

Abstract
Epithelial membrane protein-2 (EMP2) is a tetraspan protein predicted to regulate placental development. Highly expressed in secretory endometrium and trophectoderm cells, previous studies suggest that it may regulate implantation by orchestrating the surface expression of integrins and other membrane proteins. In order to test the role of EMP2 in pregnancy, mice lacking EMP2 (Emp2−/−) were generated. Emp2−/− females are fertile but have reduced litter sizes when carrying Emp2−/− but not Emp2+/− fetuses. Placentas of Emp2−/− fetuses exhibit dysregulation in pathways related to neoangiogenesis, coagulation, and oxidative stress, and have increased fibrin deposition and altered vasculature. Given that these findings often occur due to placental insufficiency resulting in an oxygen-poor environment, the expression of hypoxia-inducible factor-1 alpha (HIF-1α) was examined. Placentas from Emp2−/− fetuses had increased total HIF-1α expression in large part through an increase in uterine NK (uNK) cells, demonstrating a unique interplay between uNK cells and trophoblasts modulated through EMP2. To determine if these results translated to human pregnancy, placentas from normal, term deliveries or those complicated by placental insufficiency resulting in intrauterine growth restriction (IUGR) were stained for EMP2. EMP2 was significantly reduced in both villous and extravillous trophoblast populations in IUGR placentas. Experiments in vitro using human trophoblast cells lines indicate that EMP2 modulates angiogenesis by altering HIF-1α expression. Our results reveal a novel role for EMP2 in regulating trophoblast function and vascular development in mice and humans and suggest it may be a new biomarker for placental insufficiency.
Keywords: EMP2, placenta, homologous recombination, angiogenesis, IUGR
Introduction
Placental development is a complex process that involves successful interplay between the developing fetus and placenta, the uterus, and the maternal immune system. The reciprocal dialogue between maternal and fetal cells regulates cell invasion, tissue remodeling, as well as modulation of immune responses required to prevent rejection. Given the coordinated nature of the process, dysregulated cross-talk between maternal and fetal cells has been in implicated in a number of pregnancy disorders resulting in placental insufficiency such as recurrent miscarriages, intrauterine growth restriction (IUGR), and preeclampsia [1,2]. As pregnancy related disorders affect up to a third of all human pregnancies [3], a better understanding of the proteins involved and their ability to regulate maternal-fetal cross-talk are needed. However, significant challenges exist as while placental-related disorders in pregnancy occur relatively frequently in humans, their incidence is low in other mammalian species making models for disease scarce [4].
While a number of pathological processes can be altered, there is mounting evidence that oxidative stress plays a pivotal role in the course of placental-related diseases. For example, an imbalance in the oxidant/antioxidant activity at the utero-placental interface produces necrosis in the trophoblastic epithelium, resulting in miscarriage [3]. In pre-eclampsia, inadequate trophoblastic invasion prevents complete transformation of uterine arteries resulting in inadequate perfusion [3]. This latter process involves the interplay with maternal immune responses as uterine natural killer (uNK) cells help to regulate uterine vascular adaptations [5–7]. However, little is known regarding how the cross-talk between trophoblastic invasion and uterine natural killer recruitment occurs.
Epithelial membrane protein 2 (EMP2) is a four transmembrane protein previously identified on both the uterine epithelial cell surface as well as on trophectoderm cells of the implanting embryo [8]. On the uterine cell surface, EMP2 has been shown to regulate the expression of select integrins such αv and β3, and physiologic knock-down of EMP2 impairs successful implantation [9,10]. While the role of EMP2 in the trophectoderm is unknown, given the similarities between trophoblasts and cancer cells we predict that its role in the placenta may be similar to its role in cancer where it promotes a pro-invasive phenotype as well as the release of pro-angiogenic factors [11,12].
In this study, we show a novel role of EMP2 in placental development. Emp2−/− females carrying Emp2−/− fetuses show a significant reduction in litter size, and loss of EMP2 contributed to placental insufficiency as evidenced by an increase in oxidative stress responses. Within the human, using a combination of human trophoblast-like cell lines and patient derived samples, we demonstrate that EMP2 regulates neoangiogenesis and invasion, and loss of EMP2 within trophoblasts of the human placenta correlated with IUGR. These results suggest that EMP2 functions to regulate placentation in both humans and mice and that dysregulation of EMP2 may contribute to the development of IUGR.
Materials and Methods
Human placental collection and processing
Institutional review board approval was obtained from UCLA for the collection of placental tissue and was in accordance with the Helsinki Declaration of 1975. Collection and processing of the placentas used for this study have been described previously [13]. Placental tissue was obtained from women with normal pregnancies (n = 9) or with late-onset IUGR (n = 14) as defined by newborn weight ≤ 10th percentile for gestational age and a trajectory of fetal growth deceleration in utero, diagnosed by ultrasound in the third trimester of pregnancy.
Generation of EMP2-null mice
All animal procedures complied with NIH/NIEHS animal care guidelines. C57BL/6 mice carrying a conditional knockout allele for the Emp2 gene (floxed exon 3) were generated commercially (Taconic) by standard homologous recombination methods using C57BL/6 ES cells. Targeted clones were electroporated with a Flp recombinase-expressing plasmid to remove the neo cassette, and neo-deleted clones were injected into blastocysts derived from albino C57BL/6 mice (B6(Cg)-Tyrc-2J/J). Male chimeras were bred with C57BL/6N Tac females to generate heterozygous offspring. Germ line transmission was detected using coat color and confirmed by tail DNA genotyping. The heterozygous mice were referred to as Emp2+/f, with “f” indicating the targeted “floxed” allele. The primer sequences used for the detection of the floxed allele and to genotype animals is provided in Supplementary Table S1.
All other protocols used are described in the supplementary Materials and Methods.
Results
EMP2 expression in the mouse placenta
Previous data demonstrated the expression of EMP2 within the murine trophectoderm and decidua at E5.5-E6.5 [8]. To fully characterize its expression in the mouse at various time points during placentation, EMP2 expression was determined using placentas from E6.5-E16.5. EMP2 was detected in the maternal decidua and embryonic trophoblast-derived tissues throughout gestation in wild type mouse placentas (Figure 1). Immediately after implantation at E6.5, as previously reported [8], EMP2 was expressed in the embryo proper, trophoblastic stem cells in the ectoplacental cone, and decidual cells. In E9.5 placentas, EMP2 was strongly expressed in trophoblast giant cells and maternal decidua but was also detectable in the endometrium. No EMP2 expression was detected within the allantois. During murine placental development, four distinct trophoblast populations emerge: labyrinthine trophoblasts, glycogen trophoblasts, spongiotrophoblasts, and trophoblast giant cells [14]. By E12.5, EMP2 was most highly expressed in giant cells but was also found to be weakly expressed within labyrinthine trophoblasts and spongiotrophoblasts found in the junctional zone. At E12.5, pre-glycogen cells appear[15], and EMP2 appears to reside in a perinuclear/cytoplasmic pattern within these cells. As placentas neared term, EMP2 expression appeared to be greatly reduced in all trophoblast populations.
Figure 1.
