

Abstract
In this paper we consider a combination therapy of cancer. One drug is a vaccine which activates dendritic cells so that they induce more T cells to infiltrate the tumor. The other drug is a checkpoint inhibitor, which enables the T cells to remain active against the cancer cells. The two drugs are positively correlated in the sense that an increase in the amount of each drug results in a reduction in the tumor volume. We consider the question whether a treatment with combination of the two drugs at certain levels is preferable to a treatment by one of the drugs alone at ‘roughly’ twice the dosage level; if that is the case, then we say that there is a positive ‘synergy’ for this combination of dosages. To address this question, we develop a mathematical model using a system of partial differential equations. The variables include dendritic and cancer cells, CD4+ and CD8+ T cells, IL-12 and IL-2, GM-CSF produced by the vaccine, and a T cell checkpoint inhibitor associated with PD-1. We use the model to explore the efficacy of the two drugs, separately and in combination, and compare the simulations with data from mouse experiments. We next introduce the concept of synergy between the drugs and develop a synergy map which suggests in what proportion to administer the drugs in order to achieve the maximum reduction of tumor volume under the constraint of maximum tolerated dose.
Introduction
When cancer cells undergo necrosis, they release high mobility group box-1 (HMGB-1) which activates dendritic cells [1–3]. Activated dendritic cells (DCs) mature as APC cells and play a critical role in the communication between the innate and adaptive immune responses. Once activated, dendritic cells produce IL-12, which activates effector T cells CD4+ Th1 and CD8+ T [4, 5]. Th1 produces IL-2 which further promotes proliferation of the effector T cells. Both CD4+ Th1 and CD8+ T cells kill cancer cells [6–8]. CD8+ T cells are more effective in killing cancer cells, but the helper function of CD4+ Th1 cells improves the efficacy of tumor-reactive CD8+ T cells [9].
Cancer vaccines serve to enlarge the pool of tumor-specific T cells from the naive repertoire, and also to activate tumor specific T cells which are dormant [10]. GM-CSF can activate dendritic cells, and is commonly used as a cancer vaccine [11–13]. GVAX is a cancer vaccine composed of tumor cells genetically modified to secrete GM-CSF and then irradiated to prevent further cell division.
PD-1 is an immunoinhibitory receptor predominantly expressed on activated T cells [14, 15]. Its ligand PD-L1 is upregulated on the same activated T cells, but it is also expressed by some human cancer cells, such as in melanoma, lung cancer, colon cancer, and leukemia [15–17]. The complex PD-1-PD-L1 is known to inhibit T cell function [14]. Immune checkpoints are regulatory pathways in the immune system that inhibit its active response against specific targets. In the case of cancer, the complex PD-1-PD-L1 forms an immune checkpoint for T cells.
There has been much progress in recent years in developing checkpoint inhibitors, primarily PD-1 antibodies and PD-L1 antibodies [17]. Such drugs have been increasingly explored in single-agent studies for cancer treatment [16, 18]. The FDA recently approved several checkpoint inhibitors. However, because of lack of tumor-infiltrating effector T cells, many patients in clinical trials do not respond to checkpoint inhibitor treatment [18]. On the other hand, cancer vaccines have been shown to induce effector T-cells infiltration into tumors [19], although, to be fully effective, cancer vaccines have to overcome immune evasion [10]. It was recently suggested that the combination of a cancer vaccine and an immune checkpoint inhibitor may function synergistically to induce more effective antitumor immune responses [18, 20]. Clinical trials to test such combination therapies are currently ongoing [18, 20]; mouse experiments are also being conducted [21–27].
In the present paper we develop a mathematical model of treatment of cancer with a cancer vaccine combined with an immune checkpoint inhibitor; specifically, we combine GVAX and PD-1 inhibitor. In order to focus on the combination therapy of the two drugs, we consider in the model only the following variables: cancer cells (C), dendritic cells (DCs), CD4+ and CD8+ T cells, GM-CSF, PD-1, PD-L1, PD-1-PD-L1 complex, and cytokines IL-12 and IL-2. These species interact within the network shown in Fig 1. The mathematical model is based on Fig 1, and it is represented by a system of partial differential equations (PDEs). Simulations of the model are shown to be in qualitative agreement with the mouse experiments reported in [21–23]. The model is then used to explore the efficacy of the combined treatment. We introduce a specific concept of synergy between the vaccine and the PD-1 inhibitor, which is somewhat different from the usual definition of synergy. Roughly speaking, we compare the reduction in tumor size achieved by a combined therapy with amounts γG of GVAX and γA of PD-1 inhibitor to the reduction obtained by single-agent with either (1 + θG)γG or (1 + θA)γA with appropriately chosen 0 < θG ≤ 1 or 0 < θA ≤ 1. The larger the reduction in tumor size achieved by the combination therapy the larger the synergy is said to be. A specific choice of θG and θA, which takes into account potential negative side-effects of each drug, will be given as an example. We develop a synergy map in the (γG, γA)-plane. The map shows that, given γG, the synergy increases as γA increases as long as γA remains below a critical value γAG; thereafter, the synergy decreases as γA increases.
Fig 1. Interaction of immune cells with tumor cells.

Sharp arrows indicate proliferation/activation, blocked arrows indicate killing/blocking, and dashed lines indicate proteins on T cells. GM-CSF activates dendritic cells; activated dendritic cells produce IL-12; IL-12 activates naive CD4+ and CD8+ T cells; activated CD4+ T cells (Th1) produce IL-2 which induces proliferation of activated CD4+ and CD8+ T cells. Activated CD4+ and CD8+ T cells kill cancer cells. Activated CD4+ and CD8+ T cells express PD-1 and PD-L1, and cancer cells express PD-L1. The complex PD-1-PD-L1 inhibits the function of active CD4+ and CD8+ T cells.
A discrete-time mathematical model of the combination of radiotherapy with checkpoint inhibitors was developed in [28]. Mathematical models of immunotherapy with a cancer vaccine by a system of ordinary differential equations were developed earlier in [29, 30]; these models do not consider checkpoint inhibitors. In [29] it is shown that there is a positive correlation between the number of antigen presenting cells and prolonged cancer dormancy, and in [30] it is illustrated how the combined therapy can eliminate the cancer, though neither immunotherapy nor checkpoint inhibitors can do it alone. In this paper, we present for the first time a mathematical model which combines a cancer vaccine with a checkpoint inhibitor: the vaccine (GVAX) increases the pool of T cells and the checkpoint inhibitor (anti-PD-1) enables the T cells to remain fully active in killing cancer cells.
Mathematical model
The mathematical model is based on the network shown in Fig 1. The list of variables, with units, is given in Table 1.
Table 1. List of variables.
| Notation | Description | units |
|---|---|---|
| D | density of DCs | g/cm3 |
| T1 | density of activated CD4+ T cells | g/cm3 |
| T8 | density of activated CD8+ T cells | g/cm3 |
| C | density of cancer cells | g/cm3 |
| NC | density of necrotic cancer cells | g/cm3 |
| H | HMGB-1 concentration | g/cm3 |
| G | GM-CSF concentration | g/cm3 |
| I12 | IL-12 concentration | g/cm3 |
| I2 | IL-2 concentration | g/cm3 |
| P | PD-1 concentration | g/cm3 |
| L | PD-L1 concentration | g/cm3 |
| Q | PD-1-PD-L1 concentration | g/cm3 |
| A | anti-PD-1 concentration | g/cm3 |
We assume that the total density of cancer cells (C), active dendritic cells (D), CD4+ T cells (T1) and CD8+ T cells (T8) within the tumor remains constant in space and time:
| (1) |
It is tacitly assumed that the debris of dead cells, including cancer cells undergoing necrosis or apoptosis, is quickly cleared from the tumor tissue. It is also tacitly assumed that the densities of immature dendritic cells and naive CD4+ and CD8+ T cells are constant throughout the tumor tissue.
