

Abstract
N2 reduction by nitrogenase involves the accumulation of four reducing equivalents at the active site FeMo-cofactor to form a state with two [Fe-H-Fe] bridging hydrides (denoted E4(4H), the Janus intermediate), and we recently demonstrated that the enzyme is activated to cleave the N≡N triple bond by the reductive elimination (re) of H2 from this state. We are exploring a photochemical approach to obtaining atomic-level details of the re activation process. We have shown that when E4(4H) at cryogenic temperatures is subjected to 450 nm irradiation in an EPR cavity, it cleanly undergoes photoinduced re of H2 to give a reactive doubly-reduced intermediate, denoted E4(2H)*, which corresponds to the intermediate that would form if thermal dissociative re loss of H2 preceded N2 binding. Experiments reported here establish that photoinduced re occurs in two steps. Photolysis of E4(4H) generates an intermediate state that undergoes subsequent photoinduced conversion to [E4(2H)* + H2]. The experiments, supported by DFT calculation, indicate that the trapped intermediate is an H2 complex on the ground adiabatic potential energy suface that connects E4(4H) with [E4(2H)* + H2]. We suggest this complex, denoted E4(H2; 2H), is a thermally populated intermediate in the catalytically central re of H2 by E4(4H), and that N2 reacts with this complex to complete the activated conversion of [E4(4H) + N2] into [E4(2N2H) + H2].
TOC image
Introduction
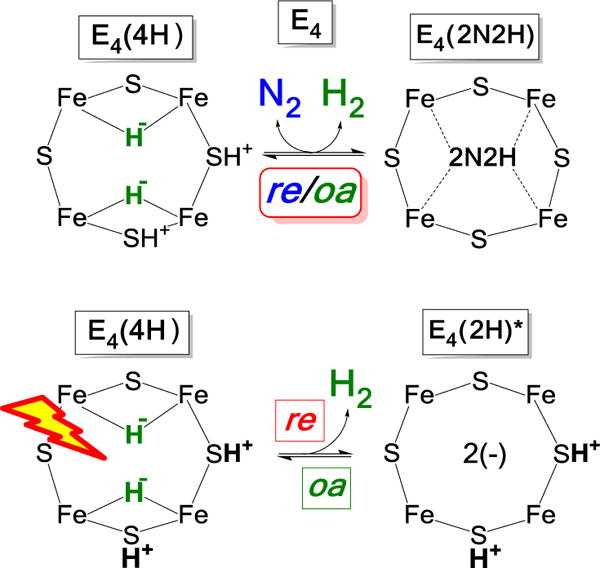
In recent years we have demonstrated1–4 that N2 reduction by nitrogenase involves the accumulation of four reducing equivalents at the active site FeMo-cofactor (FeMo-co) to form a state with two [Fe-H-Fe] bridging hydrides and two sulfur-bound protons (denoted E4(4H), the Janus intermediate), and that breaking the N≡N triple bond requires the reductive elimination (re) of H2 from E4(4H). This process corresponds to the forward direction of the equilibrium in Fig 1, upper, whose reverse is the oxidative addition (oa) of H2 with release of N2.5 This re/oa mechanism was initially founded on the identification of a freeze-trapped intermediate of the α-70Val→Ile substituted MoFe protein as E4(4H).6–8 Subsequently, EPR/ENDOR, and photophysical measurements showed that the identical Janus intermediate can be freeze-trapped during N2 reduction by wild type (WT) MoFe protein, thereby confirming the intermediate in the α-70Val→Ile variant as a reliable guide to mechanism, and establishing re/oa as a thermoneutral, kinetically accessible equilibrium process.4 The overall result of these several findings was to establish that H2 release drives the reduction of the N≡N triple bond, and thus the mechanistic requirement for the formation of one H2 per N2. This in turn implies the limiting stoichiometry of eight electrons/protons for the reduction of N2 to two NH3 (eq 1).3
| (1) |
Figure 1.
Representations of the mechanistically central re/oa equilibrium that activates nitrogenase to break the N≡N triple bond (upper), and of the photoinduced re/oa equilibrium (lower). These cartoons represent the Fe 2,3,6,7 face of FeMo-co; but we emphasize they are not meant to be ‘anatomically’ precise.
In the context of this stoichiometry, and the Lowe-Thorneley kinetic scheme that embodies it,9–11 one sees that E4(4H) sits at a transition in the N2 reduction pathway, poised to relax to E0 through sequential release of two H2, but equally poised to bind and reduce N2 through the accumulation of four more equivalents, hence the designation of E4(4H) as the Janus intermediate.2
As a part of our efforts to (i) more deeply integrate the re/oa mechanism into the organometallic chemistry of hydrides and (ii) obtain atomic-level details of the re activation process we adopted a photochemical approach inspired by the properties of inorganic dihydride complexes.12 The photolysis of transition metal dihydride complexes, primarily mononuclear centers with cis hydride ligands, commonly results in the release of H213–22 with a decrease in metal-ion oxidation state by two, “a typical example of reductive elimination”, while the thermally induced reverse reaction “is the prototype example of an oxidative addition reaction.”14 EPR and ENDOR measurements showed that when the nitrogenase E4(4H) intermediate is subjected to 450 nm irradiation in an EPR cavity, it cleanly undergoes photo-induced re of H2 to give an intermediate which we denote E4(2H)*, Figure 1, lower. It is a reactive version of the E2(2H) state that has accumulated two [e−/H+], and corresponds to the intermediate that would form upon thermal dissociative re loss of H2 prior to N2 binding. The photoinduced re of H2/D2 from E4(4H)/E4(4D) in H2O/D2O is temperature invariant in both buffers in the range of 4 – 12 K, a variation in the thermal energy (proportional to T) by a factor of ~ 3, and the photoinduced decay of Janus intermediate shows a large KIE over this range, defined as the ratio of the median decay times for D2O and H2O buffers: KIE ~ 10. These observations together imply that photoinduced release of H2 through re involves nuclear tunneling through a barrier, in contrast to the barrierless process inferred for mono-metallic metal complexes.14,18
The photoinduced state E4(2H)* relaxes to E4(4H) by the thermally activated oa of H2 to E4(2H)* (Figure 1, lower) during annealing of the frozen solid at temperatures above T ≳ 175 K. The observation of a significant KIE plus a strong temperature dependence in the oa process revealed that it involves traversal of an energy barrier associated with H2 binding and/or bond cleavage. Overall, the measurements led to proposed energy surfaces associated with photoinduced re/oa for the Janus intermediate that will be refined in the present report.