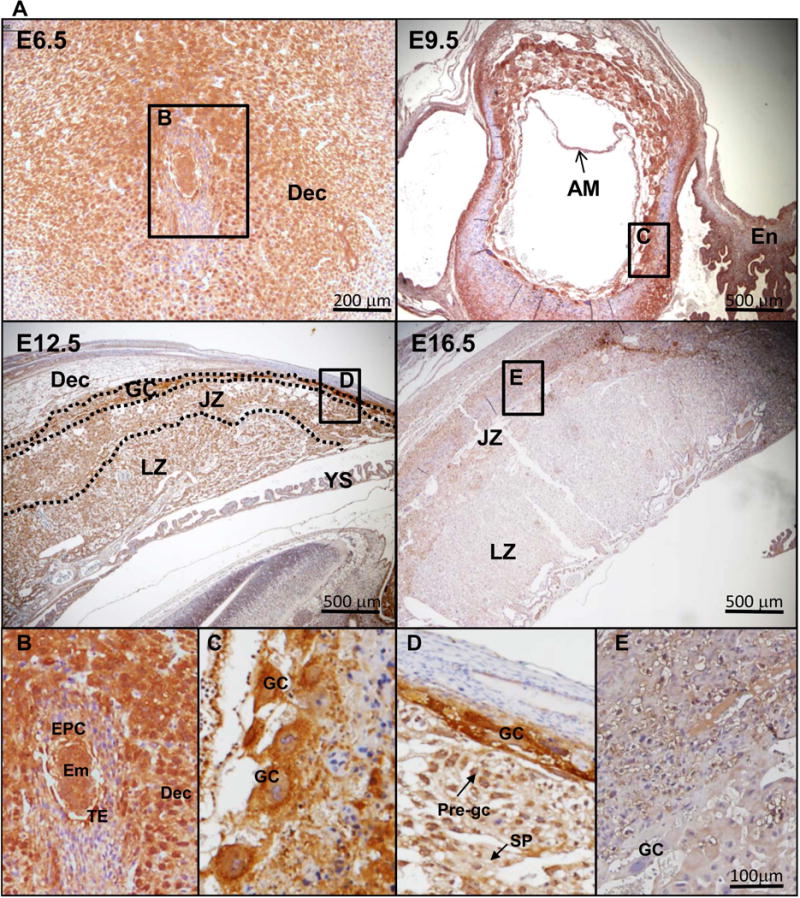
EMP2 is expressed in trophoblast layers throughout gestation. (A) Uteri or placentas were collected from wild type (Emp2+/+) mice following mating to wild type (Emp2+/+) males at E6.5, E9.5, E12.5, and E16.5 and stained for EMP2 expression. Left, Magnification=40×. Insets are provided below for E6.5 (B), E9.5 (C), E12.5 (D), and E16.5 (E). Magnification, 100×. Em, embryo; En, endometrium; AM, amnion; Ch, chorion; Dec, decidua; EPC, ectoplacental cone; GC, giant cells; JZ, Junctional zone; LZ, labyrinth zone; Pre-gc, glycogen cells; TE, trophectoderm; SP, spongiotrophoblast.
Generation and fertility phenotype of EMP2-deficient mice
To examine the function of EMP2 in implantation and placental development, a conditional knockout mouse line was generated by homologous recombination using C57BL/6 embryonic stem (ES) cells. Exon 3 of Emp2 was chosen for deletion because it encodes the majority of the first transmembrane domain and its deletion results in a frame-shift mutation in the subsequent exons. The targeting construct contained loxP sites bracketing exon 3 and Flp recombinase sites bracketing the neomycin cassette (Supplementary Figure S1A). Correct homologous recombination in the ES cells was demonstrated by southern blotting (Figure 2A). The neomycin cassette was deleted in vitro and chimeric founders were generated. Germ line transmission of the conditional knockout allele from chimeric founders was documented (Figure 2B). Deletion of Emp2 exon 3 in oocytes was accomplished using the oocyte-specific ZP3-cre transgene, which resulted in Emp2f/− offspring that were crossed to generate Emp2−/− mice. Because EMP2 is highly expressed in the lung, this tissue was used to examine EMP2 protein production. Immunoblots of lung extracts demonstrated that EMP2 was reduced by about 50% in Emp2f/− mice and was not detected at all in Emp2−/− mice (Figure 2C). Similarly, EMP2 was not detected in uterine tissue from Emp2−/− mice (Supplementary Figure S1B).
Figure 2.
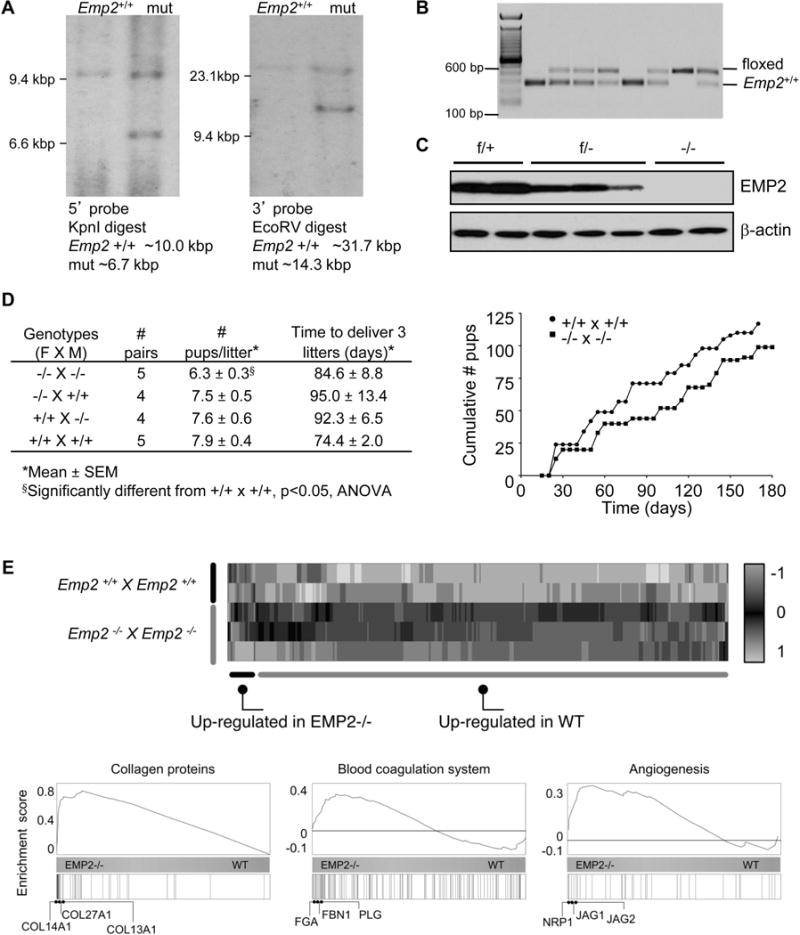
Matings of Emp2−/− × Emp2−/− mice show a reduction in litter size. (A) Recombination strategy used to target the Emp2 gene confirmed by Southern blot. (B) Documentation of germ line transmission. (C) EMP2 protein expression in lung extracts in wild-type, heterozygous, and knock-out mice. (D) Left, Summary of time to deliver 3 litters and size of the litters. Right, the number of pups from Emp2−/− × Emp2−/− and Emp2+/+ × Emp2+/+ mice were enumerated over 180 days. (E) Transcriptomic analysis of implantation sites from Emp2−/− × Emp2−/− and Emp2+/+ × Emp2+/+ mouse crosses is depicted using a heatmap and hierarchical clustering of RNA-Seq expression estimates. Shown are the Z-scores (colormap) for differentially expressed genes across the samples analyzed by RNA-Seq. Bottom, Gene Set Enrichment plots for genes annotated as “Collagens” (left), “Angiogenesis” (middle) and “Coagulation” (right). The horizontal bar in graded color from blue (left) to yellow (right) represents all mouse genes ranked from higher expression in Emp2−/− to higher expression in wild type mice. Vertical black lines represent the ranks of respective genes in each group. The curve in blue corresponds to the calculation of the enrichment score (ES), which in these cases demonstrate a high tendency of those genes to be highly expressed in Emp2−/− crosses as compared to Emp2+/+ crosses.
Crosses of Emp2+/− mice had an average litter size of 7.3 ± 0.51 s.e.m. (N=19 litters) and resulted in weaned offspring whose genotypes not different from an expected Mendelian ratio (30% wild type:51% heterozygous:19% knockout; p=0.52, Chi-square analysis). Based upon our previous findings suggesting that physiologic knockdown of EMP2 in the mouse uterus was associated with reduced implantation efficiency [8], we hypothesized that Emp2−/− females would have fertility defects. Because EMP2 is expressed in the endometrial epithelium, decidual tissue, and the trophoblast layer of the embryo, the embryo genotype was an important factor to consider in the experimental design used to test fertility. Breeding pairs were set up such that all embryos in each litter had the same genotype, while the maternal and paternal genotypes were homozygous wild type or knockout, and the average litter size was determined for 3 litters for each genotype combination (Figure 2D). There was no reduction in fecundity of Emp2−/− females when compared to Emp2+/+ females when the embryos carried were heterozygous (Emp2+/−). However, there was a significant reduction in litter size when the embryos carried were EMP2 deficient (Emp2−/−), suggesting that in the context of genetic knock-out of EMP2, fetal EMP2 has an important role in regulating placentation. It was not feasible to confirm this finding in heterozygous crosses (Emp2+/− × Emp2+/−) because if we assumed that a similar number of knockout pups failed to implant as was observed in litters from Emp2−/− females (1.2 out of 7.5), and that knockout pups would comprise 25% of each litter findings, it would take extremely large litter numbers to reach either positive or negative significance. Furthermore, it was possible that knockout fetal placentas could secrete factors that could impact the function of heterozygous or wild type placentas, or vice versa.