Under the assumption Eq (1), cancer cell proliferation, and migration of immune cells into the tumor give rise to internal pressure which results in cell movement, and we assume that all the cells move with the same velocity, u; u depends on space and time. We also assume that all the cells undergo diffusion, and that all the cytokines and drugs are diffusing within the tumor.
Equation for DCs (D). When cancer cells undergo necrosis, they release HMGB-1 [1]. We can model the dynamics of the necrotic cells and of HMGB-1 by the following equations:
where λNCC is the rate at which cancer cells become necrotic and λHNC is the rate at which necrotic cells produce HMGB-1. We note that although molecules like HMGB-1, or other proteins, may be affected by the velocity , their diffusion coefficients are several orders of magnitude larger than the diffusion coefficients of cells; hence their velocity terms may be neglected. The degradation of HMGB-1 is fast (∼0.01/day) [31], and we assume that the removal of NC is also fast. We can therefore approximate the two dynamical equations by the steady state equations λNCCC − dNCNC = 0 and λHNCNC − dHH = 0, so that H is proportional to C, i.e., H = constant × C.
Dendritic cells are activated by HMGB-1 [2, 3]. Hence, the activation rate of immature dendritic cell D0 is proportional to , where the Michaelis-Menten law is used to account for the limited rate of receptor recycling time which occurs in the process of DCs activation. In the same way, GM-CSF, produced by the cancer vaccine, activates DCs at rate proportional to . Hence, the dynamics of DCs is given by
| (2) |
where δD is the diffusion coefficient and dD is the death rate of DCs.
Equation for CD4+ T cells (T1). Naive CD4+ T cells are activated by IL-12, and IL-2 induces proliferation of activated T1 cells [4, 5]. Both processes are assumed to be inhibited by the complex PD-1-PD-L1 (Q) [14], which reduces the production of T1 cells by a factor . Hence T1 satisfies the following equation:
| (3) |
where T10 is the density of naive CD4+ T cells.
Equation for activated CD8+ T cells (T8). IL-12 activates CD8+ T cells and IL-2 induces the proliferation of CD8+ T cells [4, 5]. Hence, similarly to the equation for T1, T8 satisfies the equation
| (4) |
where T80 is the density of naive CD8+ T cells.
Equation for tumor cells (C). Cancer cells are killed by T1 and T8 [6–8]. We assume a logistic growth with carrying capacity (CM) in order to account for competition for space among the cancer cells. Hence,
| (5) |
where η1, η8 are the killing rates of cancer cells by T1 and T8, and dC is the natural death rate of cancer cells.
Equation for GM-CSF (G). We assume that GVAX is injected intradermally every 3 days for 30 days (as in mouse experiments [21]) providing a source of GM-CSF, which we represent by
where γG is the effective level of the drug; although the level of the drug varies between injections, for simplicity we take it to be constant. The concentration of GM-CSF in tissue is very small [32], and accordingly, we assume a low rate of constant source λG for GM-CSF. Hence G satisfies the following equation:
| (6) |
where dG is the degradation rate of GM-CSF.
Equation for IL-12 (I12). IL-12 is produced by activated DCs [4, 5], so that
| (7) |
Equation for IL-2 (I2). IL-2 is produced by activated CD4+ T cells (T1) [4, 5]. Hence,
| (8) |
Equation for PD-1 (P), PD-L1 (L) and PD-1-PD-L1 (Q). PD-1 is expressed on the surface of activated CD4+ T cells and activated CD8+ T cells [14, 15]. We assume that the expression level of PD-1 is the same for activated CD4+ and CD8+ T cells. Hence, P is given by
where ρP is the ratio between the mass of one PD-1 protein to the mass of one T cell. The coefficient ρP is constant when no anti-PD-1 drug is injected. In that case, to a change in T = T1 + T8, given by , there corresponds a change of P, given by . For the same reason, and ∇2P = ρP∇2T when no anti-PD-1 drug is injected. Hence, P satisfies the equation
Recalling Eqs (3) and (4) for T1 and T8, we get
When anti-PD-1 drug (A) is applied, PD-1 is depleted (or blocked) by A. In this case, the ratio may change. In order to include in the model both cases of with and without anti-PD-1, we replace ρP in the previous equation by . Hence,
| (9) |
where μPA represents the rate at which P is depleted/blocked by A.
PD-L1 is expressed on the surface of activated CD4+ and CD8+ T cells [14, 15] and on cancer cells [15, 16]. Hence, the concentration of PD-L1 (L) is proportional to (T1 + T8) and C:
| (10) |
where ε depends on the specific type of tumor.
PD-L1 from T cells or cancer cells ligands to PD-1 on the plasma membrane of T cells, thus forming a complex PD-1-PD-L1 (Q) on the T cells [15, 16]. Denoting the association and disassociation rates of Q by αPL and dQ, respectively, we can write
| (11) |
so that
The half-life of Q is less then 1 second (i.e. 1.16 × 10−5 day) [33], and hence dQ is very large, and we may approximate the dynamical equation by the steady state equation, αPLPL = dQQ, or
| (12) |
where σ = αPL/dQ.
Equation for anti-PD-1 (A). We assume that anti-PD-1 is injected intradermally every 3 days for 30 days (as in mouse experiments [21]) providing a source of anti-PD-1:
where γA is the effective level of the drug; although the level of the drug varies between injections, for simplicity we take it to be constant. The drug A is depleted in the process of blocking PD-1. Hence,
| (13) |
where μPA is the rate at which A is degraded in the process of blocking PD-1.
Equation for cell velocity (u): We assume that most of the tumor consists of the extracellular matrix, ECM (approximately, 0.6 g/cm3), and cancer cells (approximately, C = 0.4 g/cm3), and that the densities of D, T1 and T8 are approximately 4 × 10−4, 2 × 10−3 and 1 × 10−3 g/cm3, respectively (as explained in the section on parameter estimation). We further assume that all cells are approximately of the same volume and surface area, so that their diffusion coefficients are the same. For definiteness, we take the constant in Eq (1) to be 0.4034. Adding Eqs (2)–(5), we then get
| (14) |
To simplify the computations, we shall assume that the tumor is spherical and denote its moving boundary (i.e. its radius) by r = R(t). We also assume that all the densities and concentrations are radially symmetric, that is, functions of (r, t), where 0 ≤ r ≤ R(t). In particular, , where is the unit radial vector, and each of the equations of the form
with X = X(r, t), takes the form
Equation for free boundary (R): We assume that the free boundary r = R(t) moves with the velocity of cells, so that
| (15) |
Boundary conditions We assume that the naive CD4+ T cells and naive CD8+ T cells which migrated from the lymph nodes into the tumor microenvironment have constant densities and at the tumor boundary, and that they are activated by IL-12 upon entering the tumor. We represent this process by the flux conditions at the boundary r = R(t):
| (16) |
where .
We impose the no-flux boundary conditions for all the remaining variables:
| (17) |
It is tacitly assumed here that the receptors PD-1 and ligands PD-L1 become active only after the T cells are inside the tumor.
Initial conditions. We take initial values of all the variables to be constant. Later on we shall compare the simulations of the model with mouse experimental results, for 60 days. Accordingly, we take initial values whereby the average density of cancer cells has not yet increased to its steady state, 0.4 g/cm3, and, in view of Eq (1), the total density of the immune cells is initially above its steady state. We take (in unit of g/cm3):
| (18) |
We assume that initially A = 0, and
These values are close to the steady state values which are computed in the section on parameter estimation.
Results
The simulations of the model were performed by Matlab based on the moving mesh method for solving partial differential equations with free boundary [34] (see the section on computational method). All the computations are done in dimensionless form, but displayed in dimensional form.
