

ABSTRACT
The adenylate cyclase toxin-hemolysin (CyaA, ACT, or AC-Hly) of Bordetella pertussis targets phagocytic cells expressing the complement receptor 3 (CR3, Mac-1, αMβ2 integrin, or CD11b/CD18). CyaA delivers into cells an N-terminal adenylyl cyclase (AC) enzyme domain that is activated by cytosolic calmodulin and catalyzes unregulated conversion of cellular ATP into cyclic AMP (cAMP), a key second messenger subverting bactericidal activities of phagocytes. In parallel, the hemolysin (Hly) moiety of CyaA forms cation-selective hemolytic pores that permeabilize target cell membranes. We constructed the first B. pertussis mutant secreting a CyaA toxin having an intact capacity to deliver the AC enzyme into CD11b-expressing (CD11b+) host phagocytes but impaired in formation of cell-permeabilizing pores and defective in cAMP elevation in CD11b− cells. The nonhemolytic AC+ Hly− bacteria inhibited the antigen-presenting capacities of coincubated mouse dendritic cells in vitro and skewed their Toll-like receptor (TLR)-triggered maturation toward a tolerogenic phenotype. The AC+ Hly− mutant also infected mouse lungs as efficiently as the parental AC+ Hly+ strain. Hence, elevation of cAMP in CD11b− cells and/or the pore-forming capacity of CyaA were not required for infection of mouse airways. The latter activities were, however, involved in bacterial penetration across the epithelial layer, enhanced neutrophil influx into lung parenchyma during sublethal infections, and the exacerbated lung pathology and lethality of B. pertussis infections at higher inoculation doses (>107 CFU/mouse). The pore-forming activity of CyaA further synergized with the cAMP-elevating activity in downregulation of major histocompatibility complex class II (MHC-II) molecules on infiltrating myeloid cells, likely contributing to immune subversion of host defenses by the whooping cough agent.
KEYWORDS: Bordetella pertussis, adenylate cyclase toxin-hemolysin, cAMP intoxication, lung colonization, pore-forming activity, virulence
INTRODUCTION
The adenylate cyclase toxin-hemolysin is produced by all three Bordetella species pathogenic to mammals, and it plays a prominent role in the early phases of respiratory tract infection by the whooping cough agent, Bordetella pertussis (1–3). The toxin specifically binds the CD11b subunit of the complement receptor 3 (CR3) (4, 5) and exerts an array of immunosubversive and cytotoxic activities on myeloid phagocytes. CyaA delivers a cell-invasive adenylyl cyclase (AC) enzyme domain into cytosol of CD11b+ cells, where the AC is activated by calmodulin and converts cytosolic ATP to the signaling molecule cyclic AMP (cAMP). The generated supraphysiological levels of cAMP then nearly instantly ablate the bactericidal oxidative burst and opsonophagocytic killing capacities of neutrophils and macrophages (6–10). With efficacy of about 2 orders of magnitude lower, the CyaA toxin can also penetrate non-myeloid cells that lack the CR3 receptor (CD11b− cells), where due to its extremely active AC enzyme it can elevate cAMP concentrations to well detectable and physiologically relevant concentrations in epithelial and other host cells (10, 11). Indeed, amounts of CyaA detected in nasopharyngeal fluids and washes from diseased infants and infected olive baboons (12) indicate that CyaA may also be playing a prominent role in perturbation of barrier and innate immune defense functions of epithelia of airway mucosa. Recently, CyaA-bearing outer membrane vesicles (OMVs) shed by B. pertussis were shown to deliver CyaA into epithelial cells across their apical cell surface, through which the free secreted CyaA translocates very inefficiently (13, 14). Signaling of the OMV-delivered CyaA might thus be impairing the barrier function of polarized epithelial layers and enable access of the free toxin to the basolateral membrane of epithelial cells, through which CyaA invades epithelial cells more efficiently (14).
The CyaA protein consists of an N-terminal enzymatic AC domain (∼400 residues) that is fused to a pore-forming repeat-in-toxin (RTX) family hemolysin/cytolysin (Hly) moiety of about 1,300 residues (15). The Hly comprises a hydrophobic pore-forming domain (residues 500 to 700), a fatty acyl-modified domain (residues 800 to 1000), a vast calcium-binding domain characteristic of RTX proteins (residues 1000 to 1600), and a C-terminal secretion signal (15, 16). The cytotoxic activities of CyaA strictly depend on posttranslational activation of pro-CyaA by covalent palmitoylation and on functional folding of the RTX domain upon loading of ∼40 calcium ions into RTX binding sites (15–21). The Hly moiety mediates CyaA binding to CR3 (22) and enables delivery of the AC enzyme domain into the cytosol of host cells (23–25). In parallel, the Hly forms small cation-selective pores that permeabilize the cytoplasmic membrane of target cells and account for the moderate hemolytic activity of CyaA on erythrocytes (25, 26). Cell permeabilization by Hly also elicits efflux of cytosolic K+ ions and activates the p38 and Jun N-terminal protein kinase (JNK) kinases (27), thus contributing to Toll-like receptor (TLR)-induced NALP3 inflammasome formation and interleukin-1β (IL-1β) secretion by dendritic cells (DCs) (28). The pore-forming activity of CyaA further synergizes with cAMP signaling elicited by the AC enzyme in bringing about the overall cytotoxic (cytolytic) potency of CyaA (29, 30).
Upon initial interaction with N-linked oligosaccharides (31, 32), CyaA specifically binds a loop outside the I domain of the CD11b subunit of the αMβ2 integrin (CR3) (5), and the toxin delivers the AC enzyme into phagocyte cytosol in two steps (33). Compelling indirect evidence suggests that the AC-translocating and pore-forming activities of CyaA are mutually independent and are accomplished by two distinct subpopulations of CyaA conformers that employ the same transmembrane CyaA segments in an alternative manner (34). The balance between the two toxin activities can, indeed, be shifted in either direction by specific residue substitutions (35–38). Recently, we combined a glutamine (E570Q) substitution of the glutamate residue 570 (within the pore-forming domain) with an arginine substitution of the lysine 860 residue (K860R) in the second acylation site of the toxin (29, 39). This yielded a nonhemolytic (AC+ Hly−) CyaA-E570Q+K860R construct (37) that retains a full capacity to deliver the AC enzyme and elevate cAMP concentrations in CD11b-expressing (CD11b+) myeloid cells. Its potency in formation of membrane pores and permeabilization of CD11b+ cells is, however, more than 10-fold reduced (37). Further, the capacity of CyaA-E570Q+K860R to penetrate CD11b− cells is strongly decreased, and compared to intact CyaA, the AC+ Hly− toxin exhibits an about 20-fold-reduced specific capacity to deliver the AC enzyme into erythrocytes, and its specific capacity to permeabilize and lyse erythrocytes is reduced by almost 2 orders of magnitude (37). The AC+ Hly− toxin, hence, elevates cAMP concentrations in CD11b+ myeloid phagocytes without permeabilizing them any significantly, and this nonhemolytic AC toxin is almost inactive on non-myeloid CD11b− cells devoid of the CR3 receptor, such as erythrocytes or airway epithelial cells.
The capacity of CyaA to increase cAMP concentrations in host cells was previously shown to be essential for the capacity of B. pertussis to multiply in mouse lungs (2, 3, 40). It remained unclear, however, if this was solely due to cAMP elevation in phagocytes or whether the pore-forming activity of CyaA also plays a role in bacterial virulence. This issue could not be addressed before, as all previously used nonhemolytic B. pertussis mutants were producing CyaA variants with deletions in the pore-forming domain, and such toxoids are unable to translocate the AC enzyme into cells to raise cellular cAMP levels (3, 41, 42). The absence of this crucial immunomodulatory action of CyaA on CD11b+ phagocytes hence confounded the outcome of the animal challenge studies, and all previously used nonhemolytic B. pertussis mutants scored as unable to multiply in mouse lungs any efficiently (2, 3, 40–42). Here we show that neither the pore-forming (hemolytic) activity of CyaA toxin on CD11b+ phagocytes nor its capacity to elevate cAMP in CD11b− cells is per se required for persistent sublethal infection of mouse lungs by B. pertussis. These capacities, however, are involved in B. pertussis penetration across the epithelial lining and in provoking enhanced neutrophil infiltration and inflammatory damage of infected tissue.
RESULTS
The cAMP-elevating activity of CyaA is sufficient for immunomodulatory shaping of the dendritic cell phenotype by Bordetella pertussis.
We first constructed an AC+ Hly− B. pertussis mutant that secreted the cell-invasive but nonhemolytic CyaA-E570Q+K860R toxin (37). It was unable to produce any hemolytic halos around bacterial colonies on BG agar plates (Fig. 1A), while it produced normal levels of the pertussis toxin (PT), pertactin (PRN), and filamentous hemagglutinin (FHA) virulence factors (data not shown). The AC+ Hly− B. pertussis also secreted the same amounts of the CyaA protein as the parental AC+ Hly+ B. pertussis Tohama I strain (Fig. 1B). As shown in Fig. 1C, the combination of the E570Q and K860R substitutions strongly reduced the specific capacity of the AC+ Hly− CyaA to bind sheep erythrocytes (RBCs) and ablated its capacity to translocate the AC domain into these model CD11b− cells. In contrast, the AC+ Hly− toxin still elicited equally high cAMP concentrations in CD11b-expressing bone marrow-derived DCs (BMDCs) as the parental AC+ Hly+ protein (Fig. 1D). The AC+ Hly− toxin-producing B. pertussis mutant was thus fully capable of elevating cAMP levels in CR3-expressing (CD11b+) myeloid cells.
FIG 1.

The AC+ Hly− variant of CyaA is produced at normal levels and preserves the DC-binding and cAMP-elevating activity of intact CyaA. (A) Parental AC+ Hly+ B. pertussis, the nonhemolytic mutant expressing the CyaA-E570Q+K860R toxin (AC+ Hly−), and the AC− Hly+ mutant producing an enzymatically inactive CyaA-AC− toxoid were grown on Bordet-Gengou agar with 15% defibrinated sheep blood for 5 days at 37°C. (B) Bacteria were grown in liquid Stainer-Scholte (SS) medium for 18 h at 37°C, and CyaA toxin was detected on Western blots of bacterial lysates using the anti-RTX 9D4 antibody. Values below the blot indicate the total AC enzyme activities of CyaA associated with the outer surface of bacterial cells grown in SS medium with 0.13 mM Ca2+ ions. (C) CD11b− sheep erythrocytes (5 × 108/ml) were incubated at 37°C with bacterial lysates (50 ng CyaA/ml). After 30 min, aliquots were taken for determination of the cell-associated AC activity (binding) and of the AC activity internalized into erythrocytes and protected against digestion by externally added trypsin (invasive AC). Activities are expressed as percentages of intact CyaA activity and represent average values ± standard deviations from two independent determinations performed in duplicate. (D) AC domain translocation was assessed by determining the intracellular concentration of cAMP generated in cells following incubation with diluted bacterial lysates (final CyaA concentration of 7.5 ng/ml). The results represent the average of values obtained in at least two independent experiments performed in duplicate.
It has previously been shown that CyaA-elicited signaling of the cAMP-activated protein kinase A modulates maturation and cytokine production of lipopolysaccharide (LPS)-stimulated DCs (43–49). It remained unclear, however, if the cell-permeabilizing capacity of CyaA modulates the cAMP-mediated skewing of the phenotypic maturation of DCs exposed to live B. pertussis cells. We thus compared expression of maturation markers, profiles of secreted cytokines, and the antigen-presenting capacity of DCs that were coincubated in vitro with live AC+ Hly+, AC+ Hly−, and AC− Hly+ B. pertussis cells at a multiplicity of infection (MOI) of 100:1. The AC− Hly+ B. pertussis strain was used as a negative control, as it secretes the CyaA-AC− toxoid, which has the AC enzyme activity ablated by insertion of a Gly-Ser dipeptide between residues 188 and 189 of the AC domain (50). As a further control for CR3 engagement effects, the mutant producing a doubly detoxified (nonenzymatic and nonhemolytic) AC− Hly− toxoid was used. Bacteria killed by heating at 70°C for 30 min (HI-Bp) were then used as control for unspecific effects of bacterially shed TLR ligands, such as LPS and other bacterial components unrelated to CyaA.