To test whether an embryo expressing EMP2 could somehow cause expression of EMP2 in the EMP2-deficient dams, Emp2−/− females were bred with wildtype (Emp2+/+) males and the implantation sites were examined. At E9.5, EMP2 was detected within the placental giant cells, but not in the maternal endometrium (Supplementary Figure S2A). Together with the breeding data, these findings indicate that EMP2 is not required in maternal tissues for successful pregnancy.
Additional breeding studies were performed to compare outcomes in knockout pairs relative to wild type pairs. Three breeding pairs in each group were mated continuously for 6 months. While there was no difference in pup weight or length of gestation, knockout pairs were slower to achieve pregnancy over time (Figure 2D). Combined with the smaller litter size, this resulted in a lower cumulative number of pups during the breeding trial.
Transcriptome profiling of EMP2-deficient placentas
In order to understand the physiological cellular responses that may be associated with altered EMP2 levels, gene expression analysis was performed. RNA from individual E16.5 placentas were collected and subjected to RNA sequencing. For this and all subsequent experiments, pregnancies were generated from either homozygous knockout (Emp2−/− × Emp2−/−) or wild type (Emp2+/+ × Emp2+/+) breeding pairs. Overall, 348 genes showed differential gene expression in Emp2−/− as compared to wild type placentas based on a |log2(fold)|>0.8, False Discovery Rate<5% (Figure 2E). Whole-genome expression estimates and the list of differentially expressed genes are provided in Supplementary Table S2. To obtain further insight into the mechanisms of EMP2 within the placenta, gene set enrichment analysis (GSEA) was performed using data from a public repository, NCBI GEO (Figure 2E). Emp2−/− placentas showed significant alterations in genes implicated in placental insufficiency and placental deterioration. Indicative of the ischemic-reperfusion episodes that are predicted to occur during oxidative stress in the placenta [3], several gene sets were significantly enriched in the knockout placentas related to collagens, angiogenesis, and coagulation. Among these pathways, extracellular matrix Col13a1 and Col27a1 transcripts as well as factors related to coagulation such as the fibrinogen alpha chain and plasminogen were upregulated in the Emp2−/− placentas. In contrast, several transcripts related to oxidative phosphorylation were downregulated in the Emp2−/− placentas including components of the NADH oxidase and cytochrome c complex as well as ATP synthase. A summary of relevant pathways provided by GSEA is provided in Supplementary Table S3. The complete list of genes analyzed with regard to coagulation is provided in Supplementary Table S4.
Histological alterations in EMP2-deficient placentas
Transcriptional analysis suggested that the EMP2 knockout placentas may display histological changes, so we next examined the effects of EMP2-deficiency on the organization of the mouse chorioallantoic placenta. Although placentas from wild type or Emp2−/− crosses producing corresponding wild type and Emp2−/− fetuses showed similar overall architecture (Supplementary Figure S2B), careful examination of the extracellular matrix composition showed striking differences. Consistent with the transcriptional differences, collagen deposition appeared to be upregulated in Emp2−/− placentas (Figure 3A). Masson’s Trichrome staining revealed characteristic collagen around the yolk sac in both groups, but only the Emp2−/− placentas displayed collagen throughout the labyrinth at E16.5. These changes in the extracellular matrix of the placenta are intriguing given that dysregulated collagen has been associated with specific disorders of placental insufficiency, such as IUGR and preeclampsia [16–19].
Figure 3.
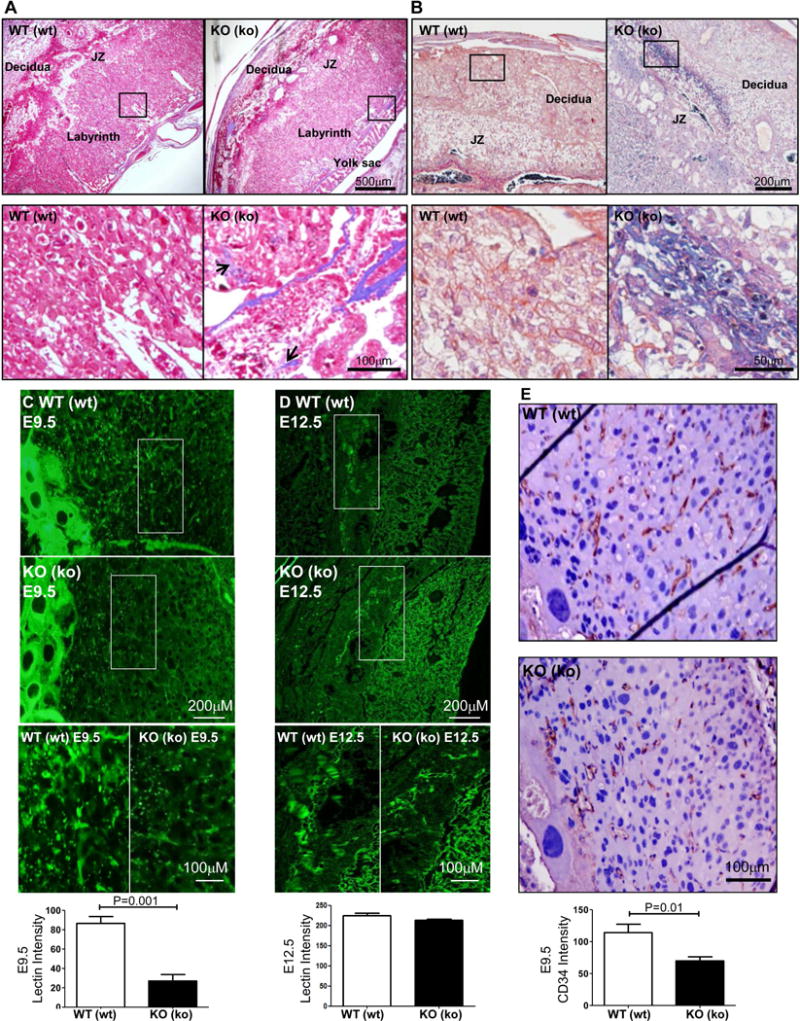
Placentas from Emp2−/− × Emp2−/− crosses show increased collagen and fibrin deposition and a reduction in placental blood vessels. (A) E16.5 placentas were stained with Masson’s Trichrome. Magnification=40×. Boxed areas are enlarged below, with black arrows used to demarcate collagen (blue) expression throughout the labyrinth. Magnification=100×. (B) At E16.5, placentas were stained using a PTAH stain to differentiate fibrin staining. Representative images from wild type mothers and embryos WT(wt) and Emp2−/− mothers and embryos KO(ko) placentas are shown. N=3 for each group with at least 2 placentas examined per mouse. Magnification=100×. Boxed areas are enlarged below, with the black arrows highlighting fibrin deposition (blue). Magnification=200×. (C, D) Placentas from E9.5 (C) and E12 (D) were stained using L. esculentum lectin and DAPI. Magnification=100×. Boxed areas are enlarged at the bottom, Magnification=400×. Bottom, Lectin staining area was quantitated using Adobe Photoshop. E. Placentas from E9.5 were stained for CD34, and representative images are shown. CD34 positive vessels were quantitated within the uterine decidua. The asterisk indicates significant differences between wildtype and KO (ko) animals (n = 5). JZ, junctional zone.
Coupled with the change in collagen, GSEA analysis suggested that Emp2−/− placentas increased expression of factors involved with coagulation. In diseases associated with placental insufficiency such as preeclampsia, it has been postulated that systemic endothelial damage causes the coagulation cascade to become activated, resulting in excess thromboses in the placenta and renal system [20]. In order to identify microscopic sites of epithelial injury in placental villi, fibrin deposition in E16.5 placentas was measured [21–23]. PTAH staining showed a striking increase in fibrin deposition throughout the decidua basalis and junctional zones in EMP2-deficient placentas as compared to controls (Figure 3B).
In humans, abundant fibrin deposition within the placenta is characteristic of the injuries often occurring due to dysregulated villous vasculogenesis [24]. To next determine if vasculogenesis was altered between the groups, placentas were stained using a fluorescein labeled Lycopersicon esculentum lectin. The intensity of lectin staining was reduced by ~3-fold within the trophoblast-decidua interface in the E9.5 EMP2-deficient placentas (Fig. 3C). At E12.5, although no quantitative differences in vascular staining intensity were measured, differences in vessel organization were prominent (Figure 3D). To confirm the changes in vasculature observed at E9.5, placentas were stained for CD34 to label vascular endothelial cells. CD34 staining was significantly reduced in EMP2-deficient compared to wild type placentas (Figure 3E).