The average density or concentration of a species is defined as its total mass in the tumor divided by the tumor volume. Fig 2 shows the average concentrations of all the species of the model over a period of 60 days in the control case, that is, when no drugs are administered; the parameter values are given in Tables 2 and 3. The radius of the tumor is increasing, from 0.01 cm to 0.0313 cm. The average density of the cancer cells is initially increasing and later it stabilizes while the densities of the immune cells are first decreasing and later stabilize. Correspondingly, the concentrations of the cytokines produced by the immune system also first decrease and later stabilize. Some of the parameters in the model were estimated by assuming the immune cells and cytokines are in steady state. Their steady states in Fig 2 approximately agree with those which we assumed in estimating the parameters, thus establishing the consistency of our assumed steady-state values.
Fig 2. Average densities/concentrations of all the variables in the model in the control case (no drugs).

Figures in the first and second panels and the first figure in the third panel show the average density or concentration of each species in the model. The last three figures in the third panel show the total average density of all cells, the growth of tumor radius and the growth of tumor volume, respectively. All parameter values are the same as in Tables 2 and 3.
Table 2. Summary of parameter values.
| Notation | Description | Value used | References |
|---|---|---|---|
| δD | diffusion coefficient of DCs | 8.64 × 10−7 cm2day−1 | [59] |
| δT1 | diffusion coefficient of CD4+ T cells | 8.64 × 10−7 cm2day−1 | [59] |
| δT8 | diffusion coefficient of CD8+ T cells | 8.64 × 10−7 cm2day−1 | [59] |
| δC | diffusion coefficient of tumor cells | 8.64 × 10−7 cm2day−1 | [59] |
| δG | diffusion coefficient of GM-CSF | 9.8495 × 10−2 cm2day−1 | estimated |
| δI12 | diffusion coefficient of IL-12 | 6.0472 × 10−2 cm2day−1 | estimated |
| δI2 | diffusion coefficient of IL-2 | 9.9956 × 10−2 cm2day−1 | estimated |
| δA | diffusion coefficient of anti-PD-1 | 7.85 × 10−2 cm2day−1 | estimated |
| σ0 | flux rate of T cells on the boundary | 1 cm−1 | [59] |
| λDG | activation rate of DCs by GM-CSF | 20.02 day−1 | [65, 66] & estimated |
| λDC | activation rate of DCs by tumor cells | 0.364 day−1 | estimated |
| λT1I12 | activation rate of CD4+ T cells by IL-12 | 4.66 day−1 | estimated |
| λT1I2 | activation rate of CD4+ T cells by IL-2 | 0.25 day−1 | estimated |
| λT8I12 | activation rate of CD8+ T cells by IL-12 | 4.15 day−1 | estimated |
| λT8I2 | activation rate of CD8+ T cells by IL-2 | 0.25 day−1 | [59] |
| λC | growth rate of cancer cells | 0.616 day−1 | [59] |
| λG | non-vaccine source of GM-CSF | 2.23 × 10−10 g/cm3 ⋅ day | estimated |
| λI12D | production rate of IL-12 by DCs | 5.18 × 10−7 day−1 | estimated |
| λI2T1 | production rate of IL-2 by CD4+ T cells | 2.82 × 10−8 day−1 | estimated |
| η1 | killing rate of tumor cells by CD4+ T cells | 11.5 day−1 ⋅ cm3/g | estmated |
| η8 | killing rate of tumor cells by CD8+ T cells | 46 day−1 ⋅ cm3/g | estimated |
| μPA | blocking rate of PD-1 by anti-PD-1 | 6.87 × 106 cm3/g ⋅ day | estimated |
| ρP | expression of PD-1 in T cells | 2.49 × 10−7 | estimated |
| ρL | expression of PD-L1 in T cells | 5.22 × 10−7 | estimated |
| ε | expression of PD-L1 in tumor cells | 0 − 0.01* | estimated |
| dD | death rate of DCs | 0.1 day−1 | [59] |
| dT1 | death rate of CD4+ T cells | 0.197 day−1 | [59] |
| dT8 | death rate of CD8+ T cells | 0.18 day−1 | [59] |
| dC | death rate of tumor cells | 0.17 day−1 | [59] |
| dG | degradation rate of GM-CSF | 1.28 day−1 | [65] |
| dI12 | degradation rate of IL-12 | 1.38 day−1 | [59] |
| dI2 | degradation rate of IL-2 | 2.376 day−1 | [59] |
| dA | degradation rate of anti-PD-1 | 0.0462 day−1 | [42] |
* In the simulations we took ε = 0.01.
Table 3. Summary of parameter values.
| KG | half-saturation of GM-CSF | 1.74 × 10−9 g/cm3 | [65] |
| KC | half-saturation of tumor cells | 0.4 g/cm3 | [59] |
| KI12 | half-saturation of IL-12 | 1.5 × 10−10 g/cm3 | [59] |
| KI2 | half-saturation of IL-2 | 2.37 × 10−11 g/cm3 | [59] |
| KT1 | half-saturation of CD4+ T cells | 2 × 10−3 g/cm3 | estimated |
| KT8 | half-saturation of CD8+ T cells | 1 × 10−3 g/cm3 | estimated |
| inhibition of function of T cells by PD-1-PD-L1 | 1.365 × 10−18 g/cm3 | estimated | |
| D0 | density of immature DCs | 2 × 10−5 g/cm3 | [59] |
| T10 | density of naive CD4+ T cells | 4 × 10−4 g/cm3 | estimated |
| T80 | density of naive CD8+ T cells | 2 × 10−4 g/cm3 | estimated |
| CM | carrying capacity of cancer cells | 0.8 g/cm3 | [59] |
| density of CD4+ T cells from lymph node | 4 × 10−3 g/cm3 | estimated | |
| density of CD8+ T cells from lymph node | 2 × 10−3 g/cm3 | estimated | |
| γG | source of GM-CSF from the vaccine | 1 × 10−10 g/cm3 ⋅ day* | estimated |
| γA | source of anti-PD-1 | 1 × 10−10 g/cm3 ⋅ day* | estimated |
* Values used in sensitivity analysis.
We can also simulate the spatial distribution of each of the variables. Fig 3 shows the distribution of cancer cells and T cells (T1 + T8) at times t = 15, 30, 60 days. We see that the density of T cells increases toward the boundary; this is a result of the influx of T cells from the lymph nodes. Correspondingly, the density of cancer cells decreases toward the boundary. In our model, we tacitly assumed avascular conditions, since we wanted to focus primarily on the difference between control and treatment. Since, however, the tumor radius reaches approximately 313 μm at day 60, hypoxic conditions may actually reduce the cancer cells’ density at the core of the tumor (both in control and treatment).
Fig 3. Spatial distribution of density of cancer cells and T1 + T8 cells at time t = 15, 30, 60 days.

All parameter values are the same as in Fig 2.
In Figs 2 and 3, we have taken ε = 0.01. Some cancer cells may express more or less PD-L1 [17, 35–37]. By decreasing or increasing ε, the radius of the tumor will decrease or increase, respectively, but the profiles of the densities/concentrations remain qualitatively the same (not shown here).
We next proceed to explore the effect of treatment with GM-CSF-secreting vaccine (GVAX) and anti-PD-1 drug. We are unable to make a direct connection between the levels of drugs administered to the patient, and their ‘effective strengths’ γG and γA in the model, since these data are not available. Based on the estimate of the concentration of GM-CSF in normal healthy tissue (see Parameter Estimations for Eq (6)), we chose the order of magnitude of GVAX to be 10−10 (g/cm3 ⋅ day). The order of magnitude for anti-PD-1 drug (γA) is chosen so as to get the best agreement with the mouse experiments. In Fig 4(a), the ‘effective strength’ of the vaccine is given by γG = 0.87 × 10−10 g/cm3 ⋅ day and the ‘effective strength’ of the anti-PD-1 is given by γA = 2 × 10−10 g/cm3 ⋅ day. We see that, as a single-agent, anti-PD-1 is more effective than GVAX, and with the combined therapy the tumor radius is still increasing. This is in agreement with the mouse experiments reported in Fig 1-(A) (with melanoma) in [21], and Figs 3-(b) (with colon carcinoma) and 3-(c) (with melanoma) in [22].