The reported results were consistent with the absence of intermediate state(s) during photoinduced re,23 but they left open that possibility. As we noted: “whether the process involves an actual intermediate H2 complex remains to be determined”. Experiments reported here not only establish the presence of an intermediate in the photoinduced re of H2, but furthermore show that this intermediate undergoes photoinduced re to E4(2H)* (Fig 1, lower). The experiments, supported by DFT calculation, indicate that the trapped intermediate is an H2 complex and lead us to suggest that it functions as an intermediate in the thermal re of H2 that activates FeMo-co to break the N≡N triple bond.
Materials and Methods
Materials and Protein purifications
All the reagents were obtained from SigmaAldrich (St. Louis, MO) or Fisher Scientific (Fair Lawn, NJ) and were used without further purification. Argon and N2 were purchased from Air Liquide America Specialty Gases LLC (Plumsteadville, PA).
Remodeling the active site of MoFe protein by the α-70Val→Ile mutation permits the freeze trapping of MoFe with high populations of E4(4H)6 identical to that in WT enzyme.4 The protein was obtained from the corresponding Azotobacter vinelandii strains as described elsewhere.24 The handling of all buffers and proteins were done anaerobically under Ar atmosphere or under Schlenk vacuum line unless stated otherwise.
EPR samples
The E4(4H) intermediate and its deuterated analogue, E4(4D), are prepared by turnover of the MoFe protein in H2O and D2O buffers, respectively. EPR samples were prepared in a dioxygen free buffer and freeze-trapped during turnover as described.12
EPR Measurements
X-band EPR spectra were recorded on a Bruker ESP 300 spectrometer equipped with an Oxford Instruments ESR 900 continuous He flow cryostat. All spectra were measured at the following conditions: temperature, 12 K; microwave frequency, 9.36 GHz; microwave power, 10 mW; modulation amplitude, 9 G; time constant, 160 ms; field sweep speed, 20 G/s. In situ photolysis of a sample held within the cryostat employed a Thorlabs Inc (Newton, New Jersey) PL450B, 450 nm, 80 mW Osram Laser Diode mounted on the cavity optical access port as described.12 Light intensity was varied through controlling the laser current.25 Data points for observed photolysis progress curves were obtained as well resolved g1 amplitudes of corresponding EPR signals, with normalization of total concentration of the intermediate species to unity.
Results and Discussion
EPR Spectra and Low-temperature Photoinduced re
As recently shown,12 intra cavity photolysis with 450 nm light of the S = ½ E4(4H) intermediate freeze-trapped during turnover of MoFe protein causes photoinduced conversion of the E4(4H) signal, g = [2.15, 2.007, 1.965], to the E4(2H)* signal with g = [2.098, 2.0, 1.956], and E4(2H)* completely relaxes back to E4(4H) by oa of H2 with annealing in the solid state at temperatures above ~ 175 K.
To simultaneously obtain the time course for both the photoinduced loss of the E4(4H) (A) and the appearance of the E4(2H)* (C) signals, rather than continuously monitoring the occupancy of E4(4H) during photolysis, we collected complete spectra at multiple times during 12 K and 50 K irradiation and measured the intensities of the two species as a function of time, Fig 2. If photoexcitation of A led directly to the A (E4(4H)) → C (E4(2H)*) conversion in a single kinetic step without buildup of a spectroscopic intermediate state, then at every time the C occupancy should equal the loss of the A occupancy, (1-A) as normalized to the value A at t = 0, and the rate parameters for the rise of C should equal those for the loss of A. However, as shown in Fig 2 (and see below), at low temperatures the rise of C lags the loss of A, which implies that photoexcitation of A creates an intermediate, B, which accumulates during the course of its conversion to C. The existence of such an on-path state B, is supported below in the characterization of B, kinetic fits, and in considerations of a potential-energy surfaces that captures the reversible [photoinduced-re/thermal-oa] process of Fig 1 lower.
Figure 2.
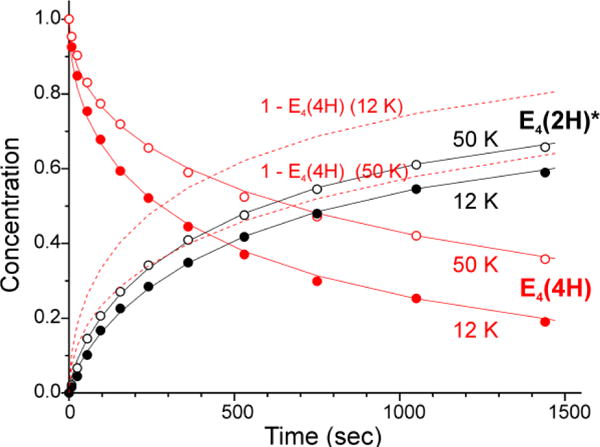
Comparison of normalized 12 K and 50 K time courses of E4(4H) (red) and E4(2H)* (black) states during irradiation of α-70Val→Ile H2O turnover sample with 450 nm laser diode light. Solid lines are to guide the eye; dotted lines show the lag in E4(2H)* formation at 12 K.
Photolysis Intermediate
To observe the B intermediate directly we carried out a variant of the photolysis cryoannealing experiment. E4(4H) was photolyzed for a fixed interval (25 min) at successively higher temperatures, then cooled back to 12 K for examination. In each step the sample was then annealed at 77 K for 2 min then restored to 12 K for another round of EPR examination. After such a process following photolysis at a given temperature, the sample was annealed at ~ 215 K, at which temperature E4(2H)* relaxes to E4(4H) within 2 minutes, thus restoring the sample to its initial state for photolysis at the next temperature (Fig 3).