HIF-1α expression and uNK cells are increased in Emp2−/− mice
The imbalance of vasculogenesis in the EMP2-deficient placentas and the enrichment in genes related to the hypoxic response determined by the GSEA analysis suggested that there may be an imbalance in placental oxygen tension. Within the uteroplacental environment, hypoxia-inducible factors (HIFs) mediate transcriptional responses to localized hypoxia by altering cellular metabolism and stimulating angiogenesis [25,26], and in particular, HIF-1α levels have been linked with placental disease and impaired trophoblast differentiation [27–29]. Within both individual and total implantation sites per dam, Hif1a expression was significantly upregulated in Emp2−/− compared with Emp2+/+ (Fig. 4A). To confirm the upregulation of the Hif1a transcript, immunohistochemistry was performed to assess protein levels. Although there appeared to be a slight decrease in cytoplasmic HIF-1α in trophoblasts in Emp2−/− placentas, strikingly, there was a net increase in HIF-1α staining within decidual leukocytes (Figure 4B). The effect was pervasive throughout all the implantation sites in a given dam, with more HIF-1α positive leukocytes at E9.5 and E12.5 present in the Emp2−/− placentas. The difference in HIF-1α expression was largely abrogated by E16.5, with minimal expression detected in both groups.
Figure 4.
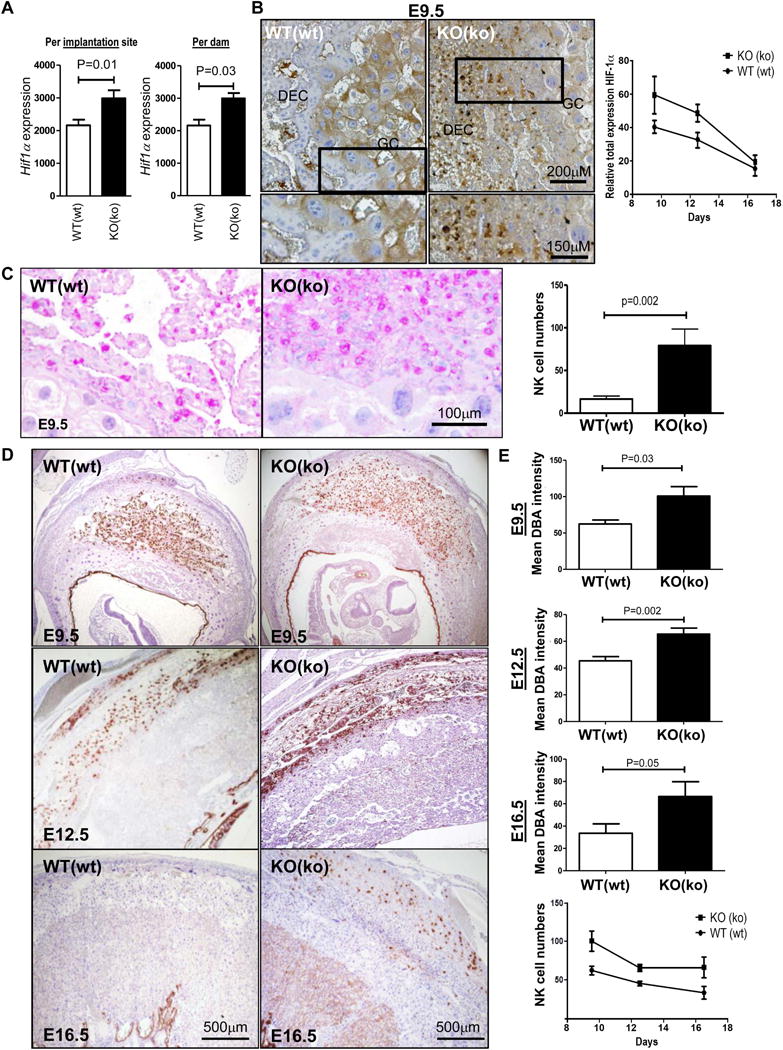
Placentas from Emp2−/− × Emp2−/− crosses show an increase in decidual leukocytes that have increased HIF-1α levels. (A) Hif-1α expression was validated by q-PCR using mRNA from whole placentas. Hif1α mRNA was increased in Emp2−/− placentas, designated KO(ko) for the mother and fetus, respectively, compared with wildtype placentas with an asterisk indicating significant differences between the two groups (n = 5; P= 0.01, for individual placentas and P=0.03 per dam using the Mann-Whitney sum rank test). A wildtype mother and placenta is denoted by WT(wt), respectively. (B) Total HIF-1α expression was determined in the chorioallantoic placenta with a representative image at E9.5 shown. Magnification=100× with insets provided of the boxed area. Far right, quantitation of total HIF-1α expression from 2 placentas from 3 different animals at E9.5, E12.5, and E16.5. (C) A Periodic acid-Schiff (PAS) stain was used to confirm the presence and staining pattern of leukocytes at E9.5. The mean intensity of PAS expression was determined from at least 2 placentas from 3 different animals using Adobe Photoshop, and results are quantitated to the right. Representative images are shown. (D) NK cells were visualized using Dolichos Biflorus Agglutinin (DBA) in E9.5, E12.5, and E16.5 placentas. Magnification=40×. E. Decidual NK cells were enumerated using Adobe Photoshop, and an increase in NK cell numbers was observed throughout gestation in the KO (ko) mice. Data represents results from at least 2 placentas from 3 independent animals. KO, knockout; DEC, decidua; GC, trophoblast giant cells.
To confirm the identification of leukocytes within the placenta, sections of E12.5 placentas were stained using a Periodic acid-Schiff stain to detect the distinct appearance of cell granules characteristic of uterine NK cells (uNK). An increase in granulated leukocytes was confirmed (Figure 4C; Supplementary Figure S3). Approximately 70% of the total leukocytes in the mid-gestational decidua are uNK cells, and these cells are thought to organize the maternal-fetal interface and regulate oxygen delivery within the trophoblast stem cell niche [30–32]. To investigate the numbers of uNK cells, a Dolichos biflorus agglutinin (DBA) lectin stain was performed on placentas from E9.5, E12.5, and E16.5 (Fig. 4D) [33]. uNK cells were significantly increased in the decidua basalis of EMP2-deficient placentas compared to controls throughout gestation (Fig. 4E), suggesting that uNK cells may be recruited to help regulate the hypoxic response.
EMP2 is expressed in human placentas and reduced in IUGR pregnancies
Our results thus far suggested that EMP2 may regulate immune cell recruitment and vascularization in the placenta. To determine if the above findings were relevant to human pregnancy, the distribution of EMP2 was initially evaluated in human placental tissue by immunohistochemistry. As in the mouse, all populations of human trophoblasts showed some expression of EMP2. In normal, term placentas, a strong EMP2 signal was present in both villous syncytiotrophobasts and cytotrophoblasts as well as on the membrane of interstitial trophoblasts (Figure 5A).
Figure 5.