Fig 4. The growth of tumor radius R(t) during the administration of GM-CSF-secreting vaccine and anti-PD-1 drug.

(a) GM-CSF-secreting vaccine is administered at rate γG = 0.87 × 10−10 g/cm3 ⋅ day and anti-PD-1 drug is administered at rate γA = 2 × 10−10 g/cm3 ⋅ day. (b) GM-CSF-secreting vaccine is administered at rate γG = 0.87 × 10−10 g/cm3 ⋅ day and anti-PD-1 drug is administered at rate γA = 3 × 10−10 g/cm3 ⋅ day.
In Fig 4(b), we increased γA by a factor 3/2. As a result, the tumor radius begins to decrease around day 50, even when administering anti-PD-1 as single-agent. This is in agreement with mouse experiments (with colon carcinoma) reported in Fig 1-(D) of [23].
In Fig 4(a), GM-CSF-secreting vaccine alone did not reduce the tumor radius as much as anti-PD-1 alone. However, if we increase the strength of the vaccine and decrease the anti-PD-1, taking for example, γG = 3.84 × 10−10 g/cm3 ⋅ day and γA = 1 × 10−10 g/cm3 ⋅ day, we then find that the vaccine decreases the tumor radius more than anti-PD-1 does; this is shown in Fig 5, and it is in agreement with mouse experiments (with melanoma) reported in Fig 1-(B) of [23].
Fig 5. The growth of tumor radius R(t) during the administration of GM-CSF-secreting vaccine and anti-PD-1 drug.
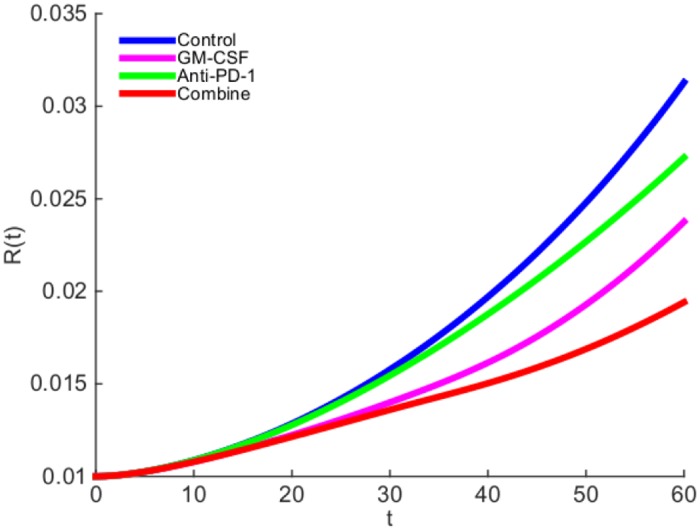
GM-CSF-secreting vaccine is administered at rate γG = 3.84 × 10−10 g/cm3 ⋅ day and anti-PD-1 drug is administered at rate γA = 1 × 10−10 g/cm3 ⋅ day.
The results of Figs 4 and 5 show that a combination therapy of GVAX and anti-PD-1 in appropriate amounts could significantly slow the growth of a tumor.
There is some uncertainty in the estimates of some of the parameters of the model. With somewhat different choices of these parameters, the simulation results will change quantitatively, and sensitivity analysis indicates the direction and intensity of the change (see the section on sensitivity analysis). In particular, the choice of γG and γA will affect the relative reduction in the growth of the tumor radius. In Figs 4 and 5 we made specific choices of γG and γA, in order to get simulations that agree with experimental results.
We next consider combination therapy for any values of GVAX and anti-PD-1. We define the efficacy of the combination therapy with (γG, γA) by the formula:
where R60 = R60(γG, γA) represents the tumor radius computed at day 60. If the tumor radius at day 60, R60(γG, γA), is smaller than the radius in the control case, R60(0, 0), then the efficacy is a positive number, and its value is between 0 and 1 (or between 0% and 100%); the efficacy increases to 1 (or to 100%) when the tumor radius R60(γG, γA) decreases to 0 by day 60. Fig 6(a) is the efficacy map of the combined therapy with γG in the range of 0 − 4.8 × 10−10) g/cm3 ⋅ day and γA in the range of 0 − 4 × 10−10 g/cm3 ⋅ day. The color column in Fig 6(a) shows the efficacy for any pair of (γG, γA); the efficacy is positive, and its maximum is 0.95 (95%). We see that an increase in either γG or γA improves the efficacy of the treatment.
Fig 6. Drug efficacy map and synergy map.

(a) Efficacy map: The color column shows the efficacy E(γG, γA) when γG varies between 0 − 4.8 × 10−10 g/cm3 ⋅ day and γA varies between 0 − 4 × 10−10 g/cm3 ⋅ day. (b) Synergy map: The color column shows the synergy σ(γG, γA) when γG varies between 0 − 2.4 × 10−10 g/cm3 ⋅ day and γA varies between 0 − 2 × 10−10 g/cm3 ⋅ day. For a given γG, the optimal synergy of the combined therapy (γG, γA) occurs when (γG, γA) lies on the solid curve.
Early stage clinical trials consider the safety of each of the drugs, GVAX and anti-PD-1, separately. In the GVAX treatment of patients with pancreatic cancer, no dose-limiting toxicity or minimal toxicity was observed [38–40]. On the other hand, in treatment with anti-PD-1, mild to moderate toxicity (grades 1 and 2) was observed in melanoma [41, 42]. In non-small-cell lung cancer, 14% of the patients were observed to have more severe toxicity (grades 3 and 4) [43]. Based on these observations we conclude that anti-PD-1 causes more toxicity than GVAX. However, clinical trials on the safety and efficacy of the combined drugs are limited [23, 44, 45].
The amount of drug in clinical trials is constrained by the maximum tolerated dose (MTD). In combination therapy this constraint may depend on the proportion between the amounts of the drugs. We note that there is a large literature on the trade-off between efficacy and toxicity [46–49]. Here we consider, as an example, two treatments, and , with and , where both satisfy the MTD requirement. The question is then which of the two treatments is more effective in reducing the tumor volume. We can use the efficacy map to address such a question. We illustrate this in one special case.
We compare treatment of combination (γG, γA) with monotherapy GVAX and monotherapy anti-PD-1. For monotherapy with GVAX, we take (1 + θG)γG, and for monotherapy with anti-PD-1 we take (1 + θA)γA, with θA < θG, to reflect the higher toxicity associated with anti-PD-1. If E(γG, γA) is larger than both E((1 + θG)γG, 0) and E(0, (1 + θA)γA), then we say that the synergy for the combination (γG, γA) is positive, and otherwise, we say that the synergy is negative. More generally, we define the synergy σ = σ(γG, γA) by the formula:
| (19) |
Thus σ(γG, γA) > 0 (positive synergy) if the combination (γG, γA) reduces tumor growth more than either one of the single agents with (1 + θG)γG or (1 + θA)γA. Negative synergy occurs in the reverse case where instead of a combination therapy with (γG, γA) we achieve better reduction of the tumor radius R60 if we apply only one drug, (1 + θG)γG or (1 + θA)γA. The above concept of synergy is somewhat different from the usual definitions of synergy. For definiteness we take θG = 1 and γA = 0.5, but this choice, which is somewhat arbitrary, could be made more precise as more clinical data become available.
Fig 6(b) is the synergy map for (γG, γA) in the same range as in Fig 6(a); the color column shows the synergy σ(γG, γA), with values that vary from -0.38 to 0.28.