DCs were exposed to live bacteria for 1 h before kanamycin (100 μg/ml) was added to kill the extracellular bacteria, and the incubation was continued for additional 23 h to allow DC maturation. When exposed to bacteria producing enzymatically active AC toxin (AC+ Hly+ and AC+ Hly−), capable of elevating intracellular cAMP concentrations, the DCs exhibited equal viability at 4 h postinfection to DCs exposed to heat-killed (HI-Bp) bacteria (Fig. 2A). At 24 h, however, the cAMP signaling already provoked a drop in the survival rate of DCs treated with the AC+ bacteria (data not shown). Following continued incubation for 24 h with kanamycin-killed bacteria as a source of TLR ligands, stimulation of expression of most of the maturation markers with DCs preincubated with heat-inactivated bacteria was then observed comparable to that upon exposure to bacteria producing the AC− toxoids (Fig. 2B). However, a specifically reduced expression of the CD54 marker and notably of the CD40 costimulatory molecule was observed with DCs that were exposed to the parental AC+ Hly+ strain and the nonhemolytic AC+ Hly− mutant, which both produce enzymatically active CyaA proteins that elevate cAMP in DCs (cf. Fig. 1D). Hence, the interference of CyaA activity with DC maturation was entirely cAMP dependent and did not involve the cell-permeabilizing (hemolytic) capacity of the secreted CyaA.
FIG 2.
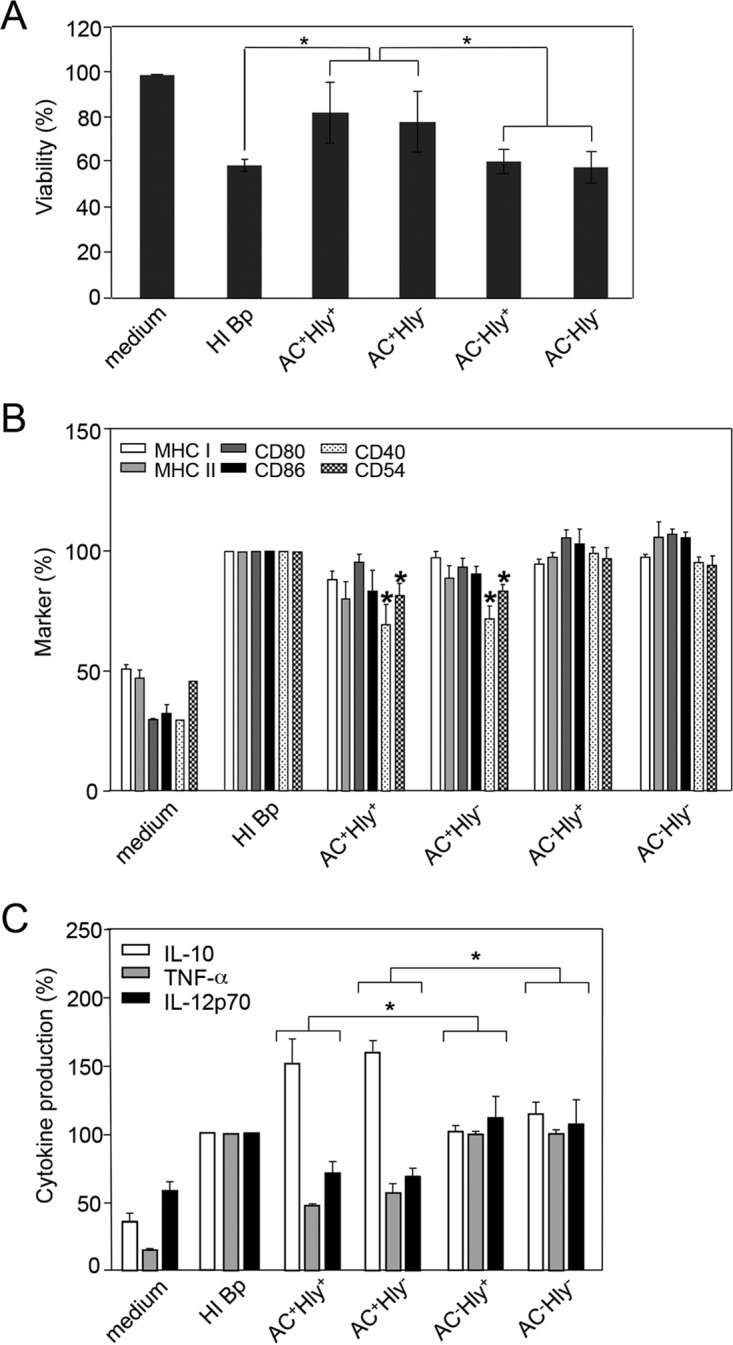

cAMP-elevating activity alone is sufficient for immunomodulatory shaping of the dendritic cell phenotype by B. pertussis-secreted CyaA. Bone marrow-derived DCs were treated with a suspension of heat-killed parental B. pertussis (HI-Bp; 70°C, 30 min) or infected with the different B. pertussis mutants at an MOI of 100:1. After 1 h of incubation, kanamycin (100 μg/ml) was added to kill the bacteria, and DCs were further incubated for 3 or 23 h. (A) DC survival after 4 h was determined by flow cytometry with TMRE and Hoechst 33258 staining. The viability (TMRE+, Hoechst 33258−) of untreated DCs (medium) was set to 100%. (B) Expression of H-2Kb, I-A/I-E, CD80, CD86, CD40, and CD54 in living CD11c+ DCs was determined by flow cytometry, and (C) the secretion of TNF-α, IL-10, and IL-12p70 at 24 h was determined in DC culture supernatants by ELISA. Expression of maturation-associated molecules and production of cytokines by DCs treated with heat-killed bacteria was set as 100%. Values represent the means ± standard errors of the mean (SEM) of n = 3 (*, P < 0.05). (D) DCs treated as indicated above were coincubated with 0.1 μM OVA protein for presentation on MHC class II molecules to OT-II CD4+ T cells or with 0.2 μM OVA for presentation on MHC class I molecules to OT-I CD8+ T cells. After 4 h of coincubation, the DCs were washed and 2 × 105 naïve OVA-specific CD8+ or CD4+ T cells were added. Production of IL-17, IFN-γ, and IL-10 in cell culture supernatants after 3 days was determined by ELISA. (E) The numbers of Foxp3+ CD4+ CD25+ T regulatory cells were determined by flow cytometry after 3 days of coculture. All experiments were reproduced at least 3 times, and representative dot plots are shown. The graph values represent means ± SEM (*, P < 0.05).
Similarly, when cytokine profiles of DCs were examined (Fig. 2C), the DCs incubated with the AC− Hly+ and AC− Hly− mutants exhibited a similar profile of cytokine production to cells incubated with heat-inactivated bacteria (primarily LPS stimulated). In contrast, a clear inhibition of TLR ligand-induced production of the proinflammatory cytokines tumor necrosis factor alpha (TNF-α) and IL-12, as well as enhanced secretion of the immunosuppressive cytokine IL-10 by DCs, was observed upon preincubation with the AC+ Hly+ and AC+ Hly− bacteria expressing the enzymatically active CyaAs.
Furthermore, exposure of DCs for 4 h to the nonhemolytic (AC+ Hly−) mutant or to the parental (AC+ Hly+) strain modulated to similar extent the ovalbumin (OVA) antigen-presenting capacity of the DCs coincubated with OVA-specific CD4+ (OT-II) and CD8+ (OT-I) T lymphocytes (Fig. 2D). Compared to treatment with heat-killed bacteria, the coincubation of DCs with bacteria expressing the AC− toxoids enhanced the induction of OVA-specific gamma interferon (IFN-γ) secretion by OVA-specific CD4+ T cells from OT-II mice, while OVA presentation by DCs exposed to AC+ Hly+ or AC+ Hly− bacteria yielded a reduced capacity to stimulate IFN-γ secretion by OVA-specific CD4+ T cells. In contrast, DCs exposed to AC+ Hly+ or AC+ Hly− bacteria exhibited clearly enhanced OVA-dependent production of IL-10 and IL-17A, compared to DCs exposed to heat-killed cells or the bacteria producing the AC− toxoid. Incubation of DCs with the parental AC+ Hly+ or AC+ Hly− bacteria also enhanced OVA-dependent production of the IL-10 and IL-17A cytokines by CD8+ OT-I T lymphocytes. As further shown in Fig. 2E, exposure of DCs to both AC+ strains (Hly+ or Hly−) but not the incubation with the two AC− mutants, strikingly enhanced the capacity of DCs to promote expansion of antigen-specific CD4+ CD25+ Foxp3+ regulatory T cells. The pore-forming (hemolytic) activity of CyaA thus did not counteract the tolerogenic skewing of the DC phenotype triggered by the cAMP-elevating CyaA toxin secreted by B. pertussis.
The sole capacity of CyaA to elevate cAMP in CD11b-expressing cells enables mouse lung infection by B. pertussis.
We next assessed if the specific capacity of CyaA to elevate cAMP in CR3-expressing cells was sufficient for virulence of B. pertussis in the intranasal infection model in mice. In total, a series of eight infection experiments was performed in outbred Swiss CD-1 mice with the parental AC+ Hly+ strain and seven and six experiments, respectively, were performed with the AC− Hly+ and AC+ Hly− B. pertussis mutants. The animals were challenged with doses ranging from 104 to 5 × 106 CFU/mouse and irrespective of the challenge dose used, the experiments yielded consistent trends. A representative result of one experiment performed with the challenge dose of 1 × 105 CFU/mice is shown in Fig. 3A. The nonhemolytic AC+ Hly− mutant infected mice as efficiently as the parental AC+ Hly+ strain, exhibiting a consistent increase in CFU counts in the lungs of infected mice by at least an order of magnitude, reaching a maximum on day 8 postchallenge. In contrast, the AC− Hly+ mutant proliferated in the lungs by about half an order of magnitude only, with CFU counts peaking already on day 5 and dropping progressively on days 8 and 12. Hence, the capacity to deliver an active AC enzyme and elevate cAMP in CD11b+ cells was sufficient to support the capacity of B. pertussis to multiply and persist in mouse lungs, while the capacities to form hemolytic pores and/or act on CD11b− cells were dispensable.
FIG 3.
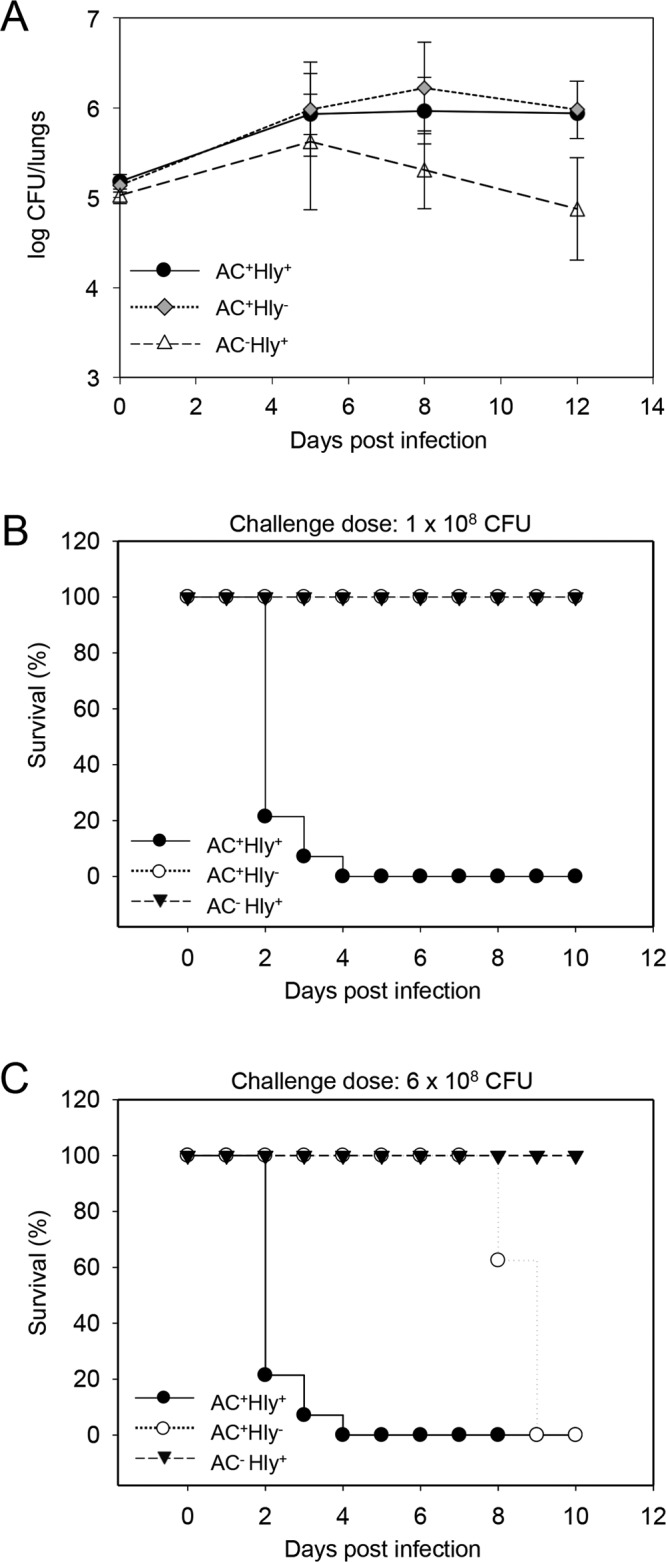
AC enzyme activity of CyaA alone supports lung infection by B. pertussis. (A) Infection of the mouse lungs. Four-week-old Swiss CD-1 mice were challenged intranasally with 105 CFU of the parental AC+ Hly+ strain or the AC− Hly+ and AC+ Hly− mutants. The mean values and standard deviations from six mice per time point were plotted (except for day 12, which is a mean value from only three mice). The results represent the average of values obtained in two experiments. (B) Survival rates of 4-week-old mice infected with 1 × 108 CFU of the WT and mutant B. pertussis strains. (C) Survival rates at the increased inoculation dose of 6 × 108 CFU. Mortality was monitored for 10 days postinfection. The results were reproduced in at least 3 experiments using 6 to 8 mice per challenged group per challenge dose.