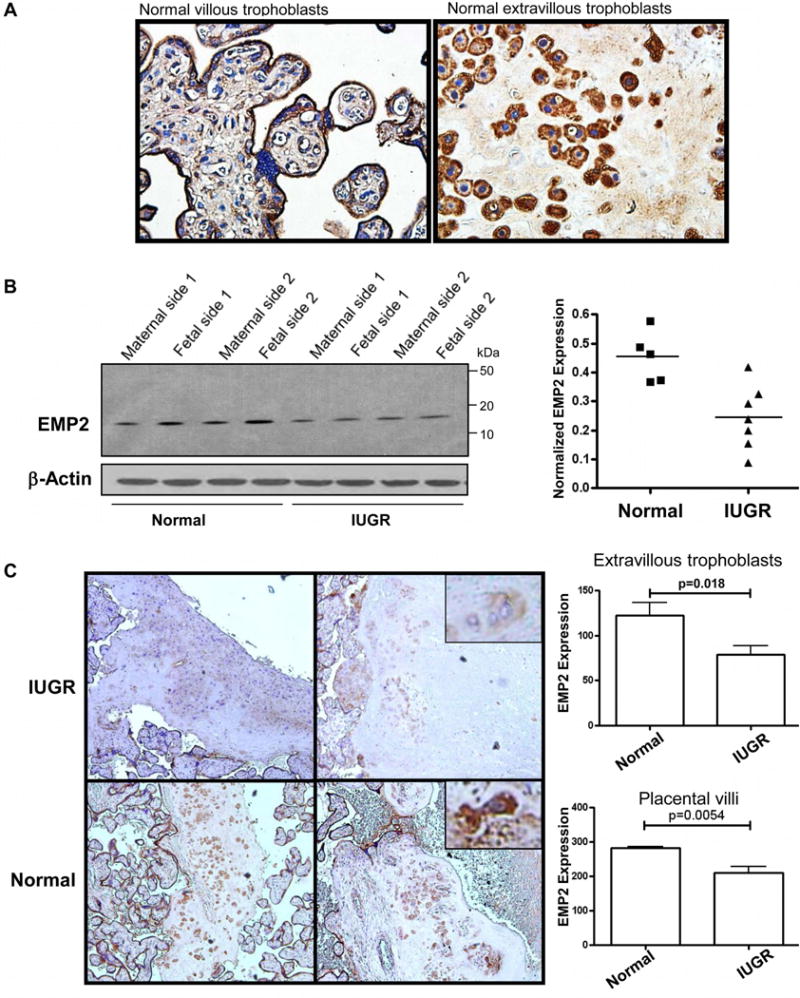
EMP2 expression in human placentas from normal and IUGR pregnancies. (A) Term, normal human placentas were stained for EMP2 expression. EMP2 expression is detected in both the placental villi as well as in extravillous trophoblasts. (B) Whole cell homogenates from normal or IUGR placentas were probed for EMP2 expression. Total EMP2 expression is reduced on both the fetal and maternal sides from IUGR placentas. Left, Blots from 2 representative patients per group are shown. Right, Combined EMP2 expression from the fetal and maternal sides relative to β-actin is shown. N=5 normal and 7 IUGR lysates. p=0.006, Student’s t-test. C. EMP2 expression from term IUGR and normal placentas. Graphs show relative expression of EMP2 in interstitial trophoblast cells (upper graph) and placental villi (lower graph). N=9 normal and 14 IUGR placentas.
Pre-eclampsia and late-onset IUGR are uteroplacental disorders resulting from inadequate trophoblast invasion and a failure to properly remodel the maternal blood supply resulting in overall placental insufficiency [34]. In placentas from pregnancies complicated by late-onset IUGR, EMP2 protein was reduced ~2-fold as indicated by immunoblot analysis (Figure 5B), with the alteration in expression present on both the fetal and maternal sides of the placenta. To confirm these results, semi-quantitative immunohistochemical analysis of placentas from normal and late onset IUGR was performed, and both placental villi and interstitial trophoblast cells showed lower EMP2 expression in placentas complicated by IUGR compared to controls (Figure 5C).
EMP2 promotes angiogenesis in human trophoblastic cells
Each population of trophoblast cells in the human has distinct functions. Extravillous trophoblasts mediate invasion into the maternal decidua while cytotrophoblasts fuse to form syncytiotrophoblasts that regulate passive diffusion between the mother and fetus as well as select hormonal function [35]. To investigate the functional role of EMP2, its expression was first determined in two cell lines of trophoblast origin, the choriocarcinoma cell lines JAR and BeWo. Both cell lines showed endogenous EMP2 protein expression, and were thus engineered to overexpress or reduce EMP2 levels (Figure 6A). To determine if EMP2 levels could alter trophoblast differentiation, BeWo cells were used as these cells have been shown to differentiate into syncytiotrophoblasts upon forskolin treatment [36]. No change in beta human chorionic gonadotropin (hCG) production was observed, suggesting the EMP2 is not necessary for differentiation (Supplementary Figure S3).
Figure 6.
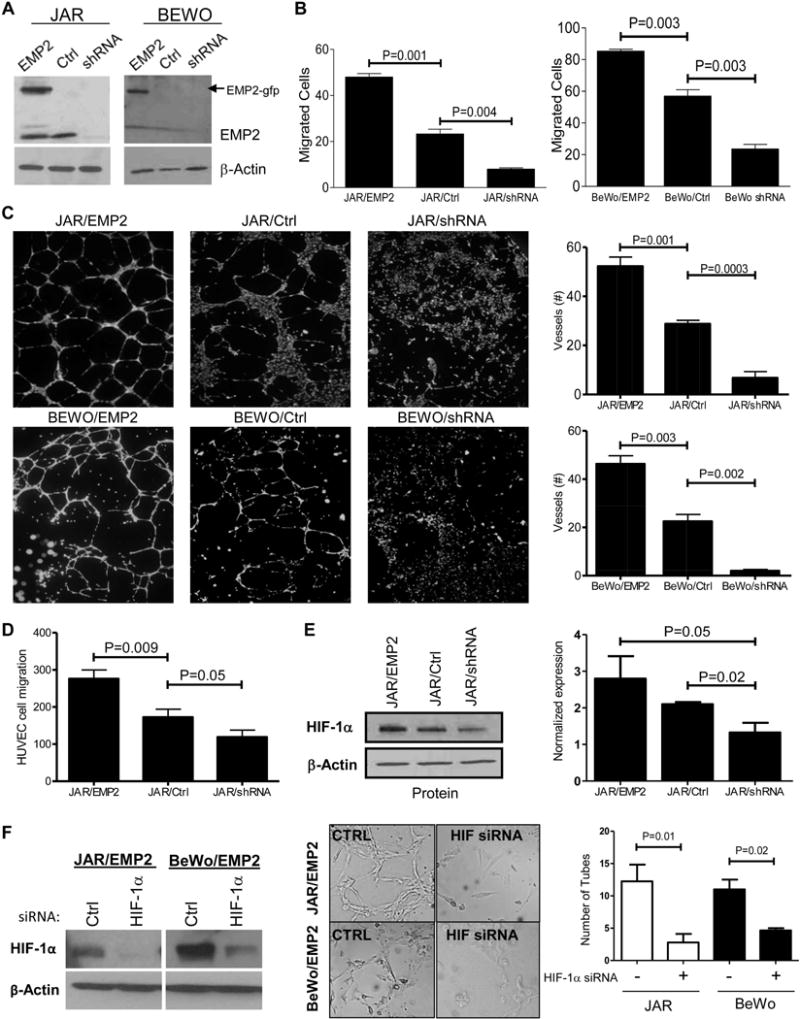
EMP2 promotes trophoblast invasion and regulates capillary tube formation. (A) JAR and BeWo cells were engineered to overexpress EMP2 (JAR/EMP2) or express a shRNA vector designed to inhibit its expression (JAR/shRNA). Additional vector control cells (JAR/Ctrl) are also included in the representative blots shown. (B) JAR and BeWo cells with altered EMP2 expression were tested for their ability to migrate through a Boyden chamber. Cells were then stained using crystal violet and manually counted using microscopy. The experiment was performed in triplicate with data expressed as the average number of cells ± SEM. (C) JAR or BeWo cells were tested for their ability to induce HUVEC capillary tube formation. Tube formation was quantitated by measuring the number of tubes formed after incubation with conditioned media from the above cells. The data in the graphs is the mean ± SEM from at least three fields of three independent experiments. (D) Chemotactic effects on the migration of HUVECs were measured using a standard Boyden chamber assay. HUVEC cells were stimulated to migrate in response to cultured media from JAR/EMP2, JAR/Ctrl or JAR/shRNA cells. Experiments were repeated three times with data representing the mean ± SEM. (E) Immunoblot of HIF-1α expression in JAR cells. Left, a representative immunoblot for HIF-1α and β-actin expression is shown. Right, quantitation of HIF-1α expression relative to β-actin from three independent experiments. (F) JAR/EMP2 or BeWo/EMP2 cells were transfected with scrambled or HIF-1α siRNA. A representative immunoblot showing knockdown in HIF-1α expression with the specific siRNA is shown on the left. Supernatants from the scrambled or HIF-1α siRNA transfected cells were tested for their ability to induce HUVEC capillary tube formation.
To next determine if EMP2 levels affect trophoblast migration, a Boyden chamber migration assay was performed. In both JAR and BeWo cells, upregulation of EMP2 correlated with increased cell migration while a downregulation in EMP2 inhibited it (Figure 6B). Previous studies have shown that trophoblast invasion regulates endothelial cell migration and vascularization[37,38]. To determine if trophoblasts with altered EMP2 levels modulate capillary vasculogenesis and neoangiogenesis, HUVEC tube formation assays were performed. Supernatants from JAR or BeWo cells with modified EMP2 protein levels dramatically increased capillary tube formation, whereas knockdown of EMP2 did the opposite (Figure 6C). While the chemotactic responses of HUVEC to supernatants from EMP2-upregulated JAR cells showed significantly enhanced directional migration compared to medium from control cells (Figure 6D), there was a non-significant reduction in directional migration when medium from JAR cells with reduced levels of EMP2 was tested. These data suggested a role for EMP2 in invasion and endothelial cell reorganization, similar to what was observed in the Emp2−/− placentas.