We first note that the synergy is negative if γG < 0.2 × 10−10 g/cm3 ⋅ day. The reason is the following: if γG is small then the numbers of T cells is also small, so instead of introducing a drug γA which blocks the relatively small number of PD-1, it is more effective to increase the number of T cells by increasing γG, i.e. E(2γG, 0) > E(γG, γA).
Next, for any fixed γG, as seen in Fig 6(b), the synergy first increases with γA and then decreases. In order to explain this occurrence, we note that with γG fixed, the number of T cells that arrive into the tumor microenvironment is limited, and so is the number of their PD-1. Thus, in order to block the PD-1 there is a need for only a limited amount of anti-PD-1 drug; i.e. it is ‘wasteful’ to administer too much of γA. We conclude that the maximum synergy is achieved when the amount of γA is appropriately dependent on the amount of γG, as indicated by the solid curve shown in Fig 6(b).
Fig 7 shows that as the amounts of γG and γA increase the average density of T cells (T1 + T8) increases, and correspondingly the average density of cancer cells decreases. We also see that the density of T cells shown in the color column increases approximately by a factor of 6, whereas the density of cancer cells decreases approximately by a factor of 1.075. The proportion of these changes (i.e. 5/0.075) is similar to the proportion of densities of cancer cells to T cells.
Fig 7. Average densities of cancer cells and T cells.

(a) The average density of cancer cells (C), and (b) the average density of T cells (T1 + T8), under the combination of the drugs with (γG, γA), where γG varies between 0 − 4.8 × 10−10 g/cm3 ⋅ day and γA varies between 0 − 4 × 10−10 g/cm3 ⋅ day.
Conclusion
The introduction of immune checkpoint inhibitors has been a very promising approach to cancer treatment. Blockage of the programmed death PD-1/PD-L1 is increasingly explored in single-agent studies [16, 18]. However, because of the lack of tumor-infiltrating effector T cells, many patients do not respond to checkpoint inhibitor treatment as single-agent [18]. On the other hand, cancer vaccines have been shown to induce effector T-cells infiltration into tumors [19], although, to be fully effective, cancer vaccines have to overcome immune evasion [10]. It was recently suggested that the combination of a cancer vaccine and an immune checkpoint inhibitor may function synergistically to induce more effective antitumor immune response [18, 20]. Clinical trials to test such combinations are currently ongoing [18, 20].
In the present paper we developed a mathematical model to study the efficacy of the combination of a GM-CSF-secreting cancer vaccine (GVAX) and an anti-PD-1 drug, and to address the following question: at what proportion should the two drugs be administered in order to achieve the best efficacy when the amount of drugs is limited by MTD. The mathematical model is represented by a system of partial differential equations, based on the interactions among cancer cells, dendritic cells, CD4+ and CD8+ T cells, cytokines IL-12 and IL-2, GM-CSF, PD-1, PD-L1, PD-1-PD-L1 complex and anti-PD-1. Simulations of the model are shown to be in qualitative agreement with mouse experiments [21–23].
The cancer vaccine and the anti-PD-1 work in collaboration: The vaccine increases the number of tumor-infiltrating effector T cells and the anti-PD-1 ensures that these cells remain active. Given a fixed amount γG of the vaccine and γA of the anti-PD-1, it is important to estimate the level of synergy between these amounts in order to administer them in the most effective proportion. We introduced a specific concept of synergy σ(γG, γA) and developed accordingly a synergy map in Fig 6(b). The map shows that if γG is small, single-agent treatment (with γA = 0) is the best treatment. Fig 6(b) also shows that, for any γG, there is a unique value γAG, such that the synergy increases as γA increases as long as γA remains smaller than γAG, but the synergy decreases as γA increases above γAG; the points (γG, γAG) form the solid curve shown in Fig 6(b). We suggest that for optimal efficacy under MTD constraint, the level of dosage of anti-PD-1 (γA) should be related to the level of dosage of GVAX (γG) by setting γA = γAG, as indicated by the solid curve in Fig 6(b).
The mathematical model presented in this paper has several limitations:
In order to focus on the combined therapy of a cancer vaccine and an anti-PD-1 drug, we did not include some other important cells and cytokines that are found in the tumor microenvironment, such as T regulatory cells, macrophages, endothelial cells, and IL-10, IL-6 and TGF-β. We also did not include blood vessels and oxygen, thus assuming that the tumor is avascular. We tacitly assumed that the effect of these omissions is not significant in comparing the results of therapy to no therapy.
We assumed that the densities of immature, or naive, immune cells remain constant throughout the progression of the cancer and that dead cells are quickly removed from the tumor.
In estimating parameters we made a steady state assumption in some of the differential equations.
In the definition of synergy, and in the synergy map, we included in a crude way the fact that anti-PD-1 causes more toxicity than GVAX. Our aim was to develop a concept that will take account not only of efficacy but also of toxicity. For this reason we compared the treatment benefits for combination (γG, γA) with the single agents (2γG, 0) and (0, 1.5γA).
We did not make any direct connection between drugs administered to the patient, and their ‘effective strengths’ γG and γA, as they appear in the differential equations, since these data are not available. The order of magnitude of GVAX (10−10) was based on the estimate of the concentration of GM-CSF in normal healthy tissue (see Parameter Estimations for Eq (6)). We simulated the model with different orders of magnitude for anti-PD-1 drug (γA) and found the best agreement with the mouse experiments (in Figs 4 and 5) when γA is also of order of magnitude 10−10.
Although our mathematical model does not presume any geometric form of the tumor, for simplicity, the simulations have been carried out only in the case of a spherical tumor. We note however that spherical cancer models have been used in research as an intermediate between in vitro cancer line cultures and in vivo cancer [50]. Furthermore, spheroids mirror the 3D cellular context and therapeutically relevant pathophysiological gradient of in vivo tumors [51].
Clinical data on efficacy and toxicity are quite limited at this time. Our model should be viewed as setting up a computational framework, to address the question of optimal efficacy in combination therapy with cancer vaccine and checkpoint inhibitor.
Materials and methods
Parameter estimation
Half-saturation. In an expression of the form where Y is activated by X, the half-saturation parameter KX is taken to be the approximate steady state concentration of species X.
Diffusion coefficients. By [52], we have the following relation for estimating the diffusion coefficients of a protein p:
where MV and δV are respectively the molecular weight and diffusion coefficient of VEGF, Mp is the molecular weight of p, and MV = 24kDa [53] and δV = 8.64 × 10−2 cm2day−1 [54]. Since MI2 = 15.5kDa, MI12 = 70kDa, MG = 16.2kDa [55] and MA = 32kDa [56], we get δI2 = 9.9956 × 10−2 cm2day−1, δI12 = 6.0472 × 10−2 cm2day−1, δG = 9.8495 × 10−2 cm2day−1, and δA = 7.85 × 10−2 cm2day−1.
Eq (2): The number of DCs in various organs (heart, kidney, pancreas and liver) in mouse varies from 1.1 × 106 cells/cm3 to 6.6 × 106 cells/cm3 [57]. Mature DCs are approximately 10 to 15 μm in diameter [58]. Accordingly, we estimate steady states of DCs by KD = 4 × 10−4 g/cm3. We assume that the density of immature DCs is smaller than the actived DCs, and take g/cm3. We also assume that the activation of DCs by GM-CSF-secreting vaccine is more effective than their activation by NCs-secreted HMGB-1, and take . From the steady state of Eq (2) in the control case (with no drug), we get
where dD = 0.1/day [59], D0 = 2 × 10−5 g/cm3, D = KD = 4 × 10−4 g/cm3, C = KC = 0.4 g/cm3, and g/cm3 and KG = 1.74 × 10−9 g/cm3 ( and KG are estimated together with other estimates of Eq (6)). Hence λDC = 0.364/day and λDG = 20.02/day.