Figure 3.
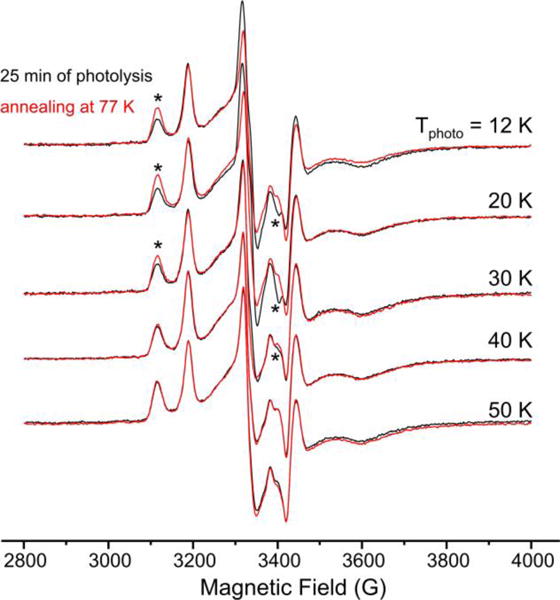
Results of 77 K annealing of α-70Val→Ile turnover irradiated at various temperatures. (*)-labeled features of EPR spectra indicate recovery of dihydride intermediate state E4(4H) upon annealing (left) in parallel with disappearance of photoinduced species (right).
Consider the results for this annealing process as applied to the samples photolyzed at 12 K (Figs 3, 4). Photolysis has converted the majority of E4(4H) to E4(2H)*, which does not relax during 77 K annealing. However, this annealing causes a small recovery of the E4(4H) signal without increase in the E4(2H)* signal. This implies another state, B, has been formed by photolysis, that B is stable in the dark at temperatures, T ≲ 30 K, and that it relaxes to E4(4H) by 77 K, even though E4(2H)* does not relax at this temperature. Essentially the same recovery of E4(4H) upon annealing is observed with 20 K photolysis, but by photolysis at 30 K, the recovery upon 77 K annealing is clearly diminished, implying a lesser accumulation of B. When photolysis is carried out above 30 K, there is no recovery of E4(4H) with 77 K annealing, indicating that B does relax at 30 K and above.
Figure 4.
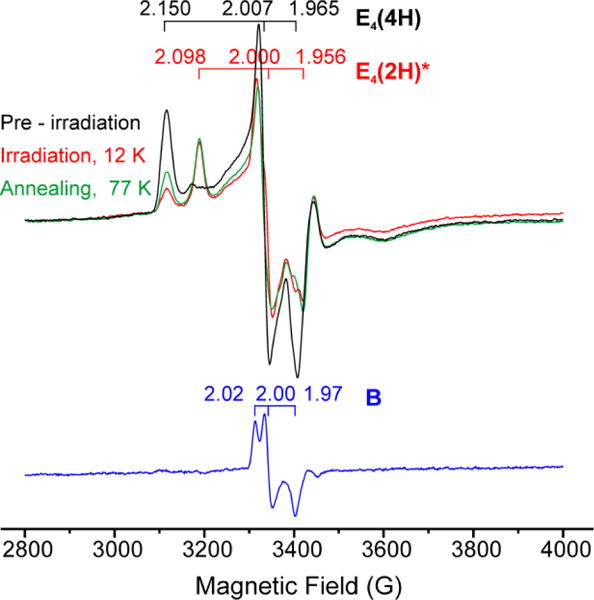
Subtraction-elimination of E4(4H) and E4(2H)* signals from EPR spectrum of α-70Val→Ile turnover irradiated at 12 K (red) with use of combination of spectra recorded before irradiation (black) and after 77 K annealing, which followed the irradiation (green). Result presents otherwise unresolved new photoinduced signal B (blue) unstable at 77K.
The measurements in Figs 3 showed the way to acquiring the spectrum of B through subtraction. The use of three spectra – before irradiation, after irradiation, after 77 K annealing – allows a subtraction-elimination of signals from all species except B. The procedure is shown in Fig S1, and as can be seen in Fig 4, reveals an S = ½ signal for B with g ~ [2.02, 2.00, 1.97]. Despite the need for this cumbersome subtraction procedure, the time course of B could actually be followed during photolysis at 12 K, as well as those of the initial A = E4(4H) and final C = E4(2H)* states to which it is kinetically linked by the kinetic scheme of eq 2.
The finding that B persists indefinitely (hours) at low temperature (T < 30 K) in the absence of the actinic light, yet converts to C during photolysis at these temperatures implies that both the A→ B and B → C conversions are photoinduced: the photoconversion of A to C thus involves two steps, each of which requires the absorption of a photon, as incorporated in the scheme of eq 2:
| (2) |
Though surprising, this result nonetheless can be analogized to the textbook photocleavage of a methyl C-H bond during photolysis of toluene, which likewise requires two consecutive photons.26–28
This finding does, however, require that we revisit our initial assignment of the photolysis product C = E4(2H)*, Fig 1, lower. In that earlier report, in addition to re mechanism of photoinduced re of H2 to yield C = E4(2H)*, now shown to be a 2-step process, eq 2, Fig 5, upper, we considered two other 2-step mechanisms that might eliminate the hydride bridges of E4(4H), each mechanism having a different assignment of the photoproduct, C, Fig 5.
Fig 5. Alternative 2-step mechanisms for photoinduced reaction.
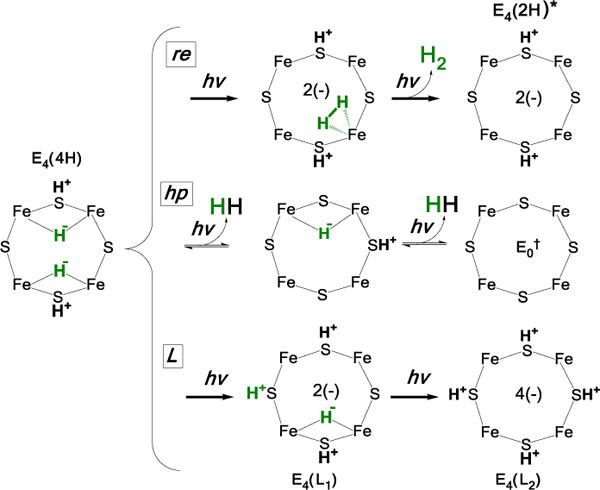
(Top) re; assignment of intermediate discussed below; (middle, bottom) alternative mechanisms, see text.