To test whether alterations in HIF-1α levels contributed to these responses, its expression was determined in these modified human trophoblast cell lines. Under normoxic conditions, JAR cells overexpressing EMP2 showed increased HIF-1α, whereas a reduction in EMP2 decreased HIF-1α levels by ~50% (Figure 6E). Next, to determine whether hypoxia could induce a reciprocal increase in EMP2 levels, JAR cells were placed under hypoxic (0.5%) or normoxic (20%) oxygen conditions for 24 h. No differences in total EMP2 protein were observed (Supplementary Figure S3B). To determine if HIF-1α controls neoangiogenesis in EMP2 upregulated cells, JAR/EMP2 and BeWo/EMP2 cells were treated with a HIF-1α or scrambled siRNA. Reduction in HIF-1α levels abrogated capillary tube formation in both cell lines (Figure 6F), further supporting the idea that EMP2 regulates trophoblast-mediated angiogenesis at least in part by altering HIF-1α levels.
Discussion
The results of the present study define an important regulatory relationship between angiogenesis and uNK regulation at the maternal-fetal interface via the tetraspan protein EMP2. Deletion of EMP2 in fetuses reduced litter sizes (and therefore, overall litter weight) by 16%, and placentas from these animals showed signs of oxidative stress and poor vascularization, phenocopying the reproductive outcomes and histological changes seen in human placental insufficiency. In an effort to translate these findings, we provide the first evidence of altered EMP2 expression in human placentas with IUGR. Significant reduction in EMP2 levels was observed in both villous and extravillous trophoblasts, and coupled with results from Emp2−/− animals, these findings suggest it has a protective role in pregnancy establishment.
The crosstalk between maternal decidual cells, extravillous trophoblasts and uNK cells sets into motion a number of events includes plugging and remodeling of maternal vessels and regulation of oxygen level [5–7]. In the human, trophoblasts differentiate along two pathways to give rise to either extravillous or villous trophoblasts that resemble giant cells or labyrinthine trophoblasts in the mouse, respectively. Within these cells, EMP2 appears to regulate a number of functions including invasion and vasculogenesis as well as playing a role in regulating the hypoxic response. Under normal conditions, the placenta adapts to changing oxygen levels to support normal placental function and fetal development by modulation of HIF-1α [39]. In humans, first trimester chorionic villi show detectable HIF-1α levels [40] while in mice, it has been observed in several trophoblast populations including giant cells and cytotrophoblasts [25,41,42]. This is similar to the results presented here as wild type human and murine trophoblasts express HIF-1α, and our results suggest that under normal, physiological conditions, this regulation occurs at least in part through EMP2. Based upon our findings in this genetic knock-out model, we cannot speculate as to whether hypoxia itself leads to altered localization of EMP2 protein in vivo, but our in-vitro studies suggest that hypoxia does not modulate total EMP2 levels (Supplementary Figure S1). Taken together with the hypovascularity seen in EMP2 knock-out, our findings indicate that EMP2 is upstream of HIF-1α and that lack of its expression leads to placental hypoxia because of a failure of appropriate placental angiogenesis. Curiously, the expression of HIF-1α in wildtype murine trophoblasts appears more cytoplasmic than nuclear. This may reflect a known dynamic whereby HIF-1α shuttles between the cytoplasm and nucleus or it may reflect an alteration in HIF-1α protein turnover [43–45]. While this study did not address the mechanism behind EMP2 regulation of HIF-1α, previous studies using normal and cancer cell lines suggest that EMP2 expression promotes FAK and Src activation [11,46]. Both of these pathways regulate HIF-1α[47], but additional studies will be needed to determine the downstream pathway governing EMP2 mediated HIF-1α expression within trophoblasts.
Moreover, our results show that the Emp2−/− placenta increased the recruitment and retention of uNK cells throughout gestation. uNK cells are distinct phenotypically from peripheral blood NK cells, exhibiting low cytotoxicity and high cytokine secreting activity [42,48]. Over the last decade, it has been proposed that uNK cells cooperate with trophoblast cells to guarantee correct arterial remodeling [49], and we thus predict that these cells may be providing a compensatory mechanism for inadequate trophoblast-mediated vascular remodeling due to loss of EMP2 in the knockout animals. In support of this hypothesis, Emp2−/− placentas had a significant increase in recruitment and subsequent expression of HIF-1α in uNK cells.
An intriguing aspect to this work is the suggestion that EMP2 may play a part in the development of placental insufficiency-related pregnancy disorders such as preeclampsia and IUGR. Importantly, it appears that in this genetic knock out of EMP2, implantation per se is not significantly affected, but that placental vascularization and maintenance is affected. Previous studies have shown that the progressive deterioration in placental function typically manifests in hypoxemia and oxidative stress as the placenta cannot provide sufficiently for the metabolic demands of the growing fetus [50,51]. While a number of signaling pathways likely contribute, it has been argued the most important ones for pregnancy are perturbations in the maternal blood supply to the placenta and excessive inflammation [3,52]. Consistent with this description, placentas from the Emp2−/− fetuses show reduced angiogenesis as documented by the differences in CD34 and lectin staining. Moreover, placental damage was evident from the gene set enrichment signature and confirmed by the increase in fibrin and collagen deposition near the maternal-fetal interface. The lack of growth restriction (e.g. birth weight; Supplementary Figure S1C) observed in the Emp2−/− offspring is not surprising given that, in contrast to humans, trophoblast invasion of the endometrium remains superficial [53].
The altered placental vascularization seen in pregnancy-related disorders such as growth restriction and preeclampsia are incompletely understood. Determining the mechanisms by which placental vascularization occurs will be important in identifying markers for high-risk pregnancies, for targeting therapies in these pregnancies, and also to better elucidate fetal effects in the short term as well as in later disease. Although we predict that altered EMP2 may be a link for the development of placental insufficiency, additional experiments will be necessary to determine its penetrance.
Supplementary Material
Figure S1. (A) Schematic of the Emp2 gene and targeting construct. (B) Uteri from Emp2−/− and Emp2+/+ were probed for EMP2 expression using western blot analysis. β-actin was used as a loading control
Figure S2. (A) Emp2−/− females were mated to EMP2+/+ males. Emp2+/− fetus and corresponding Emp2−/− uterus was collected at E9.5 and stained for EMP2 protein expression. (B) H&E stained E12.5 placentas from wildtype mother (Emp2+/+ fetus) and Emp2−/− (Emp2−/− fetus) animals. C. Weights of Emp2+/+ and Emp2−/− pups in grams ± SEM
Figure S3. Periodic acid-Schiff (PAS) stain was used to confirm the presence and staining pattern of leukocytes at E9.5. Objective magnification: 40×
Figure S4. (A) EMP2 is not required for cytotrophoblast differentiation into syncytiotrophoblasts by forskolin. (B) EMP2 expression in JAR cells is not altered by oxygen deprivation. Cells were placed in a hypoxic chamber (0.5% O2) or left in normoxia for 24 h. EMP2 levels were probed using western blot analysis.
Table S1. Primer sequences used for the detection of the floxed allele and to genotype animals
Table S2. Whole-genome expression estimates and the list of differentially expressed genes between placentas isolated from wildtype and EMP2 null mice
Table S3. Summary of relevant pathways altered in EMP2 null placentas provided by GSEA
Table S4. Differential expression of genes in wildtype versus EMP2 null placenta analyzed with regard to coagulation
Acknowledgments
The authors thank Lee Goodglick for his guidance throughout this project. We also thank Jonathan Braun for his valuable and constructive suggestions during the planning and development of this research.
Funding
This work was supported by the Intramural Research Program of the National Institutes of Health, National Institutes of Environmental Health Sciences, 1ZIAES102405, NIH/NCI R01CA163971 (M. Wadehra), and by American Heart Association Beginning Grant-in-Aid Western Affiliate States 15BGIA25710060 and NIH 5K12HD034610 (AC). SM is supported by T32 CA009056-39 (Crooks PI).
Footnotes
Conflict of interest: M.W. is a founder of Paganini Biopharma. No other authors have competing interests.
The data presented in this publication has been deposited into the NCBI Gene Expression Omnibus and is accessible through GEO Series accession number GSE81940.
Statement of author contributions
CJW and MW designed all experiments. WNJ, AC, DS, CA, CPH, NK, PP, EPB, and SM carried out experiments as well as analyzed data. CJ consented and acquired all human samples. PS scored all human tissue samples. DC analyzed the RNA sequencing data. All authors were involved in writing the paper and had final approval of the submitted and published versions.
References
* These references are cited in supplementary material only
- 1.Brosens IA. Morphological changes in the utero-placental bed in pregnancy hypertension. Clinics in obstetrics and gynaecology. 1977;4:573–593. [PubMed] [Google Scholar]
- 2.Naicker T, Khedun SM, Moodley J, et al. Quantitative analysis of trophoblast invasion in preeclampsia. Acta obstetricia et gynecologica Scandinavica. 2003;82:722–729. doi: 10.1034/j.1600-0412.2003.00220.x. [DOI] [PubMed] [Google Scholar]
- 3.Jauniaux E, Poston L, Burton GJ. Placental-related diseases of pregnancy: involvement of oxidative stress and implications in human evolution. Human Reproduction Update. 2006;12:747–755. doi: 10.1093/humupd/dml016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilmut I, Sales D, Ashworth C. Maternal and embryonic factors associated with prenatal loss in mammals. Journal of Reproduction and Fertility. 1986;76:851–864. doi: 10.1530/jrf.0.0760851. [DOI] [PubMed] [Google Scholar]
- 5.Pijnenborg R, Dixon G, Robertson WB, et al. Trophoblastic invasion of human decidua from 8 to 18 weeks of pregnancy. Placenta. 1980;1:3–19. doi: 10.1016/s0143-4004(80)80012-9. [DOI] [PubMed] [Google Scholar]
- 6.Loke YW, King A. Immunology of human implantation: an evolutionary perspective. Human reproduction. 1996;11:283–286. doi: 10.1093/humrep/11.2.283. [DOI] [PubMed] [Google Scholar]
- 7.Hanna J, Goldman-Wohl D, Hamani Y, et al. Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nature medicine. 2006;12:1065–1074. doi: 10.1038/nm1452. [DOI] [PubMed] [Google Scholar]
- 8.Wadehra M, Dayal M, Mainigi M, et al. Knockdown of the tetraspan protein epithelial membrane protein-2 inhibits implantation in the mouse. DevBiol. 2006;292:430–441. doi: 10.1016/j.ydbio.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 9.Wadehra M, Forbes A, Pushkarna N, et al. Epithelial membrane protein-2 regulates surface expression of alphavbeta3 integrin in the endometrium. DevBiol. 2005;287:336–345. doi: 10.1016/j.ydbio.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 10.Wadehra M, Dayal M, Mainigi M, et al. Knockdown of the tetraspan protein epithelial membrane protein-2 inhibits implantation in the mouse. Developmental biology. 2006;292:430–441. doi: 10.1016/j.ydbio.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 11.Fu M, Rao R, Sudhakar D, et al. Epithelial membrane protein-2 promotes endometrial tumor formation through activation of FAK and Src. PLoS One. 2011;6:e19945. doi: 10.1371/journal.pone.0019945. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Gordon LK, Kiyohara M, Fu M, et al. EMP2 regulates angiogenesis in endometrial cancer cells through induction of VEGF. Oncogene. 2013;32:5369–5376. doi: 10.1038/onc.2012.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Janzen C, Lei MYY, Cho J, et al. Placental glucose transporter 3 (GLUT3) is up-regulated in human pregnancies complicated by late-onset intrauterine growth restriction. Placenta. 2013;34:1072–1078. doi: 10.1016/j.placenta.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cross JC, Werb Z, Fisher SJ. Implantation and the placenta: key pieces of the development puzzle. Science. 1994;266:1508–1518. doi: 10.1126/science.7985020. [DOI] [PubMed] [Google Scholar]
- 15.Coan PM, Conroy N, Burton GJ, et al. Origin and characteristics of glycogen cells in the developing murine placenta. Developmental Dynamics. 2006;235:3280–3294. doi: 10.1002/dvdy.20981. [DOI] [PubMed] [Google Scholar]
- 16.Baumbusch MA, Buhimschi CS, Zhao G, et al. A study of placental collagen birefringence in normal and high risk pregnancies - Preterm delivery (PTD) and preeclampsia (PE) American Journal of Obstetrics & Gynecology. 2005;193:S69. [Google Scholar]
- 17.Li W, Mata KM, Mazzuca MQ, et al. Altered matrix metalloproteinase-2 and -9 expression/activity links placental ischemia and anti-angiogenic sFlt-1 to uteroplacental and vascular remodeling and collagen deposition in hypertensive pregnancy. Biochemical Pharmacology. 2014;89:370–385. doi: 10.1016/j.bcp.2014.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tahkola J, Räsänen J, Sund M, et al. Cardiac dysfunction in transgenic mouse fetuses overexpressing shortened type XIII collagen. Cell and Tissue Research. 2008;333:61–69. doi: 10.1007/s00441-008-0617-5. [DOI] [PubMed] [Google Scholar]
- 19.Gourgiotis D, Briana DD, Georgiadis A, et al. Perinatal collagen turnover markers in intrauterine growth restriction. The Journal of Maternal-Fetal & Neonatal Medicine. 2012;25:1719–1722. doi: 10.3109/14767058.2012.663019. [DOI] [PubMed] [Google Scholar]
- 20.Alladin AA, Harrison M. Preeclampsia: systemic endothelial damage leading to increased activation of the blood coagulation cascade. J Biotech Res. 2012;4:26–43. [Google Scholar]
- 21.Stalker A. Fibrin deposition in pregnancy. Journal of Clinical Pathology. 1976;3:70–76. [PMC free article] [PubMed] [Google Scholar]
- 22.Conrad KP, Benyo DF. Placental Cytokines and the Pathogenesis of Preeclampsia. American Journal of Reproductive Immunology. 1997;37:240–249. doi: 10.1111/j.1600-0897.1997.tb00222.x. [DOI] [PubMed] [Google Scholar]
- 23.Hernández-Núñez J, Valdés-Yong M. Utility of proteomics in obstetric disorders: a review. International Journal of Women’s Health. 2015;7:385–391. doi: 10.2147/IJWH.S79577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scifres CM, Nelson DM. Intrauterine growth restriction, human placental development and trophoblast cell death. The Journal of Physiology. 2009;587:3453–3458. doi: 10.1113/jphysiol.2009.173252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adelman DM, Gertsenstein M, Nagy A, et al. Placental cell fates are regulated in vivo by HIF-mediated hypoxia responses. Genes & Development. 2000;14:3191–3203. doi: 10.1101/gad.853700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maltepe E, Krampitz GW, Okazaki KM, et al. Hypoxia-inducible factor-dependent histone deacetylase activity determines stem cell fate in the placenta. Development. 2005;132:3393–3403. doi: 10.1242/dev.01923. [DOI] [PubMed] [Google Scholar]
- 27.Fukushima K, Murata M, Hachisuga M, et al. Hypoxia inducible factor 1 alpha regulates matrigel-induced endovascular differentiation under normoxia in a human extravillous trophoblast cell line. Placenta. 2008;29:324–331. doi: 10.1016/j.placenta.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 28.Gultice AD, Kulkarni-Datar K, Brown TL. Hypoxia-Inducible Factor 1alpha (HIF1A) Mediates Distinct Steps of Rat Trophoblast Differentiation in Gradient Oxygen. Biology of Reproduction. 2009;80:184–193. doi: 10.1095/biolreprod.107.067488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nevo O, Soleymanlou N, Wu Y, et al. Increased expression of sFlt-1 in in vivo and in vitro models of human placental hypoxia is mediated by HIF-1. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. 2006;291:R1085–R1093. doi: 10.1152/ajpregu.00794.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hofmann AP, Gerber SA, Croy BA. Uterine natural killer cells pace early development of mouse decidua basalis. Molecular Human Reproduction. 2014;20:66–76. doi: 10.1093/molehr/gat060. [DOI] [PubMed] [Google Scholar]