Eq (3): The number of lymphocytes is approximately twice the number of DCs [57]. T cells are approximately 14 to 20 μm in diameter. Assuming that the number of Th1 cells is 1/4 of the number of lymphocytes, we estimate the steady state density of Th1 cells by KT = 2 × 10−3 g/cm3. We assume the density of naive CD4+ T cells to be less than the density of Th1, and take g/cm3. We also assume that in steady state, Q/KTQ = 2 (KTQ is estimated together with other estimates of Eqs (9)–(12)). From the steady state of Eq (3), we get
where T10 = 4 × 10−4 g/cm3, T1 = KT1 = 2 × 10−3 g/cm3, and, by [59], λT1I2 = 0.25/day and dT1 = 0.197/day. Hence λT1I12 = 4.66/day.
Eq (4): The CD4/CD8 ratio in the blood is 2:1. We assume a similar ratio in the tissue, and take g/cm3. We also take the steady state of T8 to be half of the steady state of T1, i.e., g/cm3. From the steady state equation of Eq (4), we get
where T80 = 5 × 10−5 g/cm3, T8 = KT8 = 1 × 10−3 g/cm3, and, by [59], λT8I2 = 0.25/day and dT8 = 0.18/day. Hence λT1I12 = 4.15/day.
Eq (5): We take dC = 0.17 day−1 and CM = 0.8 g/cm3 [59]. In the absence of immune responses and anti-tumor drugs, the tumor grows according to
| (20) |
and with immune response, the tumor grows according to
| (21) |
Mouse experiments show that the tumor volume on average doubles within 5-15 days [21–23]. Assuming a linear growth
during the volume doubling time, we conclude from Eq (21) that
| (22) |
where . We assume that with no immune responses
so that, by Eq (20), we get
| (23) |
We take λ0 = 0.069/day, and assume that in steady state of Eqs (20) and (21), C is approximately 0.4 g/cm3, so that from Eq (23) we get
or λC = 0.616/day. From Eqs (23) and (22), we get η1T1 + η8T8 = λ0. Noting that T8 cells kill cancer cells more effectively than T1 cells, we take η8 = 4η1, so that cm3/g ⋅ day and η8 = 46 cm3/g ⋅ day (with T1 = KT1 = 2 × 10−3 g/cm3 and T8 = KT8 = 1 × 10−3 g/cm3 as in the estimates for Eqs (3) and (4)).
Eq (6): With no drugs, the steady state plasma concentration of GM-CSF is g/cm3 [60]. The half-life of GM-CSF is 13 hours [61], i.e, 0.54 day, so that /day. From the steady state of Eq (6) (with no drugs), we get g/cm3 ⋅ day. But when the GM-CSF-secreting vaccine is administered, the concentration of GM-CSF shoots up, and we assume that at steady states it reaches the level of g/cm3.
Eq (7): From the steady state of Eq (7), we get λI12DD − dI12I12 = 0, where dI12 = 1.38/day [59] and I12 = KI12 = 1.5 × 10−10 g/cm3 [59], and D = KD = 4 × 10−4 g/cm3. Hence, λI12D = 5.18 × 10−7/day.
Eq (8): From the steady state of Eq (8), we get λI2T1T1−dI2I2 = 0, where dI2 = 2.376/day [59] and I2 = KI2 = 2.37 × 10−11 g/cm3 [59], and T1 = KT1 = 2 × 10−3 g/cm3. Hence, λI2T1 = 2.82 × 10−8/day.
Eqs (9)–(12): In order to estimate the parameter KTQ (in Eqs (3) and (4)), we need to determine the steady state concentrations of P and L in the control case (no drugs). To do that, we need to estimate ρP and ρL. By [62], the mass of one PD-1 protein is mP = 8.3 × 10−8 pg = 8.3 × 10−20 g, and by [63] the mass of one PD-L1 is mL = 5.8 × 10−8 pg = 5.8 × 10−20 g. We assume that the mass of one T cell is mT = 10−9 g. By [14], there are 3000 PD-1 proteins and 9000 PD-L1 proteins on one T cell (T1 or T8). Since ρPT is the density of PD-1 (without anti-PD-1 drug), we get and .
In steady state, T1 = 2 × 10−3 g/cm3 and T8 = 1 × 10−3 g/cm3. Hence, in steady state,
The parameter ε in Eq (10) depends on the type of cancer; many cancer cells, but not all, express PD-L1 [17, 35–37]. Accordingly, we assume ε varies in the interval 0-0.01, but in the simulations we take ε = 0.01. Then, from Eq (10), we get
In steady state with , and , we have, by Eq (12), . We take . Hence, and
where g2/cm6.
Eq (13): By [42], the half-life of anti-PD-1 is 15 days, so that day−1. We assume that 10% of A is used in blocking PD-1, while the remaining 90% degrades naturally. Hence, in steady state, μPAPA/10% = dAA/90%, so that
If the percentage of A used in blocking PD-1 is changed, the value of μPA will also change, but the results of simulations do not change qualitatively (not shown here).
Sensitivity analysis
We performed sensitivity analysis, with respect to the tumor radius R at day 60, with respect to some of the production parameters of the System (2)–(13), namely, λDC, λDG, λT1I12, λT1I2, λT8I12, λT8I2, and the important parameters KTQ, η1 and η8. Following the method in [64], we performed Latin hypercube sampling and generated 1000 samples to calculate the partial rank correlation coefficients (PRCC) and the p-values with respect to the tumor radius at day 60. In sampling all the parameters, we took the range of each from 1/2 to twice its values in Tables 2 and 3. The results are shown in Fig 8.
Fig 8. Statistically significant PRCC values (p-value < 0.01) for R(t) at day 60.

Not surprisingly all the parameters are negatively correlated with the tumor radius. We note that the highest negatively correlated parameters are the activation rate of dendritic cells by cancer cells (λDC) and the inhibition of T cells activation by PD-1-PD-L1 complex (KTQ). However, with different values of γG and γA the parameter λDG can exceed λDC.
Computational method
We employ moving mesh method [34] to numerically solve the free boundary problem for the tumor proliferation model. To illustrate this method, we take Eq (2) as example and rewrite it as the following form:
| (24) |
where F represents the term in the right hand side of Eq (2). Let and denote numerical approximations of i-th grid point and , respectively, where τ is the size of time-step. The discretization of Eq (24) is derived by the fully implicit finite difference scheme:
| (25) |
where , , , and . The mesh moves by , where is solved by the velocity equation.
Acknowledgments
This work is supported by the Mathematical Biosciences Institute and the National Science Foundation (Grant DMS 0931642), and the Renmin University of China and the International Postdoctoral Exchange Fellowship Program 2016 by the Office of China Postdoctoral Council.
Data Availability
All relevant data are within the paper.
Funding Statement
This work is supported by the Mathematical Biosciences Institute and the National Science Foundation (Grant DMS 0931642), and the Renmin University of China and the International Postdoctoral Exchange Fellowship Program 2016 by the Office of China Postdoctoral Council.