However, the AC+ Hly− mutant exhibited a clearly decreased virulence when administered at increased challenge doses used for determination of the 50% lethal dose (LD50) value. As shown by a representative result in Fig. 3B, up to 80% of the mice challenged with 108 CFU of the parental AC+ Hly+ strain died within 2 days from intranasal infection, and the remaining infected mice succumbed by day 4, yielding an LD50 value of (4.1 ± 1.7) × 107 CFU/mouse. In contrast, no mice died upon infection with 108 CFU of the nonhemolytic AC+ Hly− mutant (Fig. 3B). When a higher inoculation dose of 6 × 108 CFU/mice of the AC+ Hly− mutant was used, the mice were dying only on days 8 to 9, hence 6 days later than mice infected by the lower dose of the parental AC+ Hly+ strain (Fig. 3C). Hence, an LD50 value of (3.5 ± 0.7) × 108 CFU representing an increase of about an order of magnitude was determined for the AC+ Hly− mutant, and this still represents an overestimation of the real virulence of the AC+ Hly− mutant, as the calculated LD50 value does not take into account the importantly delayed onset of mouse death (by almost a week). The AC+ Hly− mutant was, nevertheless, still significantly more virulent than the AC− Hly+ mutant that did not cause any lethality at all, even at inoculation doses exceeding 4.3 × 108 CFU/mouse (P < 0.001).
AC+ Hly− mutant infection elicits lower levels of lung inflammation.
To determine the extent of lung pathology produced by the three bacterial strains, mice were infected with 105 CFU of each strain. Animals were sacrificed on day 6, and pathological lesions were assessed by two independent examining pathologists in parallel in a blind manner, without revealing to them the identity of the samples.
Using samples from 4 animals per group, striking differences in lung pathology provoked by the three strains were observed (Fig. 4A). The lungs of control mice inoculated with sterile SS medium exhibited unaltered microscopical structure with delicate continuous alveolar septa, patent alveoli, well-preserved bronchi and bronchioles, and intact uniform respiratory epithelium. In striking contrast, inoculation of mice with 105 CFU of the parental AC+ Hly+ B. pertussis strain resulted in prominent bronchopneumonic lesions (Fig. 4A). These affected about ∼10% of the lung area (Fig. 4B) and comprised involvement of the peribronchial parenchyma. The infiltrates consisted mainly of polymorphonuclear (neutrophilic) granulocytes, increased numbers of monocytes/macrophages, scattered lymphocytes, and swollen alveolar cells. The infected bronchial respiratory epithelium exhibited a typical focal to discrete hypertrophy and prominent lobar, lobular, and septal congestion of the swollen pulmonary regions.
FIG 4.
The B. pertussis AC+ Hly− mutant elicits significantly milder inflammation of infected lungs than the parental strain. Four BALB/c mice per group were infected intranasally with 1.5 × 105 CFU in 50 μl of suspensions of the indicated B. pertussis strains. The animals were sacrificed on day 6, and lungs were processed for staining with hematoxylin and eosin (see Materials and Methods for details) and scanned. Control mice received SS medium only. (A) Longitudinal sections of the left lobes at the original magnification of 1,25× that are representative of 3 serial sections per lung lobe. The right panels show enlargements of representative images of the bronchi and peribronchial parenchyma at magnification 20×. Lungs of animals infected by the AC+ Hly+ strain exhibited bronchopneumonia affecting the regions primarily around the large lobar bronchi. Significantly milder inflammation is observed in the lungs of animals infected by the AC+ Hly− and AC− Hly+ strains. (B) Inflamed parenchyma regions were manually delimited on three consecutive lung sections for 4 infected animals per group (12 sections analyzed in total) and scanned using an AxioScan.Z1 automated slide scanner, and the ratios of inflamed (infl.) to total parenchyma areas were calculated using the ZEN software. ** and *** represent P values of <0.01 and <0.001, respectively.
At a comparable level of bacterial load (cf. Fig. 3A), the infection by the AC+ Hly− mutant caused a distinctly milder pathology (Fig. 4A), with alveoli being generally well preserved, except for some rare focal subpleural alveolar pneumonic foci. The level of respiratory epithelium damage seen in animals infected by the AC+ Hly+ strain was not observed in animals infected by the nonhemolytic AC+ Hly− mutant, which provoked markedly milder lesions that occurred mainly in the form of irregular septal thickening and peribronchial inflammation. The alveolar structure remained, however, largely preserved, and no prominent damage to the pulmonary parenchyma was observed. An evident peribronchial inflammatory infiltrate was present, extending to the vicinity of the bronchus (i.e., the peribronchial space), but it rarely affected the adjacent alveoli. Some lung congestion was also observed, but it was less pronounced than in the lungs infected by the parental AC+ Hly+ strain (cf. Fig. 4A).
Infection with the hemolytic AC− Hly+ bacteria still resulted in mild peribronchial inflammation despite the presence of bacterial counts about a half an order of magnitude lower than those upon infection by the AC+ strains (cf. Fig. 3A). Moreover, infection with the AC− Hly+ bacteria reproducibly caused more pronounced lung congestion than infection by the AC+ Hly− mutant, while the small perivascular and peribronchial clusters of lymphocytes in AC− Hly+ strain-infected lungs matched those seen in control mice.
When the overall involvement of the tissue was quantified by a median ratio of inflamed to total area of the lung section, a value of 0.091 ± 0.024 (P ≤ 0.0005) was determined for the bronchi and parenchyma of lungs infected by the AC+ Hly+ strain (Fig. 4B). Significantly lower ratios of 0.021 ± 0.081 (P < 0.005) and 0.016 ± 0.041 (P < 0.0005) were then obtained for animals infected by the AC+ Hly− and AC− Hly+ strains. Hence, at comparable levels of bacterial counts (CFU per lung), the nonhemolytic AC+ Hly− mutant provoked a significantly milder lung inflammation than the parental AC+ Hly+ strain.
Moreover, a striking difference in the locations of the AC+ Hly+ and AC+ Hly− bacteria was observed (Fig. 5). Penetration of bacteria across the epithelial lining into the underlying lung tissue was observed by immunohistological staining in the lungs infected with the wild-type AC+ Hly+ B. pertussis strain, which provoked formation of bacterial foci and focal pneumonia (Fig. 5). In contrast, the nonhemolytic AC+ Hly− bacteria were detected almost exclusively as being attached to the epithelial lining in the bronchial lumen and were only rarely observed within the underlying lung tissue.
FIG 5.
The AC+ Hly− mutant does not penetrate into lung parenchyma. Lungs of BALB/c mice infected with 105 CFU of the indicated strains in 50 μl of suspensions were examined on day 6 upon immunohistochemical (IHC) staining for B. pertussis antigens with a polyclonal rabbit serum. The parental AC+ Hly+ B. pertussis infected the bronchial tree and invaded the lung alveoli and parenchyma, resulting in massive inflammatory response and pneumonic foci with cellular infiltrate (right panel). The AC+ Hly− mutant infected the bronchial tree and was found attached to the ciliated epithelial cells of bronchi without invading the parenchyma. IHC magnifications: left panels, 1.25×; right panels, 40×.
The cell-permeabilizing activity of CyaA contributes to neutrophil recruitment into infected lungs.
Previous work suggested that the cell-permeabilizing activity of CyaA contributed to NALP3 inflammasome activation and proinflammatory IL-1β cytokine secretion by TLR-activated DCs (28). As IL-1β is known to induce production of neutrophil chemokines by nonimmune cells in infected tissues (51), we enumerated the infiltrating neutrophil and macrophage cells in the sections of infected lungs upon NASDCL staining for neutrophils and following immunostaining for the F4/80 macrophage marker (Fig. 6A). A strongly increased number of neutrophils (0.50 ± 0.067/350 μm2; P < 0.0001) was, indeed, observed in the inflamed parenchyma of lungs infected by the parental hemolytic AC+ Hly+ strain (Fig. 6B), compared to lungs infected by similar counts of the nonhemolytic AC+ Hly− mutant (neutrophil count, 0.095 ± 0.027/350 μm2; P < 0.005). Moreover, despite lower numbers of the AC− Hly+ bacteria in mice lungs on day 6 of infection, the hemolytic mutant without cAMP elevation still provoked higher neutrophil infiltration into lung tissue (0.22 ± 0.034/350 μm2; P < 0.005) than the more efficiently multiplying nonhemolytic AC+ Hly− mutant (Fig. 6B), suggesting that the cell-permeabilizing activity of CyaA played a role in neutrophil attraction.
FIG 6.