One of those alternative interpretations postulates two steps of photoinduced hydride protonolysis (hp; Fig 5, middle), each with loss of H2. This mechanism can be dismissed, for it would not lead to a photoproduct C that can regenerate E4(4H) during cryoannealing, as observed. Instead it would lead to FeMo-co at the E0 resting state level, which could not react with two released H2 to regenerate E4(4H) during cryoannealing: neither E0 nor E2(2H) react with H2.29
The second alternative shown in Fig 5, lower posits two steps of photoinduced re of a single hydride, each with transfer of the proton ‘released’ to a bridging sulfide. Instead of resulting in the release of H2, the final result of such a process would be creation of an E4(4H) ‘isomer’ with four protonated sulfur and no hydrides. Simple considerations of the expected relative stabilities of the experimentally observed B and C states rule out this mechanism. B is dark-stable at low temperatures and reverts to E4(4H) at T > 30 K, while C is more stable than B, relaxing to A only at T > 175 K. The greater stability of C is understandable within the re mechanism. The product state, C = E4(2H)*, undergoes activated ‘second-order’ binding of H2 on the ground potential-energy surface in the frozen matrix, to B as a higher-energy intermediate, which undergoes oa of H2 to generate the bridging hydrides of A = E4(4H). In contrast, in the L mechanism B is the destabilized E4(L1) ‘isomer’ of E4(4H) (Fig 5), and C would be a yet more unstable E4(L2) isomer. But this would be expected to relax to E4(L1) at a temperature lower than T ~ 30 K where B reverts to A, not at a temperature ~145 K higher.
These qualitative ideas are supported by exploratory DFT calculations (SI) on a realistic model of E4 and its local environment (Fig S4), which show that any structure with more than two protonated sulfur atoms is very high in energy relative to E4(2H)* and other relevant E4-level states, with E4(L2) much higher in energy than E4(L1) (Fig S5). Thus, the greater stability of C than B is indeed incompatible with the L mechanism.
Potential-Energy Landscape
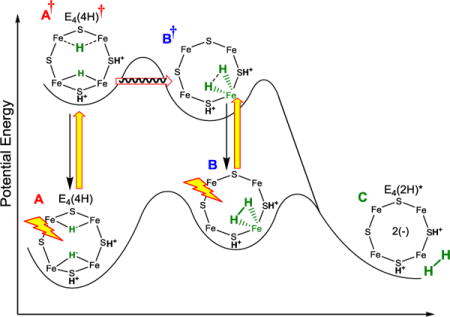
The above results can be captured by several alternative heuristic potential-energy landscapes. The most attractive of such qualitative conceptualizations is shown in Fig 6, which represents a projection of multi-dimensional potential energy surfaces that incorporate the experimental observations, along with features of re/oa of H2 associated with conventional inorganic complexes.
Figure 6.
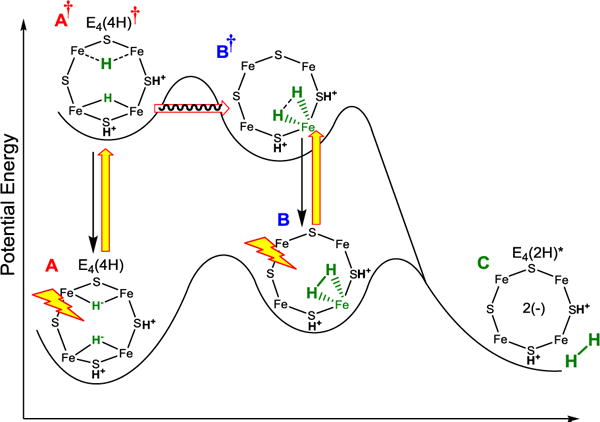
Idealized energy landscape for the low-temperature photoinduced re/oa of the Janus intermediate, E4(4H). Excited-state surface: the proposed identifications of the states A†, B, and B† are discussed in the text. Yellow arrows, photoexcitation; black arrows, relaxation, red arrow, nuclear tunneling.
The low-temperature steady-state photoinduced A → C re of H2 on the landscape of Fig 6 is visualized as following a pathway in which photoexcitation of A produces A†, which tunnels through a barrier to form an excited H2 complex, B†, which undergoes rapid relaxation to the observed S = ½, intermediate B on the ground surface. The B → C conversion then occurs by photoexcitation of B to B†, which undergoes tunneling/barrier crossing on its way to the re of H2 in state C; in the absence of direct evidence for nuclear tunneling by B†, tunneling is not indicated.
Although we as yet have no direct evidence as to the structure of the observed B intermediate (Fig 4), as visualized in Fig 6 the behavior of inorganic H2 complexes30–32 indicates that this intermediate is an H2-bound state on the ground adiabatic potential-energy surface, a possibility first noted thirty years ago.33 Firstly, thermally induced re/oa of inorganic H2 complexes generally proceed through such an intermediate,23 indicating an expectation that such a state is present on the ground adiabatic surface as shown in Fig 6. Secondly, the assignment of B as such a complex is supported by the finding that the B → C re of H2 is photoinduced: such behavior has been established for inorganic-H2 complexes.13,34–37 In support of this assignment, the DFT calculations (Fig S5 suggest that A = E4(4H) and C = E4(2H)* (+ H2) are the most stable E4 states and are very close in energy, and that they likely are connected by B = E4(H2; 2H) at a slightly higher energy.
As a final note, in SI support is provided for the assignment of B as on-path. There we show that an alternative assignment of the observed S = ½ intermediate, Figs 3, 4, to an off-path state, D, Fig S3 is inconsistent with experiment. It is shown that in general, if an off-path intermediate that both forms and decays photochemically were to be observed, then it would have to steadily accumulate during low-temperature photolysis, rather than approach a steady-state population as seen in detailed kinetic measurements presented below.