References
- 1. Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol. 2010;28:367–388. 10.1146/annurev.immunol.021908.132603 [DOI] [PubMed] [Google Scholar]
- 2. Palucka J, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. March 2012;12(4):265–277. 10.1038/nrc3258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Saenz R, Futalan D, Leutenez L, Eekhout F, Fecteau JF, Sundelius S, et al. TLR4-dependent activation of dendritic cells by an HMGB1-derived peptide adjuvant. J Transl Med. Aug 2014;12(211):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Janco JMT, Lamichhane P, Karyampudi L, Knutson KL. Tumor-infiltrating dendritic cells in cancer pathogenesis. J Immunol. Apr 2015;194(7):2985–2991. 10.4049/jimmunol.1403134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ma Y, Shurin GV, Peiyuan Z, Shurin MR. Dendritic Cells in the Cancer Microenvironment. J Cancer. 2013;4(1):36–44. 10.7150/jca.5046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fernandez NC, Lozier A, Flament C, Ricciardi-Castagnoli P, Bellet D, Suter M, et al. Dendritic cells directly trigger NK cell functions: cross-talk relevant in innate anti-tumor immune responses in vivo. Nat Med. April 1999;5(4):405–411. 10.1038/7403 [DOI] [PubMed] [Google Scholar]
- 7. Gong J, Avigan D, Chen D, Wu Z, Koido S, Kashiwaba M, et al. Activation of antitumor cytotoxic T lymphocytes by fusions of human dendritic cells and breast carcinoma cell. J Cancer. 2000;97(6):2715–2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mailliard RB, Bykovskaya SEQCAKSN, Lotze MT, et al. Complementary dendritic cell-activating function of CD8+ and CD4+ T cells: helper role of CD8+ T cells in the development of T helper type 1 responses. J Exp Med. February 2002;195(4):473–483. 10.1084/jem.20011662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Haabeth OAW, Tveita AA, Fauskanger M, Schjesvold F, Lorvik KB, Hofgaard PO, et al. How Do CD4(+) T Cells Detect and Eliminate Tumor Cells That Either Lack or Express MHC Class II Molecules? Front Immunol. April 2014;5(174):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. van der Burg SH, Arens R, Ossendorp F, van Hall T, Melief CJM. Vaccines for established cancer: overcoming the challenges posed by immune evasion. Nature Reviews Cancer. March 2016;16:219–233. 10.1038/nrc.2016.16 [DOI] [PubMed] [Google Scholar]
- 11. Kaufman HL, Ruby CE, Hughes T, Slingluff CLJ. Current status of granulocyte-macrophage colony-stimulating factor in the immunotherapy of melanoma. J Immunother Cancer. May 13, 2014;2(11):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gupta R, Emens LA. GM-CSF-secreting vaccines for solid tumors: moving forward. Discov Med. July 2010;10(50):52–60. [PMC free article] [PubMed] [Google Scholar]
- 13. Simmons AD, Li B, Gonzalez-Edick M, Lin C, Moskalenko M, Du T, et al. GM-CSF-secreting cancer immunotherapies: preclinical analysis of the mechanism of action. Cancer Immunol Immunother. October, 2007;56(10):1653–1665. 10.1007/s00262-007-0315-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cheng X, Veverka V, Radhakrishnan A, Waters LC, Muskett FW, Morgan SH, et al. Structure and interactions of the human programmed cell death 1 receptor. J Biol Chem. April 2013;288(17):11771–11785. 10.1074/jbc.M112.448126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shi L, Chen S, Yang L, Li Y. The role of PD-1 and PD-L1 in T-cell immune suppression in patients with hematological malignancies. J Hematol Oncol. September 2013;6(74). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Muppidi MR, George S. Immune Checkpoint Inhibitors in Renal Cell Carcinoma. Journal of Targeted Therapies in Cancer 2015. 2015;4:47–52. [Google Scholar]
- 17. He J, Hu Y, Hu M, Li B. Development of PD-1/PD-L1 Pathway in Tumor Immune Microenvironment and Treatment for Non-Small Cell Lung Cancer. Scientific Reports. August 2015;5(13110). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kleponis J, Skelton R, Zheng L. Fueling the engine and releasing the break: combinational therapy of cancer vaccines and immune checkpoint inhibitors. Cancer Biol Med. September 2015;12(3):201–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Guo C, Manjili MH, Subjeck JR, Sarkar D, Fisher PB, et al. Therapeutic Cancer Vaccines: Past, Present and Future. Adv Cancer Res. 2013;119:421–475. 10.1016/B978-0-12-407190-2.00007-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Burotto M, Singh N, Heery CR, Gulley JL, Mada RA. Exploiting synergy: immune-based combinations in the treatment of prostate cancer. Front Oncol. December 12, 2014;4(351):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ali OA, Lewin SA, Dranoff G, Mooney DJ. Vaccines combined with immune checkpoint antibodies promote cytotoxic T cell activity and tumor eradication. Cancer Immunology Research. Dec 15, 2015; p. [DOI] [PMC free article] [PubMed]
- 22. Tian H, Shi G, Wang Q, Li Y, Yang Q, Li C, et al. A novel cancer vaccine with the ability to simultaneously produce anti-PD-1 antibody and GM-CSF in cancer cells and enhance Th1-biased antitumor immunity. Signal Transduction and Targeted Therapy. October 15, 2016;1(16025). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li B, VanRoey M, Wang C, hui Timothy Chen T, Korman A, Jooss K. Anti-programmed death-1 synergizes with granulocyte macrophage colony-stimulating factor-secreting tumor cell immunotherapy providing therapeutic benefit to mice with established tumors. Clin Cancer Res. 2009;15(5):1623–1634. 10.1158/1078-0432.CCR-08-1825 [DOI] [PubMed] [Google Scholar]
- 24. Soares KC, Rucki AA, Wu AA, Olino K, Xiao Q, Chai Y, et al. PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T-cell infiltration into pancreatic tumors. J Immunother. January 2015;38(1):1–11. 10.1097/CJI.0000000000000062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fu J, Malm IJ, Kadayakkara DK, Levitsky H, Pardoll D, Kim YJ. Preclinical evidence that PD1 blockade cooperates with cancer vaccine TEGVAX to elicit regression of established tumors. Cancer Res. August 1, 2014;74(15):4042–4052. 10.1158/0008-5472.CAN-13-2685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rosenblatt J, Glotzbecker B, Mills H, Vasir B, Tzachanis D, Levine JD, et al. PD-1 blockade by CT-011, anti PD-1 antibody, enhances ex-vivo T cell responses to autologous dendritic/myeloma fusion vaccine. J Immunother. June 2011;34(5):409–418. 10.1097/CJI.0b013e31821ca6ce [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Duraiswamy J, Kaluza KM, Freeman GJ, Coukos G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res. June 15, 2013;73(12):3591–3603. 10.1158/0008-5472.CAN-12-4100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Serre R, Benzekry S, Padovani L, Meille C, Nicolas Andre JC, et al. Mathematical Modeling of Cancer Immunotherapy and Its Synergy with Radiotherapy. Cancer Res. September 2016;76(17):4931–40. 10.1158/0008-5472.CAN-15-3567 [DOI] [PubMed] [Google Scholar]
- 29. Joshi B, Wang X, Banerjee S, Tian H, Matzavinos A, Chaplain MAJ. On immunotherapies and cancer vaccination protocols: a mathematical modelling approach. J Theor Biol. Aguest 2009;259(4):820–827. 10.1016/j.jtbi.2009.05.001 [DOI] [PubMed] [Google Scholar]
- 30. vG de Pillis L, Gu W, Radunskay AE. Mixed immunotherapy and chemotherapy of tumors: modeling, applications and biological interpretations. J Theor Biol. February 2006;238(4):841–862. 10.1016/j.jtbi.2005.06.037 [DOI] [PubMed] [Google Scholar]
- 31. Zandarashvili L, Sahu D, Lee K, Lee YS, Singh P, Rajarathnam K, et al. Real-time kinetics of high-mobility group box 1 (HMGB1) oxidation in extracellular fluids studied by in situ protein NMR spectroscopy. J Biol Chem. April 2013;288(17):11621–11627. 10.1074/jbc.M113.449942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. van de Laar L, Coffer PJ, Woltman AM. Regulation of dendritic cell development by GM-CSF: molecular control and implications for immune homeostasis and therapy. Blood. April 2012;119(15):3383–3393. 10.1182/blood-2011-11-370130 [DOI] [PubMed] [Google Scholar]