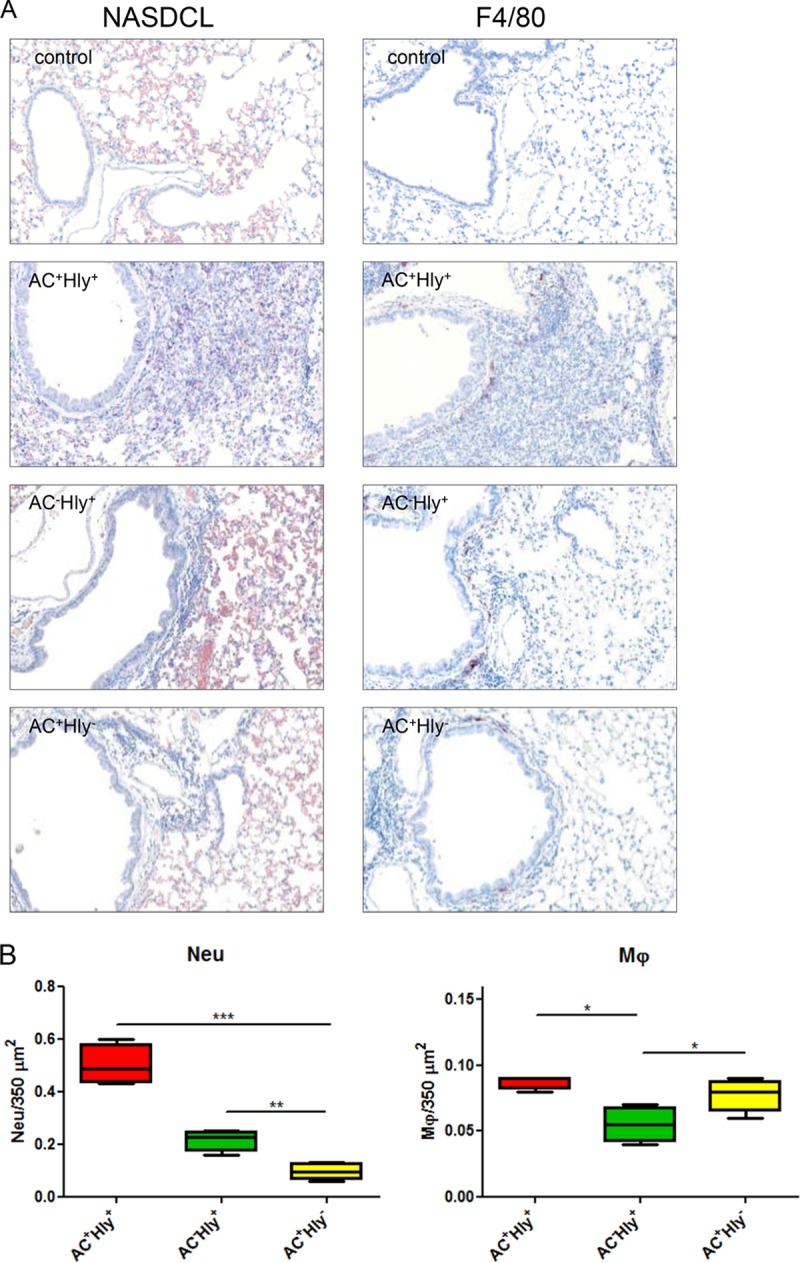
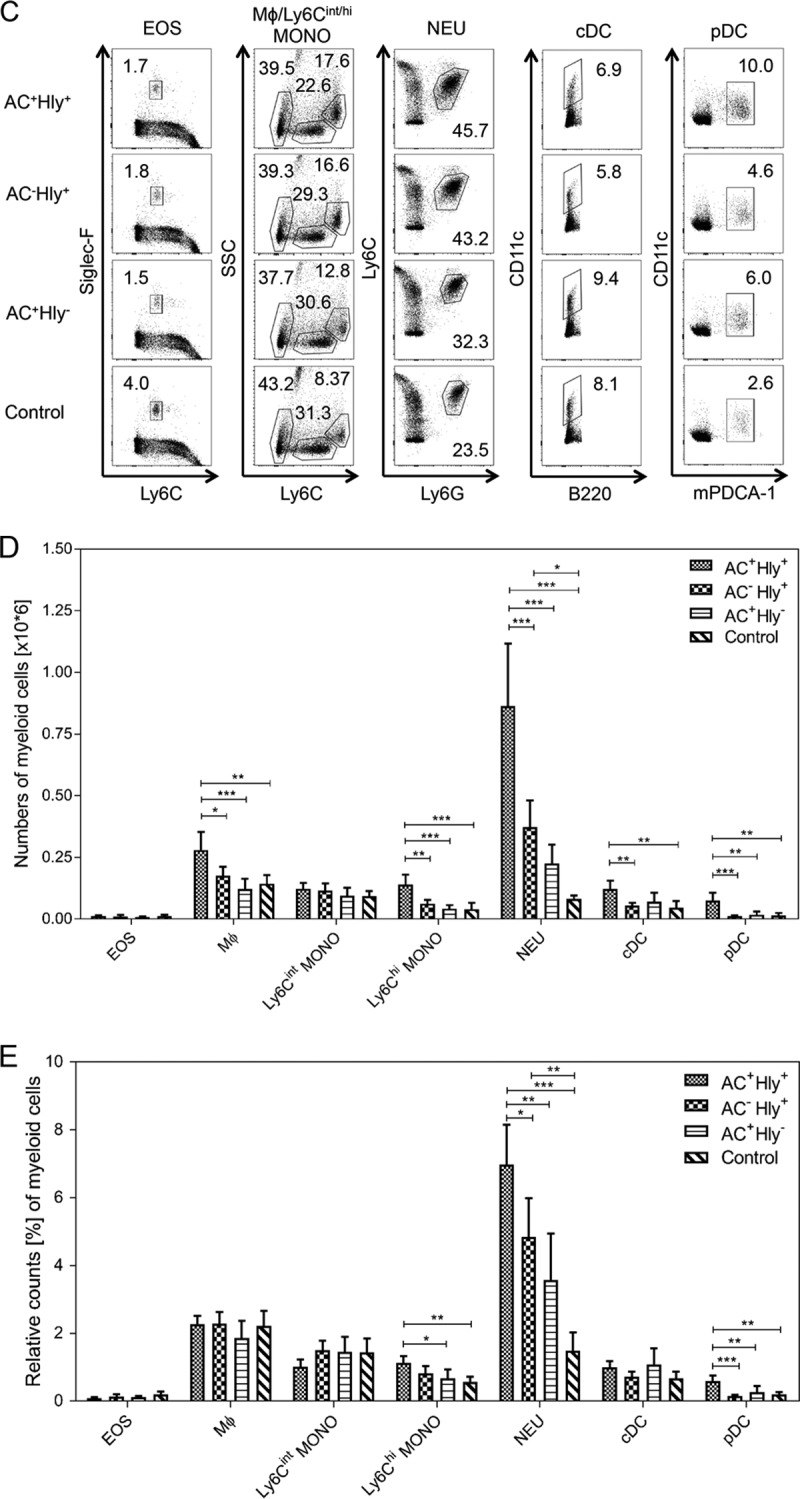
The cell-permeabilizing activity of CyaA triggers infiltration of neutrophils into B. pertussis-infected lungs. (A and B) BALB/c mice infected intranasally with 105 CFU of the indicated strains were sacrificed on day 6, and the sections of unperfused lung tissue were examined upon NASDCL histochemical and F4/80 immunohistochemical staining for neutrophils and macrophages, respectively. Representative sections documenting the main inflammatory cell components from the B. pertussis-induced lesions are shown. Neutrophil infiltration was significantly reduced in both AC+ Hly− and AC− Hly+ strain-infected lungs, while macrophage counts were reduced only in sections of lungs infected by the AC− Hly+ strain. Magnification, 20×. (B) The numbers of neutrophils and macrophages per surface unit were quantified by analysis of all inflamed parenchyma regions that were manually delimited on three consecutive lung sections for 4 infected animals per group (12 sections analyzed in total). (C to E) Distribution of cell subsets in infected mouse lungs. BALB/c mice were infected with 1.5 × 105 CFU of the various B. pertussis strains (AC+ Hly+, AC− Hly+, AC+ Hly−), using medium as a control, and suspensions of unperfused lungs were analyzed by flow cytometry. (C) Dot plots of cell subtypes in one representative mouse lung suspension per experimental group. (D) Total counts of indicated cell subsets in the infected lungs (n = 5) that were not perfused prior to homogenization. (E) Relative distribution of myeloid cell subsets in suspensions of nonperfused lungs (n = 5). Groups were compared using ANOVA followed by Tukey test for pairwise comparison of subgroups. *, **, and *** represent P values of <0.05, <0.01, and <0.001, respectively. The experiment was repeated twice with similar results. EOS, eosinophils; Mϕ, macrophages; MONO, monocytes; NEU, neutrophils; cDC, conventional dendritic cells; pDC, plasmacytoid dendritic cells.
To characterize in more detail the subpopulations of myeloid cells that infiltrated the infected lungs, immunostaining and multicolor flow cytometry were performed. Single-cell suspensions were prepared from nonperfused lungs on day 6 of infection, and the relative counts of various myeloid cell subsets were analyzed using the gating strategy outlined in Fig. S1 and S2 in the supplemental material. Dot plots obtained for one representative lung suspension for each infected mouse group are shown in Fig. 6C, and the average numbers of various myeloid cells per million for all lung cells and 5 mice per group are given in Fig. 6D. In agreement with the enumeration following NASDCL staining, the infection by the parental AC+ Hly+ strain yielded a significantly higher recruitment of neutrophils into lung tissue than the infection by comparable CFU of the nonhemolytic AC+ Hly− bacteria. While infection by the AC+ Hly− mutant caused a doubling of relative neutrophil counts compared to the control (∼3% versus ∼1.5% of all lung suspension cells [Fig. 6E]), infection by the AC+ Hly+ strain provoked more than a tripling of neutrophil counts in the lungs on day 6 of infection (∼7% of all cells in the lung suspensions being neutrophils). As already indicated by the histological examination, despite a lower capacity to multiply in the lungs, the hemolytic AC− Hly+ infection still elicited higher neutrophil infiltration than the infection by the more successfully infecting nonhemolytic AC+ Hly− mutant (cf. also Fig. 6B). This was not restricted to the infected lungs only. A systemic impact was also observed, with the relative counts of neutrophils in spleens of mice infected with the hemolytic AC+ Hly+ and AC− Hly+ bacteria being about 2 to 3 times increased in comparison to those in spleens of control mice (see Fig. S5 in the supplemental material).
Differences in relative counts of eosinophils, macrophages, Ly6Cint monocytes, and conventional dendritic cells in the lungs infected by the various strains were mostly insignificant (cf. Fig. 6E). There was also a discordance in macrophage infiltration levels as determined by histochemical analysis of manually selected segments of inflamed lung tissue (Fig. 6B) and by flow cytometry of total lung suspensions (Fig. 6C and D). The latter yielded a higher relative macrophage count for suspensions from AC− Hly+ strain-infected lungs than for suspensions from the AC+ Hly− sample, while the histology analysis of specific inflamed lung tissue regions yielded an opposite order of macrophage counts. This may be explained by enhanced counts of chemoattracted macrophages that remained in the vasculature of the AC+ Hly− strain-infected lungs that were not perfused prior to preparation of cellular suspensions for cytometry. In contrast, in the histochemical analysis, only cells that infiltrated into the inflamed parenchyma regions were counted.
The highest total counts of macrophage, Ly6Chigh monocytes and of cDCs and plasmacytoid dendritic cells (pDCs) were detected in the lungs of mice infected by the parental AC+ Hly+ strain (cf Fig. 6D). Intriguingly, these cells exhibited reduced mean levels of the major histocompatibility complex class II (MHC-II) molecule on the cell surface, compared to cells of the same type in the lungs infected by the AC− Hly+ and AC+ Hly+ mutants that are deficient in one of the two CyaA activities (Fig. 7C). Hence, the pore-forming (cell-permeabilizing) and the cAMP-elevating activities of the toxin synergized in provoking increased infiltration of MHC-II molecule-expressing cells into infected lung tissue, and the pore-forming activity contributed through an as yet unknown mechanism to reduction of the mean expression level of the MHC-II molecules on the incoming MHC-II+ myeloid cells. This effect of the cell-permeabilizing activity of CyaA manifested, however, only in situ at the site of infection. No decrease of the mean MHC-II expression levels was observed for myeloid cell populations in spleens (see Fig. S6 and S7 in the supplemental material).
FIG 7.
The cell-permeabilizing and AC enzyme activities of CyaA synergize in provoking reduction of MHC-II molecule expression on myeloid cells in infected lungs. Expression of the MHC-II molecule on selected cell subsets in lungs of BALB/c mice infected with B. pertussis 6 days after infection with 1.5 × 105 CFU of the various strains or with medium (Control). (A) Histograms for one representative mouse lung suspension per experimental group. (B) Total counts of MHC-II-expressing cells per million lung cells (n = 5). (C) Mean level (MFI) of MHC-II molecules detected on the surface of selected cell subsets in the infected lungs (n = 5). Groups were compared using ANOVA followed by Tukey test for pairwise comparison of subgroups. *, **, and *** represent P values of <0.05, <0.01, and <0.001, respectively. The experiment was repeated twice with similar results. Mϕ, macrophages; MONO, monocytes; cDC, conventional dendritic cells; pDC, plasmacytoid dendritic cells.
DISCUSSION
We used here a B. pertussis strain secreting a unique AC+ Hly− CyaA toxin construct that has the cell-permeabilizing capacity impaired and is essentially unable to bind and intoxicate non-myeloid (CD11b−) cells. Infections of mice with the AC+ Hly− B. pertussis mutant revealed that the mere capacity of CyaA to elevate cAMP concentrations in CR3-expressing myeloid (CD11b+) cells enables the pathogen to infect host airways. At comparable counts in the lungs, the nonhemolytic AC+ Hly− bacteria elicited much milder pathology than was observed upon infection by the parental AC+ Hly+ strain. Hence, the pore-forming (hemolysin) activity of CyaA and/or its capacity to elevate cAMP in non-myeloid cells, such as airway epithelial cells, accounted individually or in combination for the exacerbated pathology resulting from B. pertussis infection. Moreover, the nonhemolytic AC+ Hly− mutant was only found attached to the epithelial cells of the bronchial lumen, while the parental AC+ Hly+ B. pertussis bacteria were able to cross the epithelial lining and infiltrated the lung tissue, provoking formation of pneumonic foci. Thus, the impaired hemolytic (pore-forming) activity and/or the reduced capacity of the secreted CyaA-E570Q+K860R toxin to elevate cAMP in epithelial cells, individually or in combination, accounted for the reduced tissue invasiveness and virulence of the AC+ Hly− mutant. Whereas the parental AC+ Hly+ and the mutant AC+ Hly− bacteria multiplied in the lungs to the same extent, once inoculated at the same dose, an inoculation dose of the AC+ Hly− mutant more than an order of magnitude higher was, indeed, required to provoke death of infected mice, which still occurred almost a week later than upon infection by the AC+ Hly+ bacteria. This suggests that the lethality of B. pertussis infections in mice largely resulted from hyperactivation of the mouse innate defense, for which the synergy of the cell-permeabilizing action of the CyaA toxin with its cAMP-intoxicating capacity would account. Indeed, the cell-permeabilizing and cell-invasive AC enzyme activities of CyaA synergized in provoking neutrophil recruitment and inflammatory damage of infected lung tissue, which could be underlined by at least three already known mechanisms. First, CyaA-mediated elevation of the cAMP concentration in tracheal epithelial cells was previously shown to elicit production of the IL-6 cytokine, which is known to activate cytotoxic action of neutrophils at the site of infection (2, 40, 52). Second, cAMP signaling of CyaA was shown to induce cyclooxygenase 2 (COX-2) expression in macrophages, and this would yield release of prostaglandins that serve in chemoattraction of neutrophils (53). The third mechanism would involve permeabilization of cells by CyaA pores and potassium efflux from cells, thus enhancing assembly of the NALP3 inflammasome in TLR-primed intraepithelial dendritic cells (DCs). This was shown to trigger activation of caspase-1 and secretion of the proinflammatory cytokine IL-1β (28), which triggers neutrophil chemokine production by nonimmune bystander cells (51). Potentiation of neutrophil infiltration into lung parenchyma by these parallel mechanisms of CyaA action would then contribute to the observed extent of inflammatory damage of B. pertussis-infected lungs.
It is noteworthy that the reduced capacity of the AC− Hly+ mutant to multiply in infected lungs observed here was much less pronounced than that reported earlier by Khelef and coworkers (3). Khelef et al. used a higher challenge dose of 106.5 to 107CFU of the AC− Hly+ B. pertussis mutant, whereas we used only 105 CFU/mouse. This would suggest that at the higher inoculation dose, the AC− Hly+ bacteria provoke enhanced infiltration of neutrophils into the infected tissue and are then rapidly eliminated because they are unable to paralyze their bactericidal action by elevation of cAMP.
The nonhemolytic AC+ Hly− mutant multiplied in infected mouse lungs as efficiently as the parental AC+ Hly+ B. pertussis. This goes well with the previously reported effects of CyaA action on sentinel cells of innate immunity, where the produced cAMP signaling swiftly disarms neutrophils, promotes apoptosis of alveolar macrophages, and subverts the TLR ligand-induced dendritic cell maturation and cytokine secretion (43–49). We confirmed here that B. pertussis-produced CyaA suppresses IL-12 and TNF-α release and enhances secretion of immunosuppressive IL-10 by mouse dendritic cells that are in contact with live B. pertussis bacteria. The use of the AC+ Hly− mutant further allowed us to show in vitro that the pore-forming activity of CyaA was not contributing to the immunosuppressive shaping of DCs, which was entirely due to the capacity of CyaA to elevate cytosolic cAMP in DCs (cf. Fig. 2C). The latter interfered with presentation of the OVA antigen on DCs exposed to live B. pertussis bacteria and reduced the antigen-specific IFN-γ secretion by coincubated T cells. At the same time, the in vitro action of the B. pertussis-secreted CyaA yielded enhanced antigen-specific secretion of IL-17A and IL-10 by coincubated antigen-specific CD8+ and CD4+ T lymphocytes (cf. Fig. 2D) and promoted expansion of antigen-specific Foxp3+ CD25+ CD4+ regulatory T cells (Treg cells) by the CyaA-hijacked DCs. These results obtained with live bacteria, which also produce other B. pertussis toxins (e.g., PT) and virulence factors (dermonecrotic toxin, adhesins, tracheal cytotoxin, etc.), hence corroborate and validate our previous observations with purified recombinant CyaA. These studies showed that as little as 10 ng/ml of CyaA was able to skew the antigen-presenting capacities of LPS-activated dendritic cells toward a tolerogenic phenotype and made them expand antigen-specific Foxp3+ CD25+ CD4+ Treg cells (54). Higher CyaA concentrations were, indeed, recently found in nasal aspirates of infants with pertussis and in nasopharyngeal washes of B. pertussis-infected baboons (12). It thus appears plausible to assume that CyaA may play a rather important role in suppression of adaptive T cell immune responses during natural infections by B. pertussis and that the action of CyaA may be involved in the previously observed expansion of regulatory T cells in B. pertussis-infected lungs in vivo (55).
Intriguingly, despite comparable levels of bacterial multiplication in the lung, the parental AC+ Hly+ B. pertussis provoked a significantly higher infiltration of myeloid phagocytic cells into the infected lung tissue than did comparable counts of the AC+ Hly− bacteria. This indicates that the cell-permeabilizing activity of the CyaA toxin was contributing to attraction of neutrophil, macrophage, Ly6Chigh monocyte, and conventional and plasmacytoid dendritic cells into B. pertussis-infected tissue. Alternatively, this cellular infiltration may also have been provoked by cAMP signaling-induced chemokine production by CD11b− bystander cells. This would have been triggered only by the AC+ Hly+ B. pertussis, but not by bacteria producing the nonhemolytic AC+ Hly− toxin unable to intoxicate CD11b− cells. At present, we are unable to clearly distinguish if the much lower inflammation produced by the infection with AC+ Hly− bacteria was due to the absence of phagocyte permeabilization, the absence of cAMP elevation in CD11b− bystander cells, or both. Therefore, efforts are being undertaken to construct a next generation of nonhemolytic B. pertussis bacteria that would produce an AC+ Hly− toxin capable of elevating cAMP also in the CD11b− non-myeloid cells. If successful, such constructs will serve to address the mechanism by which the cell-permeabilizing activity synergized with the CyaA-produced cAMP signaling in causing reduced expression of the MHC-II molecules on the myeloid cells infiltrating the infected lung tissue. This was observed for several types of antigen-presenting cells, such as macrophages, monocytes, pDCs, or cDCs. It is thus likely to have a nonnegligible impact on the efficacy of antigen presentation to CD4+ T helper cells in the course of B. pertussis infection. Such subversion of intraepithelial DC function would then be plausibly expected to hamper adaptive responses of B lymphocytes as well. Delaying and restricting the efficacy of the antibody response and limiting development of the B and T cell immune memory to B. pertussis antigens may thus represent a second role of the immunosubversive activity of CyaA.
MATERIALS AND METHODS
Bacterial strains, growth conditions, and plasmids.
Escherichia coli XL1-Blue (Stratagene) was used for DNA manipulation and CyaA production. Bacteria were grown in Luria-Bertani (LB) medium supplemented with ampicillin (150 μg/ml). Escherichia coli SM10 λpir (56) was used for plasmid transfer into B. pertussis by bacterial conjugation. The E. coli SM10 λpir transformants were grown at 37°C in LB agar medium supplemented with kanamycin (60 μg/ml) and ampicillin (150 μg/ml). The B. pertussis Tohama I strain was generously provided by Nicole Guiso from Institut Pasteur, Paris (57). The parental strain and B. pertussis mutants were grown at 37°C on Bordet-Gengou agar medium (Difco, USA) supplemented with 1% glycerol and 15% defibrinated sheep blood (BG) and 5% CO2 for 72 h to visualize hemolysis. Liquid cultures were grown in modified Stainer-Scholte (SS) medium (58) (supplemented with 1 g/liter of Casamino Acids and 1 g/liter of cyclodextrin) for 18 h at 37°C. The suicide vector pSS4245, generously provided by Scott Stibitz, was used for allelic exchange on the B. pertussis chromosome, as described previously (59).
Mutagenesis of the cyaA gene on B. pertussis chromosome.
Construction of the alleles for production of the CyaA-AC− (AC− Hly+) toxoid and of the nonhemolytic CyaA-Hly− (AC+ Hly−) toxin mutant in E. coli was described in detail previously (37). The respective mutated cyaA gene segments were cloned into exchange vector plasmid pSS4245, which contains an Strr allele functional in B. pertussis but not in E. coli. Prior to mating, the B. pertussis Tohama I strain was grown for 4 days under modulating conditions on BG agar supplemented with 50 mM MgSO4 (Bvg− conditions), and mating with E. coli SM10 λpir transformed by the suicide plasmid constructs was performed on fresh BG agar plates supplemented with 10 mM MgCl2 and 50 mM MgSO4 for 3 h at 37°C. B. pertussis cells having incorporated the allelic exchange plasmid by single crossing-over into the chromosome were selected on BG agar supplemented with 50 mM MgSO4 and 500 μg/ml of streptomycin, 30 μg/ml of ampicillin, and 40 μg/ml of kanamycin for 5 days at 37°C. The resulting single colonies were streaked on the same plates for an additional 5 days before plating on BG agar lacking MgSO4 (Bvg+ conditions) to select for the second crossing-over and excision of the allelic exchange vector. For each construct, several individual colonies were characterized for phenotypic change, and the presence of introduced mutations and absence of undesired mutations were verified by sequencing of relevant portions of PCR-amplified segments of the cyaA gene. Production of PT, PRN, FHA, and CyaA antigens was verified by Western blotting of bacterial suspensions using specific mouse antibodies (60).
Preparation of urea extracts for CyaA toxin assays.
B. pertussis strains were grown for 72 h on Bordet-Gengou agar and subcultured in 2 ml of Stainer-Scholte medium to an A600 of 1, collected by centrifugation (1 min at 15,000 × g), and resuspended in 200 μl of 8 M urea, 50 mM Tris (pH 7.4), and 2 mM CaCl2 (TUC buffer). The urea extracts were cleared by centrifugation and used for determination of AC and CyaA toxin activities.
Assay of AC activity.
Adenylate cyclase enzymatic activity was measured in the presence of 1 μM calmodulin as previously described (61). One unit of AC activity corresponds to 1 μmol of cAMP formed per min at 30°C and pH 8.0.
Binding and cell-invasive activities in RBCs.
Erythrocyte binding and cell-invasive AC activities were determined as described in detail previously (25). Briefly, sheep erythrocytes were harvested for the purpose of this study, washed in TNC buffer (20 mM Tris-HCl at pH 7.4, 150 mM NaCl, 2 mM CaCl2), adjusted to 5 × 108 cells/ml, and incubated with bacterial lysates at 37°C in TNC buffer. After 30 min, cell suspensions were washed in TNC buffer to remove unbound CyaA and divided into two aliquots. One aliquot was directly used to determine the amount of cell-associated AC activity (membrane-bound CyaA). The other aliquot was treated with 20 μg/ml of trypsin for 15 min at 37°C in order to inactivate the extracellular AC enzyme that did not translocate across the cellular membrane. Soybean trypsin inhibitor (40 μg/ml) was then added to the mixture to stop the reaction before the samples were washed three times with 20 mM Tris-HCl at pH 7.4, 150 mM NaCl, and 5 mM EDTA and used to determine the amount of cell-invasive AC enzyme activity. The activity of intact CyaA was taken as 100%.
Generation of mouse BMDCs.
Bone marrow-derived dendritic cells (BMDCs) were generated according to the method of Lutz et al. (62). Briefly, bone marrow cells were flushed from femurs and tibias of C57BL/6 mice (harvested for this study) and cultured at 2 × 106/ml in 100-mm-diameter dishes in 10 ml of RPMI 1640 medium supplemented with 10% fetal calf serum (FCS) (Life Technologies, USA), 0.1 mg/ml streptomycin, 1,000 U/ml penicillin, and 0.25 μg/ml amphotericin (Sigma-Aldrich, USA), 50 μM 2-mercaptoethanol, 1% nonessential amino acids (Biochrom, Germany), 1 mM sodium pyruvate, 2 mM glutamine, and 200 U/ml granulocyte-macrophage colony-stimulating factor (GM-CSF; PeproTech). Fresh medium was added on days 3 and 6. Loosely attached cells were used for experiments on days 6 to 8. Seventy to 80% of cultured cells expressed CD11c (allophycocyanin [APC]-conjugated anti-mouse CD11c antibody, clone N418; eBioscience, USA) and 90% CD11b (phycoerythrin [PE]-conjugated anti-mouse CD11b antibody, clone M1/70; BD Pharmingen, USA).
cAMP elevation capacity of CyaA in DCs.
For determination of CyaA toxin activity (elevation of intracellular cAMP), BMDCs (106/ml) were incubated with appropriately diluted bacterial lysates (final CyaA concentration of 3 mU/ml) for 1 h at 37°C in Dulbecco's modified Eagle's medium (DMEM), and the reaction was stopped by addition of 0.2% Tween 20 in 100 mM HCl. Samples were boiled for 15 min at 100°C and neutralized by addition of 150 mM unbuffered imidazole, and cAMP concentrations were measured by enzyme-linked immunosorbent assay (ELISA) as previously described (63). The results represent the average of values obtained in at least two independent experiments performed in duplicate.
Western blotting.
Bacteria were grown in liquid Stainer-Scholte medium for 18 h at 37°C, and cells from 1 ml of bacterial suspension were collected by centrifugation (10 min, 15,000 × g). The pellet was resuspended in 100 μl of Laemmli buffer (50 mM Tris [pH 6.8], 2% SDS, 0.1% bromophenol blue, 10% glycerol, 1% β-mercaptoethanol) and dissolved for 5 min at 100°C. Ten microliters of lysates was separated by 7.5% SDS-polyacrylamide gel electrophoresis (PAGE) and transferred to a nitrocellulose membrane. CyaA was probed by the anti-RTX monoclonal antibody 9D4 (at a 1:3,000 dilution) (64) and revealed by peroxidase-conjugated secondary antibody (at a 1:10,000 dilution; GE Healthcare, UK) using a chemiluminescence detection system (Thermo Fisher Scientific, USA) and an LAS-4000 imaging system instrument (Fuji, Japan).
Ethics statement.
All animal experiments were approved by the Animal Welfare Committee of the Institute of Microbiology of the ASCR, v. v. i., in Prague, Czech Republic. Handling of animals was performed according to the Guidelines for the Care and Use of Laboratory Animals, the Act of the Czech National Assembly, Collection of Laws no. 149/2004, inclusive of the amendments, on the Protection of Animals against Cruelty, and Public Notice of the Ministry of Agriculture of the Czech Republic, Collection of Laws no. 207/2004, on Care and Use of Experimental Animals.
Intranasal infection of mice.
Four-week-old female BALB/c or Swiss CD-1 mice (AnLab, Janvier) were used in this study. Mice were anesthetized by intraperitoneal (i.p.) injection of ketamine (80 mg/kg) and xylazine (8 mg/kg) in saline and were inoculated intranasally with 50 μl of B. pertussis cell suspensions grown for 24 h in modified Stainer-Scholte liquid medium. To determine viable CFU, the suspensions were diluted and plated on BG agar plates.
Infection experiments.
Infected Swiss CD-1 mice were sacrificed by cervical dislocation 2 h after exposure to challenge suspension (day 0 + 2 h) and on the indicated days thereafter (day 5, 8, 12, and 21). The lungs were aseptically removed and homogenized in physiological solution with tissue grinders. Dilutions of lung homogenates were plated on BG agar plates, and CFU were counted after 4 days of incubation at 37°C. Three mice per time point were used, and this experiment was repeated eight times in total for the parental B. pertussis Tohama I strain, seven times for the B. pertussis AC− Hly+ mutant, and six times for the B. pertussis AC+ Hly− mutant, using challenge doses ranging from 104 to 5 × 106 CFU, yielding very similar results and courses of infection. The average value of the results obtained with a challenge dose of 105 CFU is shown.
Determination of LD50.
For determination of the LD50 values, groups of mice were challenged intranasally with serially diluted bacterial suspensions, and their survival was monitored over 10 days. The results expressed as the percentage of surviving mice represent the average of pooled values obtained in seven experiments with the parental B. pertussis Tohama I strain and B. pertussis AC− Hly+ mutant and three experiments with the B. pertussis AC+ Hly− mutant, using 6 to 8 mice per challenged group per challenge dose and individual experiment. The LD50 values were calculated by the probit analysis method of Finney using the freeware LD50/LC50 calculator downloaded at https://goo.gl/9QYcNk, as described at https://probitanalysis.wordpress.com/2016/07/07/first-blog-post/.
Histological studies.
Lung morphology was examined upon intranasal challenge of BALB/c mice with a dose of 1.5 × 105 CFU. Control mice were given 50 μl of Stainer-Scholte medium only. Mice were anesthetized by i. p. injection of ketamine (80 mg/kg) and xylazine (8 mg/kg) in saline and sacrificed by cervical dislocation. The tracheas were cannulated, and lungs were inflated with infusion of 4% (wt/vol) buffered formaldehyde solution into the thorax, excised, and immersed in 4% (wt/vol) phosphate-buffered formaldehyde for 48 h. Afterwards all samples were processed in a Leica ASP6025 tissue processor (Leica Biosystems, Nussloch, Germany), molded in paraffin, and cut at a 3-μm thickness. Hematoxylin and eosin (HE) staining was carried out using automated Ventana Symphony stainer (Ventana Medical Systems, Tucson, AZ). Naphthol AS-D chloroacetate esterase (NASDCL) histochemistry staining of neutrophils was performed using an NASDCL staining kit (Sigma-Aldrich, Germany) according to the manufacturer's instructions. For immunohistochemistry staining for the macrophage marker F4/80 and for B. pertussis cells, the histological sections were rehydrated and retrieved in the pH 9 antigen retrieval solution (Zytomed GmbH, Berlin, Germany), incubated for 1 h at room temperature (RT) with the mouse anti-F4/80 monoclonal antibody (MAb) (clone Cl:A3-1; Bio-Rad, CA) or with the rabbit anti-B. pertussis serum (kind gift of B. Vecerek), both at a 1:1,000 dilution, followed by secondary anti-mouse or anti-rabbit peroxidase-conjugated polymer (Zytomed, GmbH, Berlin, Germany) for 30 min at RT, respectively. The immune reaction was developed using 3-amino-9-ethylcarbazole (AEC) solution (Dako, Glostrup, Denmark) for 10 min at RT. Slides were mounted with Aquatex mounting medium (Merck-Millipore, Darmstadt, Germany) and scanned using an AxioScan.Z1 automated slide scanner (Carl Zeiss, Göttingen, Germany). Representative images were generated using the ZEN software (Carl Zeiss, Göttingen, Germany). Quantification of F4/80-stained cell (macrophages) and neutrophils was performed on full-slide scans where the two cell types were counted only in the inflammatory areas. The significance of the differences between groups was determined by unpaired two-tailed Student's t test. Differences were considered statistically significant at P < 0.05 (*) and P < 0.01 (**).
MAbs for cell phenotyping.
The following anti-mouse MAbs were used for flow cytometry analysis: CD3-V500, CD4-V500, CD8-V500, CD45R (B220)-V500, CD45-peridinin chlorophyll protein (PerCP), Ly6G-A700, Siglec-F–phycoerythrin (PE) (BD Biosciences); CD45R (B220)-A488, CD11b-eF450, CD11c-eF450, Ly6C-A488, MHC-II (I-A/I-E)–allophycocyanin (APC) (eBioscience), and mPDCA-1–PE (Miltenyi Biotec). Nonspecific binding of cells was blocked by anti-mouse CD16/CD32 MAb (eBiosciences).
Surface staining and flow cytometry analysis.
Single-cell suspensions were prepared from unperfused lungs and spleens of BALB/c mice by homogenization in a GentleMACS dissociator (Miltenyi Biotech) and digestion with collagenase D (1 mg/ml; Roche) in Hanks' balanced salt solution (HBSS) (Sigma-Aldrich) for 30 min at 37°C, followed by incubation with 50 mM EDTA (Gibco) for 5 min, repeated homogenization and filtration through a 70-μm nylon cell strainer (BD Falcon). After RBC lysis (ACK lysing buffer; Gibco), cells were resuspended in fluorescence-activated cell sorter (FACS) buffer (phosphate-buffered saline [PBS], 2% fetal calf serum [FCS; Gibco], 2 mM EDTA), blocked by 10% BALB/c mouse serum and anti-mouse CD16/CD32 MAb (0.5 μg/sample; eBioscience) for 20 min on ice, and stained with fluorochrome-labeled MAbs for 30 min on ice in the dark. Cells were washed twice after each step in FACS buffer, fixed in BD Cytofix/Cytoperm solution (BD Biosciences) for 1 h on ice, and resuspended in FACS buffer. Flow cytometric analysis was performed on an LSRII flow cytometer (BD Biosciences), and data were analyzed using FlowJo X software (Tree Star). In the case of lung samples, 200,000 CD45+ cells with excluded doublets and debris were run. The depicted data represent single-cell populations after exclusion of doublets and debris and being gated from the CD45+ population (for the eosinophil population) or CD45+ DUMP− population: i.e., CD45+ CD3− CD4− CD8− B220− for the monocyte, macrophage, and neutrophil population or CD45+ CD3− for the dendritic cell population (Fig. S1 and S2). In the case of spleen samples, 200,000 lymphocytes were analyzed. The depicted data represent single-cell populations after exclusion of doublets and debris (for the eosinophil population) and cell populations gated from the DUMP− population: i.e., CD3− CD4− CD8− B220− for the monocyte, macrophage, and neutrophil population or CD3− for the dendritic cell population (see Fig. S3 and S4 in the supplemental material). All dot plots show 10,000 representative cells for better clarity.
Mice and cell lines for analysis of DC maturation.
Six- to 8-week-old C57BL/6 mice were purchased from the specific-pathogen-free (SPF) breeding facility of the Institute of Molecular Genetics of the ASCR, v.v.i., in Prague, Czech Republic. OT-I mice are transgenic for the T cell receptor recognizing the ovalbumin (OVA) epitope OVA257–264 in the context of H-2Kb. The OT-II mice bear the transgenic T cell receptor recognizing OVA323–339 in the context of I-Ab and were a generous gift from Pavel Otahal, Institute of Molecular Genetics of the ASCR, v.v.i., in Prague, Czech Republic.
T cells were cultured in RPMI 1640 medium supplemented with 10% FCS (Life Technologies, USA), 0.1 mg/ml streptomycin, 1,000 U/ml penicillin, 0.25 μg/ml amphotericin (Sigma-Aldrich, USA), 50 μM 2-mercaptoethanol, 1% nonessential amino acids (Biochrom, Germany), 1 mM sodium pyruvate, 2 mM glutamine, and 6.5 g/liter glucose. RPMI 1640 medium without antibiotics was used for the infection experiments with B. pertussis.
Infection of DCs.
DCs (106/ml) in DMEM without antibiotics were exposed to the suspension of heat-killed (70°C, 30 min) parental B. pertussis bacteria or were infected with live suspensions of the indicated B. pertussis strains at a multiplicity of infection (MOI) of 100:1 bacteria to DCs. After addition of bacteria to DCs, plates were gently centrifuged (700 × g for 5 min) to enable bacterial attachment to DCs. After 1 h of incubation, extracellular bacteria were killed by addition of kanamycin (100 μg/ml), and incubation with DCs continued for the indicated times. Control cell suspensions were supplemented with kanamycin (100 μg/ml) as well.
Detection of DC viability.
DCs (5 × 105/well) were infected as described above. After 1 h of bacterial infection at 100:1, killing of bacteria by kanamycin, and 3 h of further incubation, the DCs were incubated with tetramethylrhodamine ethyl ester (TMRE; 40 nM [Invitrogen]) for 15 min at 37°C and subsequently stained with Hoechst 33258 (0.5 μg/ml; Invitrogen, USA). Live (TMRE+, Hoechst 33258−) cells were detected by flow cytometry using the FACS LSR II instrument (BD Bioscience, USA) and analyzed by the flow cytometry software FlowJo version 7.2.1 (Treestar, Inc.).
Maturation of DCs.
DCs (2 × 106/well) were incubated with B. pertussis bacteria at an MOI of 100:1, as outlined above, for 24 h. DCs were next stained with APC-conjugated anti-mouse CD11c (clone N418) and one of the following antibodies: fluorescein isothiocyanate (FITC)-conjugated anti-mouse I-A/I-E (clone M5/114.15.2), FITC–anti-mouse CD80 (clone 16-10A1), FITC–anti-mouse CD86 (clone GL1), FITC–anti-mouse CD54 (clone YN1/1.7.4) from eBioscience (USA), or FITC–anti-mouse CD40 (clone 3.23) and FITC–anti-mouse H-2Kb (clone AF6-88.5) from BD Pharmingen (USA), respectively. The expression of cell surface markers by live CD11c+ cells was detected by flow cytometry using 106 cells. Mean fluorescence intensity (MFI) values of samples were determined using FlowJo X software (Tree Star). The expression of surface markers of live DCs treated with HI-Bp was set to 100%.
Cytokine production by DCs.
The cell culture supernatants of DCs (2 × 106/well) were taken after 24 h of incubation with bacteria, and the concentrations of IL-10, IL-12p70, and TNF-α were determined using a mouse IL-10 and TNF-α ELISA Max standard kit (BioLegend, USA) and mouse IL-12p70 Duo Set ELISA kit (RD Systems, USA) according to the manufacturer's instructions, taking induction by HI-Bp as 100%.
OVA-specific T cell responses and expansion of CD4+ CD25+ Foxp3+ T cells.
Naive OVA-specific CD8+ or CD4+ T cells were isolated from lymph nodes and spleen of OT-I or OT-II transgenic mice, respectively, using magnetic cell separation (MACS) with CD8+ and CD4+ T cell isolation kits (Miltenyi, Germany) according to the manufacturer's instructions. DCs (5 × 104/well) were seeded in a 96-well plate and coincubated with the OVA protein (albumin from chicken egg white; Calbiochem, USA) and bacteria as outlined above. After 1 h of incubation, bacteria were killed by addition of kanamycin (100 μg/ml) and incubation of DCs was continued for another 4 h in the presence of ovalbumin (OVA). For subsequent MHC class II presentation to OT-II CD4+ T cells, the OVA antigen at a concentration of 0.1 μM was used. OVA at 0.2 μM was used to assess MHC class I presentation to OT-I CD8+ T cells. After 4 h, the DCs were washed and 2 × 105/well naive OVA-specific CD8+ or CD4+ T cells were added and coincubated with pretreated DCs. The production of IL-17, IFN-γ, and IL-10 (RD Systems, USA) in cell culture supernatant after 3 days was determined by ELISA. The remaining CD4+ T cells were collected and stained with APC–anti-mouse CD3, Alexa 488–anti-mouse CD25, and PE–anti-mouse Foxp3 (eBioscience, USA) using fixation and permeabilization buffers from eBioscience, USA. The expression of Foxp3 in CD4+ CD25+ T cells was determined by flow cytometry.
Statistical analysis.
The significance of the differences between groups was in general determined by unpaired two-tailed Student's t test. Differences were considered statistically significant at P < 0.05 (*) and P < 0.01 (**). Where appropriate (as indicated in the figure legends), the data were analyzed by analysis of variance (ANOVA) followed by Tukey test for pairwise comparison of subgroups: *, **, and *** represent P values of <0.05, <0.01, and <0.001, respectively. Data are representative of at least two experiments.
Supplementary Material
ACKNOWLEDGMENTS
Help with mouse challenge experiments by Anne-Sophie Paris is gratefully acknowledged. K. Skopova is a doctoral student of the University of Chemistry and Technology, and M. Svedova is a doctoral student of the Charles University in Prague.
This work, including the efforts of Peter Sebo, Marek Kovar, Radim Osicka, Jiri Masin, and Karolina Skopova, was funded by grants NV16-28126A, GA13-14547S, GA13-12885S, GA15-09157S, GAP302/12/0460, and RVO 61388971 and the Czech National Node to EATRIS project LM2015064. The efforts of Nicole Guiso were funded by the institutional funding of Institut Pasteur and CNRS URA-3012. The efforts of Radislav Sedlacek and Ivan Kanchev were funded by the MYES of the CR from the Czech Centre for Phenogenomics project LM2015040.
The authors made the following contributions to this article: P.S., idea generation and manuscript preparation; K.S., B.T., I.K., P.R., M.S., I.A., I.B., J.T., and J.M., performing experiments; and P.S., K.S., B.T., I.K., P.R., M.S., I.A., I.B., J.T., J.M., N.G., R.O., R.S., and M.K., study design and figure design.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00937-16.
REFERENCES
- 1.Goodwin MS, Weiss AA. 1990. Adenylate cyclase toxin is critical for colonization and pertussis toxin is critical for lethal infection by Bordetella pertussis in infant mice. Infect Immun 58:3445–3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khelef N, Bachelet CM, Vargaftig BB, Guiso N. 1994. Characterization of murine lung inflammation after infection with parental Bordetella pertussis and mutants deficient in adhesins or toxins. Infect Immun 62:2893–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khelef N, Sakamoto H, Guiso N. 1992. Both adenylate cyclase and hemolytic activities are required by Bordetella pertussis to initiate infection. Microb Pathog 12:227–235. doi: 10.1016/0882-4010(92)90057-U. [DOI] [PubMed] [Google Scholar]
- 4.Guermonprez P, Khelef N, Blouin E, Rieu P, Ricciardi-Castagnoli P, Guiso N, Ladant D, Leclerc C. 2001. The adenylate cyclase toxin of Bordetella pertussis binds to target cells via the alpha(M)beta(2) integrin (CD11b/CD18). J Exp Med 193:1035–1044. doi: 10.1084/jem.193.9.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Osicka R, Osickova A, Hasan S, Bumba L, Cerny J, Sebo P. 2015. Bordetella adenylate cyclase toxin is a unique ligand of the integrin complement receptor 3. eLife 4:e10766. doi: 10.7554/eLife.10766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Confer DL, Eaton JW. 1982. Phagocyte impotence caused by an invasive bacterial adenylate cyclase. Science 217:948–950. doi: 10.1126/science.6287574. [DOI] [PubMed] [Google Scholar]
- 7.Kamanova J, Kofronova O, Masin J, Genth H, Vojtova J, Linhartova I, Benada O, Just I, Sebo P. 2008. Adenylate cyclase toxin subverts phagocyte function by RhoA inhibition and unproductive ruffling. J Immunol 181:5587–5597. doi: 10.4049/jimmunol.181.8.5587. [DOI] [PubMed] [Google Scholar]
- 8.Pearson RD, Symes P, Conboy M, Weiss AA, Hewlett EL. 1987. Inhibition of monocyte oxidative responses by Bordetella pertussis adenylate cyclase toxin. J Immunol 139:2749–2754. [PubMed] [Google Scholar]
- 9.Prior S, Corbel MJ, Xing DK. 2005. Development of an approach for the laboratory toxicological evaluation of Bordetella pertussis adenylate cyclase genetic toxoid constructs as multipurpose vaccines. Hum Vaccin 1:151–159. doi: 10.4161/hv.1.4.1972. [DOI] [PubMed] [Google Scholar]
- 10.Vojtova J, Kamanova J, Sebo P. 2006. Bordetella adenylate cyclase toxin: a swift saboteur of host defense. Curr Opin Microbiol 9:69–75. doi: 10.1016/j.mib.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 11.Hanski E. 1989. Invasive adenylate cyclase toxin of Bordetella pertussis. Trends Biochem Sci 14:459–463. doi: 10.1016/0968-0004(89)90106-0. [DOI] [PubMed] [Google Scholar]
- 12.Eby JC, Gray MC, Warfel JM, Paddock CD, Jones TF, Day SR, Bowden J, Poulter MD, Donato GM, Merkel TJ, Hewlett EL. 2013. Quantification of the adenylate cyclase toxin of Bordetella pertussis in vitro and during respiratory infection. Infect Immun 81:1390–1398. doi: 10.1128/IAI.00110-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Donato GM, Goldsmith CS, Paddock CD, Eby JC, Gray MC, Hewlett EL. 2012. Delivery of Bordetella pertussis adenylate cyclase toxin to target cells via outer membrane vesicles. FEBS Lett 586:459–465. doi: 10.1016/j.febslet.2012.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eby JC, Ciesla WP, Hamman W, Donato GM, Pickles RJ, Hewlett EL, Lencer WI. 2010. Selective translocation of the Bordetella pertussis adenylate cyclase toxin across the basolateral membranes of polarized epithelial cells. J Biol Chem 285:10662–10670. doi: 10.1074/jbc.M109.089219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Masin J, Osicka R, Bumba L, Sebo P. 2015. Bordetella adenylate cyclase toxin: a unique combination of a pore-forming moiety with a cell-invading adenylate cyclase enzyme. Pathog Dis 73:ftv075. doi: 10.1093/femspd/ftv075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sebo P, Osicka R, Masin J. 2014. Adenylate cyclase toxin-hemolysin relevance for pertussis vaccines. Expert Rev Vaccines 13:1215–1227. doi: 10.1586/14760584.2014.944900. [DOI] [PubMed] [Google Scholar]
- 17.Rose T, Sebo P, Bellalou J, Ladant D. 1995. Interaction of calcium with Bordetella pertussis adenylate cyclase toxin. Characterization of multiple calcium-binding sites and calcium-induced conformational changes. J Biol Chem 270:26370–26376. [DOI] [PubMed] [Google Scholar]
- 18.Bumba L, Masin J, Macek P, Wald T, Motlova L, Bibova I, Klimova N, Bednarova L, Veverka V, Kachala M, Svergun DI, Barinka C, Sebo P. 2016. Calcium-driven folding of RTX domain β-rolls ratchets translocation of RTX proteins through type I secretion ducts. Mol Cell 62:47–62. doi: 10.1016/j.molcel.2016.03.018. [DOI] [PubMed] [Google Scholar]
- 19.Hackett M, Guo L, Shabanowitz J, Hunt DF, Hewlett EL. 1994. Internal lysine palmitoylation in adenylate cyclase toxin from Bordetella pertussis. Science 266:433–435. doi: 10.1126/science.7939682. [DOI] [PubMed] [Google Scholar]
- 20.Hackett M, Walker CB, Guo L, Gray MC, Van Cuyk S, Ullmann A, Shabanowitz J, Hunt DF, Hewlett EL, Sebo P. 1995. Hemolytic, but not cell-invasive activity, of adenylate cyclase toxin is selectively affected by differential fatty-acylation in Escherichia coli. J Biol Chem 270:20250–20253. doi: 10.1074/jbc.270.35.20250. [DOI] [PubMed] [Google Scholar]
- 21.Hewlett EL, Gray L, Allietta M, Ehrmann I, Gordon VM, Gray MC. 1991. Adenylate cyclase toxin from Bordetella pertussis. Conformational change associated with toxin activity. J Biol Chem 266:17503–17508. [PubMed] [Google Scholar]
- 22.El-Azami-El-Idrissi M, Bauche C, Loucka J, Osicka R, Sebo P, Ladant D, Leclerc C. 2003. Interaction of Bordetella pertussis adenylate cyclase with CD11b/CD18: role of toxin acylation and identification of the main integrin interaction domain. J Biol Chem 278:38514–38521. doi: 10.1074/jbc.M304387200. [DOI] [PubMed] [Google Scholar]
- 23.Hanski E, Farfel Z. 1985. Bordetella pertussis invasive adenylate cyclase. Partial resolution and properties of its cellular penetration. J Biol Chem 260:5526–5532. [PubMed] [Google Scholar]
- 24.Wolff J, Cook GH, Goldhammer AR, Berkowitz SA. 1980. Calmodulin activates prokaryotic adenylate cyclase. Proc Natl Acad Sci U S A 77:3841–3844. doi: 10.1073/pnas.77.7.3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bellalou J, Sakamoto H, Ladant D, Geoffroy C, Ullmann A. 1990. Deletions affecting hemolytic and toxin activities of Bordetella pertussis adenylate cyclase. Infect Immun 58:3242–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Benz R, Maier E, Ladant D, Ullmann A, Sebo P. 1994. Adenylate cyclase toxin (CyaA) of Bordetella pertussis. Evidence for the formation of small ion-permeable channels and comparison with HlyA of Escherichia coli. J Biol Chem 269:27231–27239. [PubMed] [Google Scholar]
- 27.Svedova M, Masin J, Fiser R, Cerny O, Tomala J, Freudenberg M, Tuckova L, Kovar M, Dadaglio G, Adkins I, Sebo P. 2016. Pore-formation by adenylate cyclase toxoid activates dendritic cells to prime CD8(+) and CD4(+) T cells. Immunol Cell Biol 94:322–333. doi: 10.1038/icb.2015.87. [DOI] [PubMed] [Google Scholar]
- 28.Dunne A, Ross PJ, Pospisilova E, Masin J, Meaney A, Sutton CE, Iwakura Y, Tschopp J, Sebo P, Mills KH. 2010. Inflammasome activation by adenylate cyclase toxin directs Th17 responses and protection against Bordetella pertussis. J Immunol 185:1711–1719. doi: 10.4049/jimmunol.1000105. [DOI] [PubMed] [Google Scholar]
- 29.Basler M, Masin J, Osicka R, Sebo P. 2006. Pore-forming and enzymatic activities of Bordetella pertussis adenylate cyclase toxin synergize in promoting lysis of monocytes. Infect Immun 74:2207–2214. doi: 10.1128/IAI.74.4.2207-2214.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hewlett EL, Donato GM, Gray MC. 2006. Macrophage cytotoxicity produced by adenylate cyclase toxin from Bordetella pertussis: more than just making cyclic AMP! Mol Microbiol 59:447–459. doi: 10.1111/j.1365-2958.2005.04958.x. [DOI] [PubMed] [Google Scholar]
- 31.Hasan S, Osickova A, Bumba L, Novak P, Sebo P, Osicka R. 2015. Interaction of Bordetella adenylate cyclase toxin with complement receptor 3 involves multivalent glycan binding. FEBS Lett 589:374–379. doi: 10.1016/j.febslet.2014.12.023. [DOI] [PubMed] [Google Scholar]
- 32.Morova J, Osicka R, Masin J, Sebo P. 2008. RTX cytotoxins recognize beta2 integrin receptors through N-linked oligosaccharides. Proc Natl Acad Sci U S A 105:5355–5360. doi: 10.1073/pnas.0711400105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bumba L, Masin J, Fiser R, Sebo P. 2010. Bordetella adenylate cyclase toxin mobilizes its beta2 integrin receptor into lipid rafts to accomplish translocation across target cell membrane in two steps. PLoS Pathog 6:e1000901. doi: 10.1371/journal.ppat.1000901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vojtova-Vodolanova J, Basler M, Osicka R, Knapp O, Maier E, Cerny J, Benada O, Benz R, Sebo P. 2009. Oligomerization is involved in pore formation by Bordetella adenylate cyclase toxin. FASEB J 23:2831–2843. doi: 10.1096/fj.09-131250. [DOI] [PubMed] [Google Scholar]
- 35.Basler M, Knapp O, Masin J, Fiser R, Maier E, Benz R, Sebo P, Osicka R. 2007. Segments crucial for membrane translocation and pore-forming activity of Bordetella adenylate cyclase toxin. J Biol Chem 282:12419–12429. doi: 10.1074/jbc.M611226200. [DOI] [PubMed] [Google Scholar]
- 36.Fiser R, Masin J, Basler M, Krusek J, Spulakova V, Konopasek I, Sebo P. 2007. Third activity of Bordetella adenylate cyclase (AC) toxin-hemolysin. Membrane translocation of AC domain polypeptide promotes calcium influx into CD11b+ monocytes independently of the catalytic and hemolytic activities. J Biol Chem 282:2808–2820. doi: 10.1074/jbc.M609979200. [DOI] [PubMed] [Google Scholar]
- 37.Osickova A, Masin J, Fayolle C, Krusek J, Basler M, Pospisilova E, Leclerc C, Osicka R, Sebo P. 2010. Adenylate cyclase toxin translocates across target cell membrane without forming a pore. Mol Microbiol 75:1550–1562. doi: 10.1111/j.1365-2958.2010.07077.x. [DOI] [PubMed] [Google Scholar]
- 38.Osickova A, Osicka R, Maier E, Benz R, Sebo P. 1999. An amphipathic alpha-helix including glutamates 509 and 516 is crucial for membrane translocation of adenylate cyclase toxin and modulates formation and cation selectivity of its membrane channels. J Biol Chem 274:37644–37650. [PubMed] [Google Scholar]
- 39.Basar T, Havlicek V, Bezouskova S, Halada P, Hackett M, Sebo P. 1999. The conserved lysine 860 in the additional fatty-acylation site of Bordetella pertussis adenylate cyclase is crucial for toxin function independently of its acylation status. J Biol Chem 274:10777–10783. doi: 10.1074/jbc.274.16.10777. [DOI] [PubMed] [Google Scholar]
- 40.Gueirard P, Druilhe A, Pretolani M, Guiso N. 1998. Role of adenylate cyclase-hemolysin in alveolar macrophage apoptosis during Bordetella pertussis infection in vivo. Infect Immun 66:1718–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ehrmann IE, Weiss AA, Goodwin MS, Gray MC, Barry E, Hewlett EL. 1992. Enzymatic activity of adenylate cyclase toxin from Bordetella pertussis is not required for hemolysis. FEBS Lett 304:51–56. doi: 10.1016/0014-5793(92)80587-7. [DOI] [PubMed] [Google Scholar]
- 42.Gross MK, Au DC, Smith AL, Storm DR. 1992. Targeted mutations that ablate either the adenylate cyclase or hemolysin function of the bifunctional cyaA toxin of Bordetella pertussis abolish virulence. Proc Natl Acad Sci U S A 89:4898–4902. doi: 10.1073/pnas.89.11.4898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bagley KC, Abdelwahab SF, Tuskan RG, Fouts TR, Lewis GK. 2002. Pertussis toxin and the adenylate cyclase toxin from Bordetella pertussis activate human monocyte-derived dendritic cells and dominantly inhibit cytokine production through a cAMP-dependent pathway. J Leukoc Biol 72:962–969. [PubMed] [Google Scholar]
- 44.Boschwitz JS, Batanghari JW, Kedem H, Relman DA. 1997. Bordetella pertussis infection of human monocytes inhibits antigen-dependent CD4 T cell proliferation. J Infect Dis 176:678–686. doi: 10.1086/514090. [DOI] [PubMed] [Google Scholar]
- 45.Fedele G, Spensieri F, Palazzo R, Nasso M, Cheung GY, Coote JG, Ausiello CM. 2010. Bordetella pertussis commits human dendritic cells to promote a Th1/Th17 response through the activity of adenylate cyclase toxin and MAPK-pathways. PLoS One 5:e8734. doi: 10.1371/journal.pone.0008734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hickey FB, Brereton CF, Mills KH. 2008. Adenylate cyclase toxin of Bordetella pertussis inhibits TLR-induced IRF-1 and IRF-8 activation and IL-12 production and enhances IL-10 through MAPK activation in dendritic cells. J Leukoc Biol 84:234–243. doi: 10.1189/jlb.0208113. [DOI] [PubMed] [Google Scholar]
- 47.Njamkepo E, Pinot F, Francois D, Guiso N, Polla BS, Bachelet M. 2000. Adaptive responses of human monocytes infected by Bordetella pertussis: the role of adenylate cyclase hemolysin. J Cell Physiol 183:91–99. doi:. [DOI] [PubMed] [Google Scholar]
- 48.Ross PJ, Lavelle EC, Mills KH, Boyd AP. 2004. Adenylate cyclase toxin from Bordetella pertussis synergizes with lipopolysaccharide to promote innate interleukin-10 production and enhances the induction of Th2 and regulatory T cells. Infect Immun 72:1568–1579. doi: 10.1128/IAI.72.3.1568-1579.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spensieri F, Fedele G, Fazio C, Nasso M, Stefanelli P, Mastrantonio P, Ausiello CM. 2006. Bordetella pertussis inhibition of interleukin-12 (IL-12) p70 in human monocyte-derived dendritic cells blocks IL-12 p35 through adenylate cyclase toxin-dependent cyclic AMP induction. Infect Immun 74:2831–2838. doi: 10.1128/IAI.74.5.2831-2838.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ladant D, Glaser P, Ullmann A. 1992. Insertional mutagenesis of Bordetella pertussis adenylate cyclase. J Biol Chem 267:2244–2250. [PubMed] [Google Scholar]
- 51.Miller LS, O'Connell RM, Gutierrez MA, Pietras EM, Shahangian A, Gross CE, Thirumala A, Cheung AL, Cheng G, Modlin RL. 2006. MyD88 mediates neutrophil recruitment initiated by IL-1R but not TLR2 activation in immunity against Staphylococcus aureus. Immunity 24:79–91. doi: 10.1016/j.immuni.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 52.Bassinet L, Gueirard P, Maitre B, Housset B, Gounon P, Guiso N. 2000. Role of adhesins and toxins in invasion of human tracheal epithelial cells by Bordetella pertussis. Infect Immun 68:1934–1941. doi: 10.1128/IAI.68.4.1934-1941.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perkins DJ, Gray MC, Hewlett EL, Vogel SN. 2007. Bordetella pertussis adenylate cyclase toxin (ACT) induces cyclooxygenase-2 (COX-2) in murine macrophages and is facilitated by ACT interaction with CD11b/CD18 (Mac-1). Mol Microbiol 66:1003–1015. doi: 10.1111/j.1365-2958.2007.05972.x. [DOI] [PubMed] [Google Scholar]
- 54.Adkins I, Kamanova J, Kocourkova A, Svedova M, Tomala J, Janova H, Masin J, Chladkova B, Bumba L, Kovar M, Ross PJ, Tuckova L, Spisek R, Mills KH, Sebo P. 2014. Bordetella adenylate cyclase toxin differentially modulates Toll-like receptor-stimulated activation, migration and T cell stimulatory capacity of dendritic cells. PLoS One 9:e104064. doi: 10.1371/journal.pone.0104064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Higgs R, Higgins SC, Ross PJ, Mills KH. 2012. Immunity to the respiratory pathogen Bordetella pertussis. Mucosal Immunol 5:485–500. doi: 10.1038/mi.2012.54. [DOI] [PubMed] [Google Scholar]
- 56.Miller VL, Mekalanos JJ. 1988. A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J Bacteriol 170:2575–2583. doi: 10.1128/jb.170.6.2575-2583.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Caro V, Hot D, Guigon G, Hubans C, Arrive M, Soubigou G, Renauld-Mongenie G, Antoine R, Locht C, Lemoine Y, Guiso N. 2006. Temporal analysis of French Bordetella pertussis isolates by comparative whole-genome hybridization. Microbes Infect 8:2228–2235. doi: 10.1016/j.micinf.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 58.Stainer DW, Scholte MJ. 1970. A simple chemically defined medium for the production of phase I Bordetella pertussis. J Gen Microbiol 63:211–220. doi: 10.1099/00221287-63-2-211. [DOI] [PubMed] [Google Scholar]
- 59.Inatsuka CS, Xu Q, Vujkovic-Cvijin I, Wong S, Stibitz S, Miller JF, Cotter PA. 2010. Pertactin is required for Bordetella species to resist neutrophil-mediated clearance. Infect Immun 78:2901–2909. doi: 10.1128/IAI.00188-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weber C, Boursaux-Eude C, Coralie G, Caro V, Guiso N. 2001. Polymorphism of Bordetella pertussis isolates circulating for the last 10 years in France, where a single effective whole-cell vaccine has been used for more than 30 years. J Clin Microbiol 39:4396–4403. doi: 10.1128/JCM.39.12.4396-4403.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ladant D, Brezin C, Alonso JM, Crenon I, Guiso N. 1986. Bordetella pertussis adenylate cyclase. Purification, characterization, and radioimmunoassay. J Biol Chem 261:16264–16269. [PubMed] [Google Scholar]
- 62.Lutz MB, Kukutsch N, Ogilvie AL, Rossner S, Koch F, Romani N, Schuler G. 1999. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods 223:77–92. doi: 10.1016/S0022-1759(98)00204-X. [DOI] [PubMed] [Google Scholar]
- 63.Karimova G, Pidoux J, Ullmann A, Ladant D. 1998. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc Natl Acad Sci U S A 95:5752–5756. doi: 10.1073/pnas.95.10.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee SJ, Gray MC, Guo L, Sebo P, Hewlett EL. 1999. Epitope mapping of monoclonal antibodies against Bordetella pertussis adenylate cyclase toxin. Infect Immun 67:2090–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.