Kinetic Scheme
In the context of the landscape of Fig 6, the finding that the photoconversion of A to C involves the dark-stable intermediate B and the absorption of two photons consecutively, with B reverting solely to A for T > 30 K, implies the minimum kinetics of Scheme 1. During steady-state photoexcitation of A = E4(4H), one photon forms the excited state, A† = E4(4H)†. This can decay back to A or tunnel through the energy barrier to form B†, without return at low temperature (kb ≈ 0). This excited state branches: it can proceed directly with the re of H2 to form C, but, primarily decays to B (kd’ > kB†C). B is dark-stable at low temperatures (kBA ≈ 0), so would accumulate, but reaches a steady-state population because photoexcitation of B re-forms B†, and cycling through this process ultimately allows A to convert entirely to C during prolonged steady-state photolysis. At higher temperatures, T ≳ 30 K, B is no longer dark-stable, which implies that the A†⥨ B† reaction becomes reversible and/or direct reversion of B to A becomes operative: [kb and/or kBA] > 0. At temperatures, below ~ 175 K, C is indefinitely stable, kCB ≈ 0. Upon warming the frozen solid to T ≳ 175 K, C relaxes to A, [kCB, kBA] > 0, namely E4(2H)* undergoes oa of H2 to regenerate E4(4H), passing through the H2 intermediate B on the ground adiabatic surface of Fig 6.
Scheme 1.
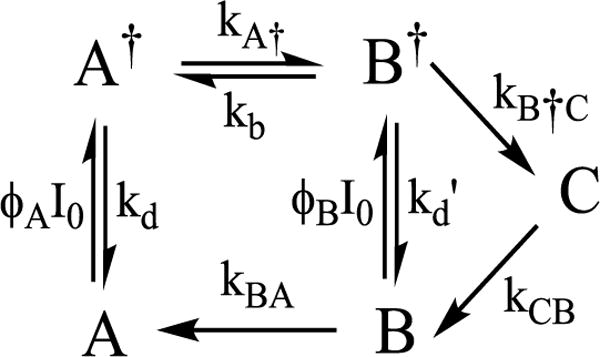
Considering in more detail the predictions of Scheme 1 in the steady-state photolysis measurements for temperatures T < 175 K, where C accumulates (kCB ≈ 0), the rate constant for formation of the photoexcited A† state is the product of the temperature-independent quantum yield, ϕA (taken to include the extinction coefficient) with the light intensity, I0, and kd is the rate constant for direct relaxation of A† back to ground. It can be presumed that kd is by far the largest rate constant in this scheme, and as a result, A† will enter a photostationary state during the intra-cavity photolysis, with instantaneous population proportional to the instantaneous concentration of A, with a temperature-independent proportionality constant determined by the photophysical constants,
| (3) |
and analogously for B†. As a result, Scheme 1 maps onto the far-simpler Scheme 2, an elaboration of eq 2, with a two photoinduced reaction steps involving observable states, but with provision for the first step to become reversible at higher temperatures. The net rate constant for the A → B conversion in Scheme 2 is given by
| (4) |
where kA† is the (tunneling) rate constant for A† → B† conversion. The rate constant for the second step, k2, takes an analogous form; we show shortly that this is augmented by an activated, thermally induced contribution.
Scheme 2.
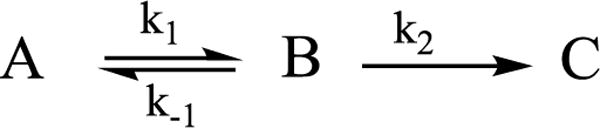
Kinetic Measurements
Progress curves for photolysis at 12 K and 50 K were collected for the frozen sample in H2O buffer, Fig 7, and D2O buffer, Fig S2. A fit to Scheme 2 using stretched exponentials, a decay function written, exp(-(t/τ)m), with median time constant, τ and ‘stretch parameter’, 0 < m ≤ 1, which decreases with deviations from simple exponential (m = 1). This description is needed to incorporate variation in I0 over the sample tube caused by light-scattering by the ‘frozen snow’ samples,12 and yields rate parameters listed on Fig 7. As shown in this figure, at 12 K the occupancy of B quickly rises to a value of roughly 20% occupancy that persists over the time of the experiment. Within the errors of determining the occupancy of intermediate B by the subtraction procedure described above, and of using stretched exponentials to model the distribution in apparent rate constants caused by light scattering, the measured time course of B at 12 K is in satisfactory agreement with the time course predicted by an ‘omit-fit’, namely a fit of the progress curves of A and C alone to eq 2, the low-temperature limit of the more elaborate Scheme 2.
Figure 7.
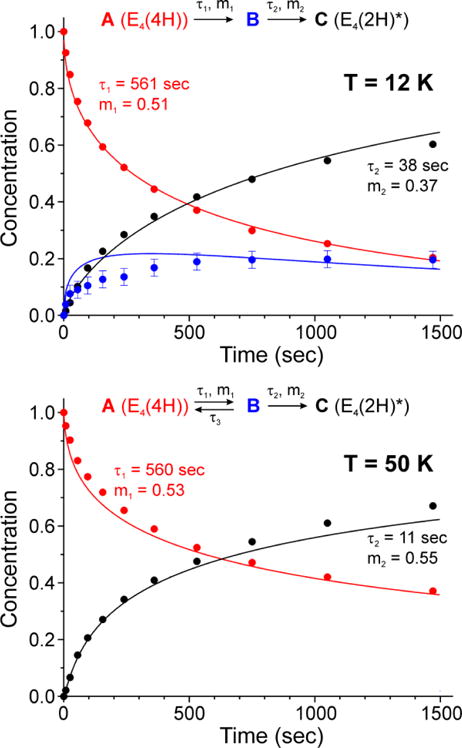
Normalized time courses of E4(4H) (red) and E4(2H)* (black) states measured during photolysis at 12 K (upper) and 50 K (lower) are shown fitted as previously described7 in accordance with corresponding low and high temperature kinetic schemes. Data points for photoinduced intermediate B at 12 K were obtained with subtraction procedure described in Figs 4, S1, and are shown in comparison with the kinetic scheme prediction (blue trace). Low accumulation of B state at 50 K is due to back reaction step with time constant τ3 = 160 sec.
In discussing the stretched-exponential rate parameters that describe the progress curves in Fig 7, there are different possible ways to define the correspondence of a rate constant defined in Scheme 2 with the stretched-exponential time-constant τ and ‘stretch parameter, 0 < m ≤ 138 For present purposes it is adequate to discuss the results as though rate constant and time constant are inverses: k ~ 1/τ.
Considering the 12 K measurements, B is stable at this temperature, which implies that at this temperature the rate constants for B† → A† and direct B → A reversion are vanishingly small, k−1 ~ 0. As a result, the photoinduced decay of A as measured by steady-state intracavity photolysis corresponds directly to k1 (eq 4). The fit to the present measurements at 12 K shown in Fig 7 confirm the previously reported low-temperature kinetic parameters for the A → B as well as the large isotope effect associated with k1, KIE ~ 13 (Fig S2). It was shown in the earlier measurements from 3.8 – 12 K that this process exhibits A† → B tunneling associated with the factor, kA†, that is incorporated into k1 (eq 4). Importantly, experiments in which the intensity of the actinic light is varied we find that k1 exhibits the expected linear dependence, eq 4. In the fits to Scheme 2 the time constant for A → B is unchanged by warming from 12 → 50 K (Fig 7), consistent with a persistence of the A† → B tunneling and continuation of the temperature invariance of the tunneling rate constant previously seen between 4 and 12K.12 However, the overall reversion of B to A (k−1) has become significant by 50 K.
The rate constant, k2, for the photoinduced B → C conversion is much greater than k1 at 12 and 50 K (Fig 7). As with k1, at 12 K it exhibits the expected linear dependence on actinic light intensity and a large isotope effect, KIE ≳ 10. The significant KIE plus an observed increase with increasing temperature dependence (Fig 7) is consistent with the B† → C with loss of H2 involving traversal of or tunneling through an energy barrier, rather than excitation of B to a dissociative state.
On the Structure of A†, and the mechanism of A†→B conversion
Regarding the nature of the excited state A†, as previously,12 we suggest that photoinduced re is initiated by excitation of E4(4H) into a (σ*) antibonding ligand-field excited state of one of the two bridging hydrides, as in the orbital sketch of Fig 8, which leads to the cartoon representation of A† in Fig 6. We suggest that this anti-bonding state could undergo reductive elimination, liberating a proton that tunnels the short distance to the second hydride bridge and protonates that hydride, thereby generating the intermediate B†, the excited state of H2-bound FeMo-co (B).
Figure 8.
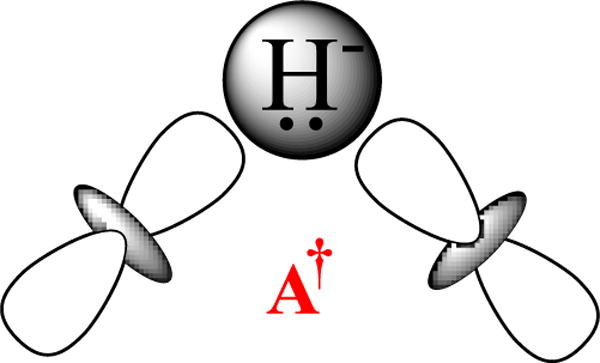
Antibonding excited state of bridging hydride.
Conclusions
This report describes a detailed study of the photoinduced re of H2 by E4(4H) to form the activated E4(2H)* state, and the thermal regeneration of E4(4H) by the oa of H2 to E4(2H)*, Fig 1, lower. We have examined the progress curves for loss of E4(4H) and appearance of E4(2H)* at 12 K and 50 K with samples prepared in H2O and D2O buffer. The key advance of these measurements is the discovery and characterization of a photogenerated intermediate, B, whose properties assign it as an H2 complex on the pathway for thermal oa of H2 by E4(2H)*, as visualized in Fig 6. This state accumulates during 12 K photolysis, and is dark-stable at this temperature, but can undergo photoinduced re to release H2.
The present study furthers our effort to obtain atomic-level details of the central mechanistic step in nitrogen fixation, re of H2 by E4(4H) coupled to the binding/reduction of N2, by which the nitrogenase MoFe protein is activated to carry out one of the most challenging chemical transformation in biology, the reduction of the N≡N triple bond. In the present work we extend the use of a photochemical approach inspired by the photoinduced re of H2 by inorganic dihydride complexes. Our initial report showed that E4(4H) exhibits the photoinduced re/oa equilibrium of Fig 1,12 and we subsequently showed that the E4(4H) in the α-70Val→Ile variant and the WT enzyme are identical in properties and behavior.4
The initial study of E4(4H) photolysis12 did not detect an intermediate state during photoinduced re of H2 from E4(4H). However, the more detailed experiments reported here now establish the presence of an intermediate, and furthermore show that this photogenerated intermediate can proceed to form E4(2H)* through subsequent photoinduced re with loss of H2. These findings, supported by DFT calculations, lead us to conclude that the trapped intermediate is an H2 complex on the ground adiabatic potential energy suface that connects E4(4H) with [E4(2H)* + H2], Fig 6. As such, we further suggest that this H2 complex is a thermally populated intermediate in the catalytically central re of H2 by E4(4H), and that N2 reacts with this complex to complete the conversion of [E4(4H) + N2] into [E4(2N2H) + H2], Fig 9. Whether there are additional intermediates in this catalytic conversion remains to be determined. We do not at this point favor the idea that E4(2H)* itself is such an intermediate. If H2 were thermally released from FeMo-co before N2 binds, forming E4(2H)*, then it is hard to see why D2 would not undergo oa by this FeMo-co state, contrary to the observation that D2 does not react with MoFe protein during turnover in the absence of N2.9
Figure 9.
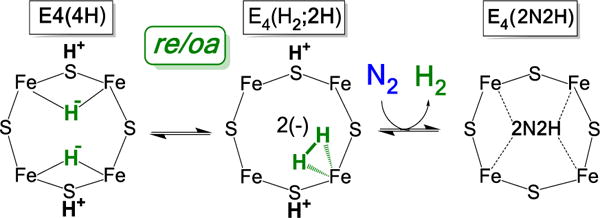
Cartoon version of suggested catalytic pathway for re/oa activation of FeMo-co for N2 reduction.
Supplementary Material
Synopsis.
N2 reduction by nitrogenase involves the accumulation of four reducing equivalents at the active site FeMo-cofactor to form a state with two [Fe-H-Fe] bridging hydrides (denoted E4(4H), the Janus intermediate), which is activated to cleave the N≡N triple bond by the reductive elimination (re) of H2. We report that photolysis of E4(4H) induces re of H2 in two steps, each photoinduced. Results indicate the intermediate state in this process is an H2 complex of FeMo-cofactor.
Acknowledgments
This work was supported by the NIH (GM 111097; BMH), NSF (MCB 1515981 to BMH) and U.S. Department of Energy, Office of Science, Basic Energy Sciences (BES) (DESC0010687 and DE-SC0010834; LCS and DRD), and the Division of Chemical Sciences, Geosciences, and Bio-Sciences (S.R.).
Footnotes
Supporting Information Available: Kinetics data; EPR decomposition protocol; discussion of energy landscapes; details of DFT calculations; 5 figures in all.
References
- 1.Hoffman BM, Lukoyanov D, Yang ZY, Dean DR, Seefeldt LC. Mechanism of Nitrogen Fixation by Nitrogenase: The Next Stage. Chem Rev. 2014;114:4041–4062. doi: 10.1021/cr400641x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoffman BM, Lukoyanov D, Dean DR, Seefeldt LC. Nitrogenase: A Draft Mechanism. Acc Chem Res. 2013;46:587–595. doi: 10.1021/ar300267m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lukoyanov D, Yang ZY, Khadka N, Dean DR, Seefeldt LC, Hoffman BM. Identification of a Key Catalytic Intermediate Demonstrates That Nitrogenase Is Activated by the Reversible Exchange of N2 for H2. J Am Chem Soc. 2015;137:3610–3615. doi: 10.1021/jacs.5b00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lukoyanov D, Khadka N, Yang ZY, Dean DR, Seefeldt LC, Hoffman BM. Reductive Elimination of H2 activates Nitrogenase to Reduce the N≡N Triple Bond: Characterization of the E4(4H) Janus Intermediate in Wild-Type Enzyme. J Am Chem Soc. 2016;138:10674–10683. doi: 10.1021/jacs.6b06362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.A knowledgeable reviewer noted that a more common notation uses capitals in the abbreviations (RE, OA), but we feel that it is beneficial to retain consistency with our earlier reports.
- 6.Igarashi RY, Laryukhin M, Dos Santos PC, Lee HI, Dean DR, Seefeldt LC, Hoffman BM. Trapping H- Bound to the Nitrogenase FeMo-Cofactor Active Site During H2 Evolution: Characterization by Endor Spectroscopy. J Am Chem Soc. 2005;127:6231–6241. doi: 10.1021/ja043596p. [DOI] [PubMed] [Google Scholar]
- 7.Lukoyanov D, Yang Z-Y, Dean DR, Seefeldt LC, Hoffman BM. Is Mo Involved in Hydride Binding by the Four-Electron Reduced (E4) Intermediate of the Nitrogenase MoFe Protein? J Am Chem Soc. 2010;132:2526–2527. doi: 10.1021/ja910613m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lukoyanov D, Barney BM, Dean DR, Seefeldt LC, Hoffman BM. Connecting Nitrogenase Intermediates with the Kinetic Scheme for N2 Reduction by a Relaxation Protocol and Identification of the N2 Binding State. PNAS. 2007;104:1451–1455. doi: 10.1073/pnas.0610975104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burgess BK, Lowe DJ. Mechanism of Molybdenum Nitrogenase. Chem Rev. 1996;96:2983–3012. doi: 10.1021/cr950055x. [DOI] [PubMed] [Google Scholar]
- 10.Thorneley RNF, Lowe DJ. Kinetics and Mechanism of the Nitrogenase Enzyme System. Metal Ions in Biology. 1985;7:221–284. [Google Scholar]
- 11.Wilson PE, Nyborg AC, Watt GD. Duplication and Extension of the Thorneley and Lowe Kinetic Model for Klebsiella Pneumoniae Nitrogenase Catalysis Using a Mathematica Software Platform. Biophysical Chemistry. 2001;91:281–304. doi: 10.1016/s0301-4622(01)00182-x. [DOI] [PubMed] [Google Scholar]
- 12.Lukoyanov D, Khadka N, Yang ZY, Dean DR, Seefeldt LC, Hoffman BM. Reversible Photoinduced Reductive Elimination of H2 from the Nitrogenase Dihydride State, the E4(4H) Janus Intermediate. J Am Chem Soc. 2016;138:1320–1327. doi: 10.1021/jacs.5b11650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perutz RN, Procacci B. Photochemistry of Transition Metal Hydrides. Chem Rev. 2016 doi: 10.1021/acs.chemrev.6b00204. [DOI] [PubMed] [Google Scholar]
- 14.Perutz RN. Metal Dihydride Complexes: Photochemical Mechanisms for Reductive Elimination. Pure and Applied Chemistry. 1998;70:2211–2220. [Google Scholar]
- 15.Colombo M, George MW, Moore JN, Pattison DI, Perutz RN, Virrels IG, Ye TQ. Ultrafast Reductive Elimination of Hydrogen from a Metal Carbonyl Dihydride Complex; a Study by Time-Resolved IR and Visible Spectroscopy. Journal of the Chemical Society-Dalton Transactions. 1997:2857–2859. [Google Scholar]
- 16.Whittlesey MK, Mawby RJ, Osman R, Perutz RN, Field LD, Wilkinson MP, George MW. Transient and Matrix Photochemistry of Fe(Dmpe)2H2 (Dmpe = Me2PCH2CH2PMe2) - Dynamics of C-H and H-H Activation. J Am Chem Soc. 1993;115:8627–8637. [Google Scholar]
- 17.Ballmann J, Munha RF, Fryzuk MD. The Hydride Route to the Preparation of Dinitrogen Complexes. Chem Commun. 2010;46:1013–1025. doi: 10.1039/b922853e. [DOI] [PubMed] [Google Scholar]
- 18.Ozin GA, Mccaffrey JG. The Photoreversible Oxidative-Addition, Reductive-Elimination Reactions Fe+H2-Reversible-FeH2 in Low-Temperature Matrices. J Phys Chem. 1984;88:645–648. [Google Scholar]
- 19.Dugan TR, Holland PL. New Routes to Low-Coordinate Iron Hydride Complexes: The Binuclear Oxidative Addition of H2. J Organomet Chem. 2009;694:2825–2830. doi: 10.1016/j.jorganchem.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu Y, Smith JM, Flaschenriem CJ, Holland PL. Binding Affinity of Alkynes and Alkenes to Low-Coordinate Iron. Inorg Chem. 2006;45:5742–5751. doi: 10.1021/ic052136+. [DOI] [PubMed] [Google Scholar]
- 21.Smith JM, Sadique AR, Cundari TR, Rodgers KR, Lukat-Rodgers G, Lachicotte RJ, Flaschenriem CJ, Vela J, Holland PL. Studies of Low-Coordinate Iron Dinitrogen Complexes. J Am Chem Soc. 2006;128:756–769. doi: 10.1021/ja052707x. [DOI] [PubMed] [Google Scholar]
- 22.Yu Y, Sadique AR, Smith JM, Dugan TR, Cowley RE, Brennessel WW, Flaschenriem CJ, Bill E, Cundari TR, Holland PL. The Reactivity Patterns of Low-Coordinate Iron-Hydride Complexes. J Am Chem Soc. 2008;130:6624–6638. doi: 10.1021/ja710669w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mas-Balleste R, Lledos A. From Dihydrogen to Dihydride: Homolytic Splitting of H2. In: Alvarez S, editor. Comprehensive Inorganic Chemistry Ii. Vol. 9. Elsevier Ltd; 2013. pp. 736–740. [Google Scholar]
- 24.Christiansen J, Goodwin PJ, Lanzilotta WN, Seefeldt LC, Dean DR. Catalytic and Biophysical Properties of a Nitrogenase Apo-MoFe Protein Produced by a NifB-Deletion Mutant of Azotobacter Vinelandii. Biochemistry. 1998;37:12611–12623. doi: 10.1021/bi981165b. [DOI] [PubMed] [Google Scholar]
- 25.https://www.thorlabs.com/thorproduct.cfm?partnumber=PL450B, see MFG Spec pdf file, page 4.
- 26.Michl J, Bonacic-Koutecky V. Electronic Aspects of Organic Photochemistry. John Wiley and Sons; 1990. [Google Scholar]
- 27.Devaquet A. Avoided Crossings in Photochemistry. Pure Appl Chem. 1975;41:455–473. [Google Scholar]
- 28.Schwartz FP, Albrecht AC. One and Two-Photon Photochemistry in Rigid Solutions of Durene in 3-Methylpentane at 77 Degrees K. Chem Phys Lett. 1971;9:163–165. [Google Scholar]
- 29.Simpson FB, Burris RH. Nitrogen Pressure of 50 Atmospheres Does Not Prevent Evolution of Hydrogen by Nitrogenase. Science. 1984;224:1095–1097. doi: 10.1126/science.6585956. [DOI] [PubMed] [Google Scholar]
- 30.Kubas GJ. Fundamentals of H2 Binding and Reactivity on Transition Metals Underlying Hydrogenase Function and H2 Production and Storage. Chem Rev. 2007;107:4152–4205. doi: 10.1021/cr050197j. [DOI] [PubMed] [Google Scholar]
- 31.Kubas GJ. Activation of Dihydrogen and Coordination of Molecular H2 on Transition Metals. J Organomet Chem. 2014;751:33–49. [Google Scholar]
- 32.Crabtree RH. Dihydrogen Complexation. Chem Rev. 2016;116:8750–8769. doi: 10.1021/acs.chemrev.6b00037. [DOI] [PubMed] [Google Scholar]
- 33.Crabtree RH. Dihydrogen Binding in Hydrogenase and Nitrogenase. Inorg Chim A-Bioinor. 1986;125:L7–L8. [Google Scholar]
- 34.Sweany RL. Photolysis of Hexacarbonylchromium in Hydrogen-Containing Matrices - Evidence of Simple Adducts of Molecular-Hydrogen. J Am Chem Soc. 1985;107:2374–2379. [Google Scholar]
- 35.Sweany RL. Photolysis of (Cyclopentadienyl) Tricarbonylhydridometal and (Pentamethylcyclopentadienyl) Tricarbonylhydridometal Complexes of Tungsten and Molybdenum in Dihydrogen-Containing Matrices - Evidence for Adducts of Molecular-Hydrogen. J Am Chem Soc. 1986;108:6986–6991. [Google Scholar]
- 36.Loges B, Boddien A, Junge H, Noyes JR, Baumann W, Beller M. Hydrogen Generation: Catalytic Acceleration and Control by Light. Chem Commun. 2009:4185–4187. doi: 10.1039/b908121f. [DOI] [PubMed] [Google Scholar]
- 37.Doherty MD, Grills DC, Huang KW, Muckerman JT, Polyansky DE, van Eldik R, Fujita E. Kinetics and Thermodynamics of Small Molecule Binding to Pincer-Pcp Rhodium(I) Complexes. Inorg Chem. 2013;52:4160–4172. doi: 10.1021/ic300672g. [DOI] [PubMed] [Google Scholar]
- 38.Phillips JC. Stretched Exponential Relaxation in Molecular and Electronic Glasses. Reports on Progress in Physics. 1996;59:1133–1207. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.