- 33. Mautea RL, Gordona SR, Mayere AT, McCrackena MN, Natarajane A, Ring NG, et al. Engineering high-affinity PD-1 variants for optimized immunotherapy and immuno-PET imaging. Proc Natl Acad Sci USA. November 2015;112(47):E6506–14. 10.1073/pnas.1519623112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. D’Acunto B. Computational Methods for PDE in mechanics Series on Advances in Mathematics for Applied Sciences-Vol.67. Singapore: Word Scientific; 2004. [Google Scholar]
- 35. Buttea MJ, Pena-Cruzc V, Kima MJ, Freemanc GJ, Sharpe AH. Interaction of human PD-L1 and B7-1. Mol Immunol. August 2008;45(13):3567–3572. 10.1016/j.molimm.2008.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. March 2012;4(127):127ra37 10.1126/scitranslmed.3003689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therap. Clin Cancer Res. October 2014;20(19):5064–5074. 10.1158/1078-0432.CCR-13-3271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jaffee EM, Hruban RH, Biedrzycki B, Laheru D, Schepers K, Sauter PR, et al. Novel allogeneic granulocyte-macrophage colony-stimulating factor-secreting tumor vaccine for pancreatic cancer: a phase I trial of safety and immune activation. J Clin Oncol. 2001;19:145–156. 10.1200/JCO.2001.19.1.145 [DOI] [PubMed] [Google Scholar]
- 39. Laheru D, Lutz E, Burke J, Biedrzycki B, Solt S, Onners B, et al. Allogeneic granulocyte macrophage colony-stimulating factor-secreting tumor immunotherapy alone or in sequence with cyclophosphamide for metastatic pancreatic cancer: a pilot study of safety, feasibility, and immune activation. Clin Cancer Res. 2008;14:1455–1463. 10.1158/1078-0432.CCR-07-0371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lutz E, Yeo CJ, Lillemoe KD, Biedrzycki B, Kobrin B, Herman J, et al. A lethally irradiated allogeneic granulocyte-macrophage colony stimulating factor-secreting tumor vaccine for pancreatic adenocarcinoma. A Phase II trial of safety, efficacy, and immune activation. Ann Surg. 2011;253:328–335. 10.1097/SLA.0b013e3181fd271c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134–44. 10.1056/NEJMoa1305133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28:3167–3175. 10.1200/JCO.2009.26.7609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. 10.1056/NEJMoa1200690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Swart M, Verbrugge I, Beltman JB. Combination Approaches with Immune-Checkpoint Blockade in Cancer Therapy. Front Oncol. 2016;6(233):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Soares KC, Rucki AA, Wu AA, Olino K, Xiao Q, Chai Y, et al. PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T-cell infiltration into pancreatic tumors. J Immunother. 2015;38(1):1–11. 10.1097/CJI.0000000000000062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Owonikoko TK, Ramalingam SS. Minimize toxicity or preserve efficacy? A delicate trade-off in the management of older patients with lung cancer. J Clin Oncol. February 2015;33(6):534–536. 10.1200/JCO.2014.59.5033 [DOI] [PubMed] [Google Scholar]
- 47. Lee CK, Gebski VJ, Coates AS, Veillard AS, Harvey V, Tattersall MH, et al. Trade-offs in quality of life and survival with chemotherapy for advanced breast cancer: mature results of a randomized trial comparing single-agent mitoxantrone with combination cyclophosphamide, methotrexate, 5-fluorouracil and prednisone. Springerplus. Augest 2013;21(391):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Marshall DA, Deal K, Bombard Y, Leighl N, MacDonald KV, Trudeau M. How do women trade-off benefits and risks in chemotherapy treatment decisions based on gene expression profiling for early-stage breast cancer? A discrete choice experiment. BMJ Open. June 2016;6(6):e010981 10.1136/bmjopen-2015-010981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Blinman P, King M, Norman R, Viney R, Stockler MR. Preferences for cancer treatments: an overview of methods and applications in oncology. Ann Oncol. 2012;23(5):1104–1110. 10.1093/annonc/mdr559 [DOI] [PubMed] [Google Scholar]
- 50. Weiswald LB, Bellet D, Dangles-Marie V. Spherical cancer models in tumor biology. Neoplasia. 2015;17(1):1–15. 10.1016/j.neo.2014.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hirschhaeuser F, Menne H, Dittfeld C, West J, Mueller-Klieser W, Kunz-Schughart LA. Multicellular tumor spheroids: An underestimated tool is catching up again. J Biotechnol. 2010;148(1):3–15. 10.1016/j.jbiotec.2010.01.012 [DOI] [PubMed] [Google Scholar]
- 52. Young ME. Estimation of diffusion coefficients of proteins. Biotechnology and Bioengineering. 1980;XXII:947–955. 10.1002/bit.260220504 [DOI] [Google Scholar]
- 53. Shui YB, Wang X, Hu JS, Wang SP, Garcia CM, et al. Vascular endothelial growth factor expression and signaling in the lens. Invest Ophthalmol Vis Sci. 2003;44(9):3911–3919. 10.1167/iovs.02-1226 [DOI] [PubMed] [Google Scholar]
- 54. Liao KL, Bai XF, Friedman A. Mathematical modeling of interleukin-27 induction of anti-tumor T cells response. PLoS ONE. 2014;9(3):e91844 10.1371/journal.pone.0091844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.PhosphoSitePlus/. GM-CSF, http://www.phosphosite.org/proteinAction?id=2454248&showAllSites=true;
- 56.Abcam/. Anti-PD1 antibody (ab89828), http://www.abcam.com/pd1-antibody-ab89828.html;
- 57. Steptoe RJ, Patel RK, Subbotin VM, Thomson AW. Comparative analysis of dendritic cell density and total number in commonly transplanted organs: morphometric estimation in normal mice. Transpl Immunol. 2000;8(1):49–56. 10.1016/S0966-3274(00)00010-1 [DOI] [PubMed] [Google Scholar]
- 58. Dumortier H, van Mierlo GJD, Egan D, van Ewijk W, Toes REM, Offringa R, et al. Antigen presentation by an immature myeloid dendritic cell line does not cause CTL deletion in vivo, but generates CD8+ central memory-like T cells that can be rescued for full effector function. J Immunol. 2005;175(2):855–863. 10.4049/jimmunol.175.2.855 [DOI] [PubMed] [Google Scholar]
- 59.Hao W, Friedman A. The role of exosomes in pancreatic cancer microenvironment. Bull Math Biol. 2017;accepted. [DOI] [PubMed]
- 60. Fiehn C, Wermann M, Pezzutto A, Hufner M, Heilig B. Plasma GM-CSF concentrations in rheumatoid arthritis, systemic lupus erythematosus and spondyloarthropathy. Z Rheumatol. 1992;51(3):121–126. [PubMed] [Google Scholar]
- 61. Dale DC, Liles WC, Llewellyn C, Price TH. Effects of granulocyte-macrophage colony-stimulating factor (GM-CSF) on neutrophil kinetics and function in normal human volunteers. Am J Hematol. 1998;57(1):7–15. [DOI] [PubMed] [Google Scholar]
- 62. Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubat T, Yagita H, et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol. 1996;8(5):765–772. 10.1093/intimm/8.5.765 [DOI] [PubMed] [Google Scholar]
- 63.Human PD-L1/B7-H1/CD274 Protein. Sino Biological Inc, http://wwwsinobiologicalcom/PD-L1-B7-H1-CD274-Protein-g-533html;
- 64. Marino S, Hogue I, Ray C, Kirschner D. A methodology for performing global uncertainty and sensitivity analysis in systems biology. J Theor Biol. 2008;254(1):178–196. 10.1016/j.jtbi.2008.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hao W, Schlesinger LS, Friedman A. Modeling Granulomas in Response to Infection in the Lung. PLoS ONE. March 17, 2016;11(3):e0148738 10.1371/journal.pone.0148738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rini B. Future approaches in immunotherapy. Semin Oncol. October 2014;41(5, Suppl 5):S30–S40. 10.1053/j.seminoncol.2014.09.005 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper.