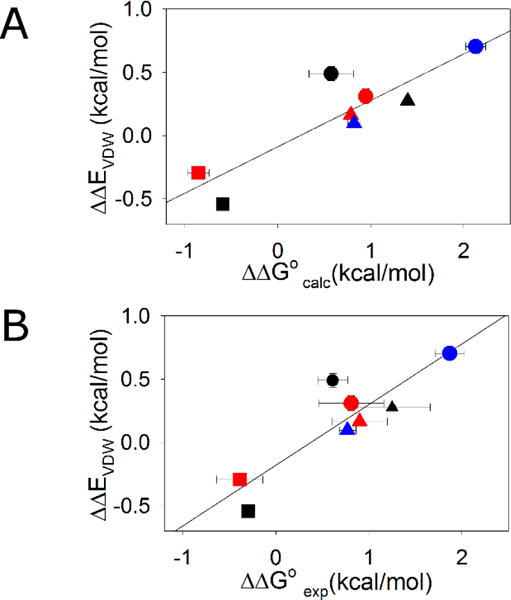
Scatter plot of ΔΔEvdw and ΔΔG° with solid line showing the linear fit. (A) Correlation of ΔΔEvdw and ΔΔG° values calculated by thermodynamic integration. r=0.89, p-value=0.0033 (B) Correlation of ΔΔEvdw and experimental ΔΔG° values. r=0.89, p-value =0.0033. EH ●; GA
 ; HP35
; HP35
 ; NTL9
; NTL9
 ; PSBD ▲; Trp-cage
; PSBD ▲; Trp-cage
 ; UBA
; UBA
 ; Ubiquitin ■. Positive ΔΔG° values indicate stabilization.
; Ubiquitin ■. Positive ΔΔG° values indicate stabilization.
In order to examine potential correlations between the extent of backbone desolvation and the ΔΔG° values, the first shell water molecules around backbone atoms in both the folded and unfolded states were counted. The difference in the number of water molecules blocked by D-Ala relative to Gly in the unfolded states and folded states (unfolded-folded) provides an estimate of the net desolvation effect of the new sidechain. Since the methyl group in D-Ala is non-polar, the mutation from Gly-to-D-Ala only changes the water accessibility of the backbone and counting the number of water around backbone is a reasonable metric for measuring desolvation effects. The calculations were performed by averaging over the last 160 ns of 12 independent MD simulations for the folded state and 144 ns of MD simulations for the unfolded state of each protein. No significant correlation is observed with ΔΔG° values. The correlation coefficient for the number of waters blocked by D-Ala and ΔΔG°calc is only 0.16 and is just 0.17 for the correlation with ΔΔG°exp (Figure 4). If the desolvation effects in the unfolded state are disregarded and only the number of blocked waters in the folded state are counted, the correlation between ΔΔG°calc or ΔΔG°exp and the number of waters blocked by D-Ala relative to Gly is not improved, with correlation coefficients of 0.20 and 0.16 respectively. For three of the proteins (EH, HP35 and GA) the uncertainty, defined here as the standard deviation of the three sets of simulations with 4 independent simulations in each set, in the number of waters blocked by D-Ala in the unfolded and folded states is relatively large. However, this does not affect the conclusion that desolvation effects are not correlated with ΔΔG°. The good convergence in the ΔΔG°calc values in the absence of good convergence in the number of blocked waters reinforces that there is unlikely to be a significant net contribution of desolvation to ΔΔG° for the systems studied here.
Figure 4.
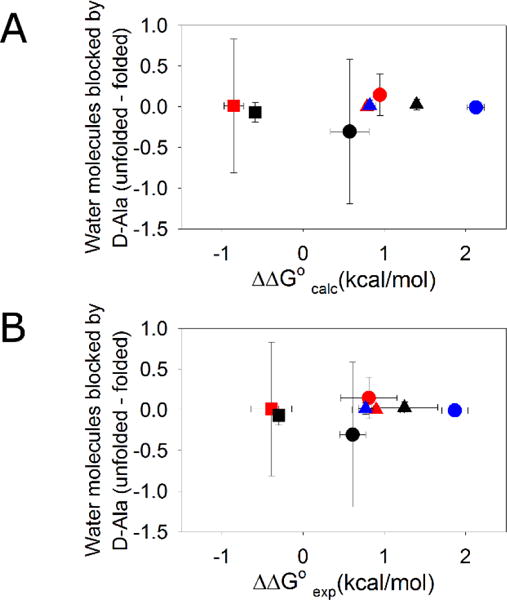
Changes in backbone solvation do not correlate with ΔΔG°. The difference in the number of water molecules blocked by D-Ala relative to Gly (Unfolded-folded) is plotted vs (A) calculated ΔΔG° values (r=0.16). (B) experimental ΔΔG° values (r=0.17). EH ●; GA
 ; HP35
; HP35
 ; NTL9
; NTL9
 ; PSBD ▲; Trp-cage
; PSBD ▲; Trp-cage
 ; UBA
; UBA
 ; Ubiquitin ■. Positive ΔΔG° values indicate stabilization.
; Ubiquitin ■. Positive ΔΔG° values indicate stabilization.
In principle, Poisson-Boltzmann (PB) based calculations could be used to estimate desolvation effects43, however we observed during the 200 ns MD simulations of the folded states that subtle changes in conformation can lead to a significant change in the calculated PB desolvation energy of the backbone atoms owing to the long range nature of electrostatic interactions. This results in poor convergence for the PB calculations if the fluctuations in conformation are on the same time scale of the MD simulations and leads to large error bars for PB based calculations of desolvation effects. EH and HP35 showed poor convergence in the PB calculations. The other six proteins have relatively good convergence, but no correlation between the desolvation effects calculated by PB and ΔΔG°exp was observed (r=0.28, p=0.58, slope=0.2) (Figure S3). The small slope indicates that differences in the PB desolvation energy do not make a contribution to the differences in ΔΔG°. The good convergence in the ΔΔG°calc values in the absence of convergence in the PB calculated solvation energy for all proteins further reinforces our conclusion that it is unlikely that desolvation makes a significant contribution to the range of ΔΔG° values observed for the systems studied here.
The rapid screening of target proteins for D-Ala substitutions; a designed negative control helps to demonstrate proof of principle
It is prohibitively expensive to generate entire ensembles from an MD trajectory in explicit solvent in order to calculate ΔΔEvdw values for a large set of proteins. Instead, a method which estimates ΔΔEvdw in a time-efficient manner was developed in order to enable rapid screening of proteins for sites suitable for D-Ala substitution. The method was used to identify the HP35 D-Ala11 mutant as a negative control. The approach exploits the strong correlation between ΔΔEvdw and ΔΔG° identified above and uses a more rapid method to calculate ΔΔEvdw. We calculated ΔΔEvdw_gb, which like ΔΔEvdw, quantifies the contribution of the intramolecular van der Waals energy to ΔΔG°, but is obtained by running a short implicit-solvent simulation44 instead of using a large ensemble from a long explicit-solvent MD simulation. The correlation between ΔΔEvdw and ΔΔEvdw_gb is 0.84 (Figure S4) for the 8 systems in Figure 3. Although the implicit-solvent model is more coarse-grained than the explicit-solvent model and the length of simulation is significantly decreased, calculation of ΔΔEvdw_gb for a range of proteins should allow one to predict trends of ΔΔG°exp for hundreds of proteins in a time-efficient manner, provided the correlation between ΔΔEvdw_gb and the known ΔΔG°exp values is good. If desired, one can conduct further analysis of promising sites using longer MD simulations with explicit solvent or TI.
As shown in Figure 5, ΔΔEvdw_gb values (positive values represent net stabilization) are strongly correlated with the known values of ΔΔG°exp (r=0.94) (Figure 5). The strong correlation between ΔΔEvdw_gb and ΔΔG°exp further supports our hypothesis that the perturbation of van der Waals interactions are correlated with the effect of Gly-to-D-Ala substitutions on stability.
Figure 5.
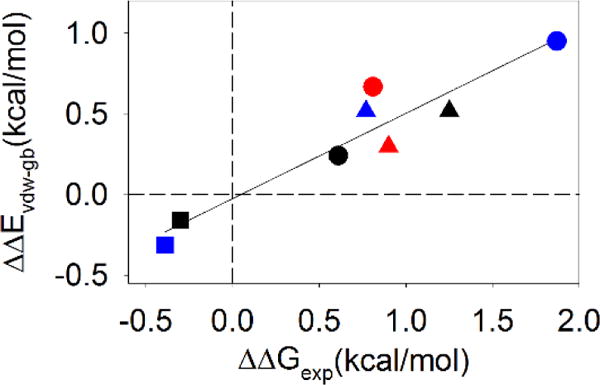
There is a strong correlation between ΔΔEvdw-gb and ΔΔG°exp. Positive values of ΔΔG° indicate stabilizing effects. EH ●; GA
 ; HP35
; HP35
 ; NTL9
; NTL9
 ; PSBD ▲; Trp-cage
; PSBD ▲; Trp-cage
 ; UBA
; UBA
 ; Ubiquitin ■; r=0.94 and p=0.0004. Positive ΔΔG° values indicate stabilization.
; Ubiquitin ■; r=0.94 and p=0.0004. Positive ΔΔG° values indicate stabilization.
The strong correlation between ΔΔEvdw_gb and ΔΔG°exp (Figure 5) indicates that linear regression can be used to predict the ΔΔG°exp values from their ΔΔEvdw_gb values using the empirical function:
We examined a set of 120 monomeric proteins of less than 130 residues, which have structures determined at 2.0 Å resolution or better and at least one helix with a C-capping Gly. ΔΔEvdw_gb values were calculated for proteins with high sequence diversity. In all, 160 C-capping sites were analyzed (Table S3) and ΔΔEvdw_gb values ranging from −0.35 to 1.67 kcal/mol were obtained (Figure 6A). Here, negative values indicate a net destabilization and positive values reflect a net stabilization. The distribution of predicted ΔΔG° values is plotted as a histogram in Figure 6B. Overall, 95% of the substitutions are predicted to lead to increased stability. Furthermore, ~80% of C-capping Gly-to-D-Ala substitutions in monomeric proteins will result in significant stabilization larger than 1kT.
Figure 6.
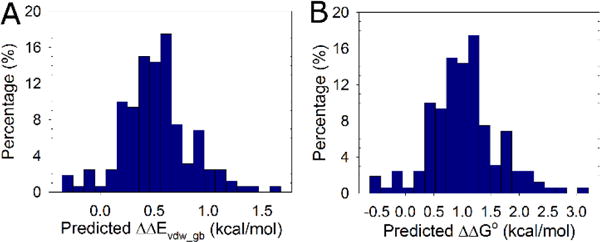
Proteins are stabilized by D-Ala substitutions. The distribution of ΔΔEvdw_gb and ΔΔG° values for the 160 C-capping sites in the 120 non-redundant proteins is shown as a histogram. (A) Distribution of ΔΔEvdw_gb values. (B) Distribution of predicted ΔΔG° values. Positive ΔΔG° values represent a stabilizing effect.
From this distribution we selected the helical subdomain of the villin headpiece (HP35) as a negative test case, since it was one of the few proteins for which a D-Ala substitution was predicted to be destabilizing (Figure S5). ΔΔEvdw_gb for the replacement of Gly by D-Ala in HP35 was −0.31 kcal/mol (negative values represent net destabilization), which is comparable to the value for ubiquitin (Figure 5). As noted above, HP35 G11D-Ala has an experimentally measured stability 0.39 kcal/mol lower than wild-type HP35 (Table 1), confirming the computational prediction made prior to experiments.
Conclusions
Our analysis indicates that the energetics of C-capping interactions involve an interplay between two competing factors. Glycine residues are selected for such sites because they are able to adopt positive values of φ, but the choice of glycine introduces packing defects in the native state. The extremely high conservation of C-capping sites indicates that the evolutionary pressure to maintain the ability to adopt a positive value of phi at these sites leads to tolerance of packing defects in the structure. This highlights that protein stability includes compromises between competing interactions. Our results clearly show that Gly-to-D-Ala substitutions in C-capping motifs stabilize proteins when the folded state is not perturbed by unfavorable van der Waals interactions. The stability of EH, GA, NTL9, PSBD, Trp-cage and UBA were improved by 0.59 to 1.87 kcal/mol by Gly-to-D-Ala substitutions. Van der Waals interactions make a significant contribution to the observed spread in ΔΔG° values. The fact that TI calculations quantitatively reproduced the experimentally observed effects, including the destabilization of HP35 and ubiquitin, argues that the range of reported ΔΔG° values are not caused by variation in experimental protocols or complex effects in the unfolded state. The D-Ala variants of HP35 and ubiquitin were destabilized due to new unfavorable folded state van der Waals interactions that counteract the entropic stabilization. The systems studied here are two state folding but the general principles, unfolded state destabilization via entropic effects and native state stabilization by new favorable Van der Waals interaction also apply to proteins that fold via intermediates.
An important practical observation from this work is that steric clashes may still be generated by D-Ala substitution even if a C-capping glycine is identified as solvent exposed by measuring its solvent accessible surface area (SASA) (Table S2). The effect arises because the repulsive part of the van der Waals potential energy has a strong distance dependence, with the potential energy increasing rapidly as the distance between two atoms decreases. For example, moving a β-carbon from 3.2 Å to 2.8 Å from a carboxyl oxygen results in an increase in van der Waals potential energy of 1.9 kcal/mol using the Lennard-Jones potential in the Amber ff14SB force field42. This indicates that a more quantitative method than measuring SASA should be used when predicting the consequence of Gly-to-D-Ala substitutions at C-caps of α-helices.
Does the observation that the effects of the D-Ala substitutions can be predicted accurately using a highly simplified model of the unfolded state imply that the unfolded state is devoid of structure or long range contacts or residual structure? The answer is no; the data simply argues that the substitutions do not significantly impact the energetics of other unfolded state interactions; indeed residual structure has been detected in the unfolded states of several of the proteins studied45–48.
In this study, experimental values of ΔΔG° have been successfully reproduced by using molecular modelling for all proteins tested. These examples show that in silico molecular modelling and design serve as an excellent complement to experimental studies, and can allow one to rationally target unfolded state interactions. Predicted ΔΔG° values of a large data set of structures indicate that most proteins will be stabilized by Gly-to-D-Ala substitutions at C-capping sites, opening the door to mirror image protein design.
C-capping glycines are strongly conserved in protein structures and can be identified by multiple sequence alignments, thus they can often be identified in the absence of structural information. The analysis presented here demonstrates that the replacement of such glycines is expected to be stabilizing 95% of the cases and to be significantly stabilizing 80% of the cases. This expected success rate is considerably better than has been observed with consensus method based on multiple sequence alignment and is comparable to the most successful consensus method which take into account co-variation, suggesting that rational protein design is possible in the absence of structural information49–50.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge Prof. Rohit Pappu for numerous helpful discussions and Prof. Robert C. Rizzo for helpful suggestions. The authors also thank the Laufer Center for Physical and Quantitative Biology at Stony Brook University for access to computational resources and support, and Feng Zhang, Dr. James Maier and Koushik Kasavajhala for their administration of computational resources.
Funding sources. This work was supported by NSF grant MCB -1330259 to DPR and NIH grant GM107104 and an NSF Petascale Computational Resource (PRAC) Award from the NSF (OCI-1036208) to CS. We gratefully acknowledge support from Henry and Marsha Laufer. J.Z. was supported in part by a fellowship from the Laufer Center.
Footnotes
Supporting information
Experimental and computational methods. Additional figures and tables as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Chi EY, Krishnan S, Randolph TW, Carpenter JF. Pharm Res. 2003;20:1325–1336. doi: 10.1023/a:1025771421906. [DOI] [PubMed] [Google Scholar]
- 2.Daniel RM, Cowan DA, Morgan HW, Curran MP. Biochem J. 1982;207:641–644. doi: 10.1042/bj2070641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parsell DA, Sauer RT. J Biol Chem. 1989;264:7590–7595. [PubMed] [Google Scholar]
- 4.Chi EY, Krishnan S, Kendrick BS, Chang BS, Carpenter JF, Randolph TW. Protein Sci. 2003;12:903–913. doi: 10.1110/ps.0235703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McLendon G, Radany EJ. Biol Chem. 1978;253:6335–6337. [PubMed] [Google Scholar]
- 6.Binz HK, Amstutz P, Pluckthun A. Nat Biotechnol. 2005;23:1257–1268. doi: 10.1038/nbt1127. [DOI] [PubMed] [Google Scholar]
- 7.Skrlec K, Strukelj B, Berlec A. Trends Biotechnol. 2015;33:408–418. doi: 10.1016/j.tibtech.2015.03.012. [DOI] [PubMed] [Google Scholar]
- 8.Matthews BW, Nicholson H, Becktel WJ. Proc Natl Acad Sci U S A. 1987;84:6663–6667. doi: 10.1073/pnas.84.19.6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nicholson H, Becktel WJ, Matthews BW. Nature. 1988;336:651–656. doi: 10.1038/336651a0. [DOI] [PubMed] [Google Scholar]
- 10.Matsumura M, Becktel WJ, Levitt M, Matthews BW. Proc Natl Acad Sci U S A. 1989;86:6562–6566. doi: 10.1073/pnas.86.17.6562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spector S, Wang M, Carp SA, Robblee J, Hendsch ZS, Fairman R, Tidor B, Raleigh DP. Biochemistry. 2000;39:872–879. doi: 10.1021/bi992091m. [DOI] [PubMed] [Google Scholar]
- 12.Anil B, Song B, Tang Y, Raleigh DP. J Am Chem Soc. 2004;126:13194–13195. doi: 10.1021/ja047119i. [DOI] [PubMed] [Google Scholar]
- 13.Sauer RT, Hehir K, Stearman RS, Weiss MA, Jeitler-Nilsson A, Suchanek EG, Pabo CO. Biochemistry. 1986;25:5992–5998. doi: 10.1021/bi00368a024. [DOI] [PubMed] [Google Scholar]
- 14.Wells JA, Powers DB. J Biol Chem. 1986;261:6564–6570. [PubMed] [Google Scholar]
- 15.Wetzel R, Perry LJ, Baase WA, Becktel WJ. Proc Natl Acad Sci U S A. 1988;85:401–405. doi: 10.1073/pnas.85.2.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tidor B, Karplus M. Proteins. 1993;15:71–79. doi: 10.1002/prot.340150109. [DOI] [PubMed] [Google Scholar]
- 17.Rodriguez-Granillo A, Annavarapu S, Zhang L, Koder RL, Nanda V. J Am Chem Soc. 2011;133:18750–18759. doi: 10.1021/ja205609c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Betz SF, Pielak GJ. Biochemistry. 1992;31:12337–12344. doi: 10.1021/bi00164a007. [DOI] [PubMed] [Google Scholar]
- 19.Camarero JA, Fushman D, Sato S, Giriat I, Cowburn D, Raleigh DP, Muir TW. J Mol Biol. 2001;308:1045–1062. doi: 10.1006/jmbi.2001.4631. [DOI] [PubMed] [Google Scholar]
- 20.Stites WE, Meeker AK, Shortle D. J Mol Biol. 1994;235:27–32. doi: 10.1016/s0022-2836(05)80008-7. [DOI] [PubMed] [Google Scholar]
- 21.Richardson JS, Richardson DC. Science. 1988;240:1648–1652. doi: 10.1126/science.3381086. [DOI] [PubMed] [Google Scholar]
- 22.Aurora R, Rose GD. Protein Sci. 1998;7:21–38. doi: 10.1002/pro.5560070103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gunasekaran K, Nagarajaram HA, Ramakrishnan C, Balaram PJ. Mol Biol. 1998;275:917–932. doi: 10.1006/jmbi.1997.1505. [DOI] [PubMed] [Google Scholar]
- 24.Hutchinson EG, Thornton JM. Protein Sci. 1994;3:2207–2216. doi: 10.1002/pro.5560031206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haque TS, Gellman SH. J Am Chem Soc. 1997;119:2303–2304. [Google Scholar]
- 26.Sibanda BL, Thornton JM. Nature. 1985;316:170–174. doi: 10.1038/316170a0. [DOI] [PubMed] [Google Scholar]
- 27.Bystroff C, Baker DJ. Mol Biol. 1998;281:565–577. doi: 10.1006/jmbi.1998.1943. [DOI] [PubMed] [Google Scholar]
- 28.Bang D, Gribenko AV, Tereshko V, Kossiakoff AA, Kent SB, Makhatadze GI. Nat Chem Biol. 2006;2:139–143. doi: 10.1038/nchembio766. [DOI] [PubMed] [Google Scholar]
- 29.Chiu TK, Kubelka J, Herbst-Irmer R, Eaton WA, Hofrichter J, Davies DR. Proc Natl Acad Sci U S A. 2005;102:7517–7522. doi: 10.1073/pnas.0502495102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clarke ND, Kissinger CR, Desjarlais J, Gilliland GL, Pabo CO. Protein Sci. 1994;3:1779–1787. doi: 10.1002/pro.5560031018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johansson MU, de Chateau M, Wikstrom M, Forsen S, Drakenberg T, Bjorck LJ. Mol Biol. 1997;266:859–865. doi: 10.1006/jmbi.1996.0856. [DOI] [PubMed] [Google Scholar]
- 32.Kalia YN, Brocklehurst SM, Hipps DS, Appella E, Sakaguchi K, Perham RN. J Mol Biol. 1993;230:323–341. doi: 10.1006/jmbi.1993.1145. [DOI] [PubMed] [Google Scholar]
- 33.Religa TL, Johnson CM, Vu DM, Brewer SH, Dyer RB, Fersht AR. Proc Natl Acad Sci U S A. 2007;104:9272–9277. doi: 10.1073/pnas.0703434104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang T, Zhu YJ, Gai F. J Phys Chem B. 2004;108:3694–3697. doi: 10.1021/jp037272j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spector S, Kuhlman B, Fairman R, Wong E, Boice JA, Raleigh DP. J Mol Biol. 1998;276:479–489. doi: 10.1006/jmbi.1997.1522. [DOI] [PubMed] [Google Scholar]
- 36.Humphrey W, Dalke A, Schulten K. J Mol Graph. 1996;14:33–38. 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 37.Scott KA, Alonso DO, Sato S, Fersht AR, Daggett V. Proc Natl Acad Sci U S A. 2007;104:2661–2666. doi: 10.1073/pnas.0611182104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nemethy G, Leach SJ, Scheraga HA. J Phys Chem. 1966;70:998–&. [Google Scholar]
- 39.DAquino JA, Gomez J, Hilser VJ, Lee KH, Amzel LM, Freire E. Proteins: Struct Funct Genet. 1996;25:143–156. doi: 10.1002/(SICI)1097-0134(199606)25:2<143::AID-PROT1>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 40.Zaman MH, Shen MY, Berry RS, Freed KF, Sosnick TR. J Mol Biol. 2003;331:693–711. doi: 10.1016/s0022-2836(03)00765-4. [DOI] [PubMed] [Google Scholar]
- 41.Baxa MC, Haddadian EJ, Jumper JM, Freed KF, Sosnick TR. Proc Natl Acad Sci U S A. 2014;111:15396–15401. doi: 10.1073/pnas.1407768111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling CJ. Chem Theory Comput. 2015;11:3696–3713. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE., 3rd Acc Chem Res. 2000;33:889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 44.Nguyen H, Roe DR, Simmerling C. J Chem Theory Comput. 2013;9:2020–2034. doi: 10.1021/ct3010485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cho JH, Meng W, Sato S, Kim EY, Schindelin H, Raleigh DP. Proc Natl Acad Sci U S A. 2014;111:12079–12084. doi: 10.1073/pnas.1402054111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meng W, Lyle N, Luan B, Raleigh DP, Pappu RV. Proc Natl Acad Sci U S A. 2013;110:2123–2128. doi: 10.1073/pnas.1216979110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mok KH, Kuhn LT, Goez M, Day IJ, Lin JC, Andersen NH, Hore PJ. Nature. 2007;447:106–109. doi: 10.1038/nature05728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spector S, Rosconi M, Raleigh DP. Biopolymers. 1999;49:29–40. doi: 10.1002/(SICI)1097-0282(199901)49:1<29::AID-BIP4>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 49.Magliery TJ. Curr Opin Struct Biol. 2015;33:161–168. doi: 10.1016/j.sbi.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Steipe B, Schiller B, Pluckthun A, Steinbacher SJ. Mol Biol. 1994;240:188–192. doi: 10.1006/jmbi.1994.1434. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.