- 31.Chakraborty D, Rumi MAK, Konno T, et al. Natural killer cells direct hemochorial placentation by regulating hypoxia-inducible factor dependent trophoblast lineage decisions. Proceedings of the National Academy of Sciences. 2011;108:16295–16300. doi: 10.1073/pnas.1109478108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ashkar AA, Di Santo JP, Croy BA. Interferon γ contributes to initiation of uterine vascular modification, decidual integrity, and uterine natural killer cell maturation during normal murine pregnancy. The Journal of experimental medicine. 2000;192:259–270. doi: 10.1084/jem.192.2.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang JH, Yamada AT, Croy BA. DBA-lectin reactivity defines natural killer cells that have homed to mouse decidua. Placenta. 2009;30:968–973. doi: 10.1016/j.placenta.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 34.Mayhew TM, Charnock-Jones DS, Kaufmann P. Aspects of Human Fetoplacental Vasculogenesis and Angiogenesis. III. Changes in Complicated Pregnancies. Placenta. 25:127–139. doi: 10.1016/j.placenta.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 35.Malassiné A, Frendo JL, Evain‐Brion D. A comparison of placental development and endocrine functions between the human and mouse model. Human Reproduction Update. 2003;9:531–539. doi: 10.1093/humupd/dmg043. [DOI] [PubMed] [Google Scholar]
- 36.Al-Nasiry S, Spitz B, Hanssens M, et al. Differential effects of inducers of syncytialization and apoptosis on BeWo and JEG-3 choriocarcinoma cells. Hum Reprod. 2006;21:193–201. doi: 10.1093/humrep/dei272. [DOI] [PubMed] [Google Scholar]
- 37.Red-Horse K, Rivera J, Schanz A, et al. Cytotrophoblast induction of arterial apoptosis and lymphangiogenesis in an in vivo model of human placentation. J Clin Invest. 2006;116:2643–2652. doi: 10.1172/JCI27306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aldo PB, Krikun G, Visintin I, et al. A novel three-dimensional in vitro system to study trophoblast-endothelium cell interactions. Am J Reprod Immunol. 2007;58:98–110. doi: 10.1111/j.1600-0897.2007.00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tuuli MG, Longtine MS, Nelson DM. Review: Oxygen and trophoblast biology – A source of controversy. Placenta. 2011;32(Supplement 2):S109–S118. doi: 10.1016/j.placenta.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Caniggia I, Mostachfi H, Winter J, et al. Hypoxia-inducible factor-1 mediates the biological effects of oxygen on human trophoblast differentiation through TGFβ3. The Journal of clinical investigation. 2000;105:577–587. doi: 10.1172/JCI8316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rajakumar A, Conrad KP. Expression, ontogeny, and regulation of hypoxia-inducible transcription factors in the human placenta. Biol Reprod. 2000;63:559–569. doi: 10.1095/biolreprod63.2.559. [DOI] [PubMed] [Google Scholar]
- 42.Wallace AE, Fraser R, Gurung S, et al. Increased angiogenic factor secretion by decidual natural killer cells from pregnancies with high uterine artery resistance alters trophoblast function. Human Reproduction. 2014 doi: 10.1093/humrep/deu017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nguyen LK, Cavadas M, Scholz CC, et al. A dynamic model of the hypoxia-inducible factor 1a (HIF-1a) network. J Cell Sci. 2013;126:1454–1463. doi: 10.1242/jcs.119974. [DOI] [PubMed] [Google Scholar]
- 44.Chachami G, Paraskeva E, Mingot J-M, et al. Transport of hypoxia-inducible factor HIF-1α into the nucleus involves importins 4 and 7. Biochemical and Biophysical Research Communications. 2009;390:235–240. doi: 10.1016/j.bbrc.2009.09.093. [DOI] [PubMed] [Google Scholar]
- 45.Mazure NM, Brahimi-Horn MC, Berta MA, et al. HIF-1: master and commander of the hypoxic world: a pharmacological approach to its regulation by siRNAs. Biochemical Pharmacology. 2004;68:971–980. doi: 10.1016/j.bcp.2004.04.022. [DOI] [PubMed] [Google Scholar]
- 46.Morales SA, Mareninov S, Wadehra M, et al. FAK activation and the role of epithelial membrane protein 2 (EMP2) in collagen gel contraction. Invest Ophthalmol Vis Sci. 2009;50:462–469. doi: 10.1167/iovs.07-1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Skuli N, Monferran S, Delmas C, et al. αvβ3/αvβ5 Integrins-FAK-RhoB: A novel pathway for hypoxia regulation in glioblastoma. Cancer Research. 2009;69:3308–3316. doi: 10.1158/0008-5472.CAN-08-2158. [DOI] [PubMed] [Google Scholar]
- 48.Wallace AE, Goulwara SS, Whitley GS, et al. Oxygen modulates decidual natural killer cell surface receptor expression and interactions with trophoblast. Biology of Reproduction. 2014 doi: 10.1095/biolreprod.114.121566. biolreprod. 114.121566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moffett A, Colucci F. Uterine NK cells: active regulators at the maternal-fetal interface. J Clin Invest. 2014;124:1872–1879. doi: 10.1172/JCI68107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McCarthy C, Cotter FE, McElwaine S, et al. Altered gene expression patterns in intrauterine growth restriction: Potential role of hypoxia. American Journal of Obstetrics and Gynecology. 2007;196:70.e71–70.e76. doi: 10.1016/j.ajog.2006.08.027. [DOI] [PubMed] [Google Scholar]
- 51.Tal R, Shaish A, Barshack I, et al. Effects of hypoxia-inducible factor-1alpha overexpression in pregnant mice: possible implications for preeclampsia and intrauterine growth restriction. The American Journal of Pathology. 2010;177:2950–2962. doi: 10.2353/ajpath.2010.090800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Burton GJ, Jauniaux E. Oxidative stress. Best Practice & Research Clinical Obstetrics & Gynaecology. 2011;25:287–299. doi: 10.1016/j.bpobgyn.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carter A. Animal models of human placentation–a review. Placenta. 2007;28:S41–S47. doi: 10.1016/j.placenta.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 54*.Wadehra M, Sulur GG, Braun J, et al. Epithelial Membrane Protein-2 is expressed in discrete anatomical regions of the eye. Experimental and Molecular Pathology. 2003;74:106–112. doi: 10.1016/s0014-4800(03)00009-1. [DOI] [PubMed] [Google Scholar]
- 55*.Agley CC, Velloso CP, Lazarus NR, et al. An image analysis method for the precise selection and quantitation of fluorescently labeled cellular constituents: application to the measurement of human muscle cells in culture. The journal of histochemistry and cytochemistry: official journal of the Histochemistry Society. 2012;60:428–438. doi: 10.1369/0022155412442897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56*.Barber EM, Pollard JW. The uterine NK cell population requires IL-15 but these cells are not required for pregnancy nor the resolution of a Listeria monocytogenes infection. The Journal of Immunology. 2003;171:37–46. doi: 10.4049/jimmunol.171.1.37. [DOI] [PubMed] [Google Scholar]
- 57*.Mellinghoff IK, Wang MY, Vivanco I, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353:2012–2024. doi: 10.1056/NEJMoa051918. [DOI] [PubMed] [Google Scholar]
- 58*.Orendi K, Gauster M, Moser G, et al. The choriocarcinoma cell line BeWo: syncytial fusion and expression of syncytium-specific proteins. Reproduction. 2010;140:759–766. doi: 10.1530/REP-10-0221. [DOI] [PubMed] [Google Scholar]
- 59*.Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60*.Anders S, Pyl PT, Huber W. HTSeq–A Python framework to work with high-throughput sequencing data. Bioinformatics. 2014:btu638. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61*.Anders S, Huber W. Differential expression analysis for sequence count data. Genome biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62*.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63*.Pfaffl MW. A new mathematical model for relative quantification in real-time RT–PCR. Nucleic acids research. 2001;29:e45–e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. (A) Schematic of the Emp2 gene and targeting construct. (B) Uteri from Emp2−/− and Emp2+/+ were probed for EMP2 expression using western blot analysis. β-actin was used as a loading control
Figure S2. (A) Emp2−/− females were mated to EMP2+/+ males. Emp2+/− fetus and corresponding Emp2−/− uterus was collected at E9.5 and stained for EMP2 protein expression. (B) H&E stained E12.5 placentas from wildtype mother (Emp2+/+ fetus) and Emp2−/− (Emp2−/− fetus) animals. C. Weights of Emp2+/+ and Emp2−/− pups in grams ± SEM
Figure S3. Periodic acid-Schiff (PAS) stain was used to confirm the presence and staining pattern of leukocytes at E9.5. Objective magnification: 40×
Figure S4. (A) EMP2 is not required for cytotrophoblast differentiation into syncytiotrophoblasts by forskolin. (B) EMP2 expression in JAR cells is not altered by oxygen deprivation. Cells were placed in a hypoxic chamber (0.5% O2) or left in normoxia for 24 h. EMP2 levels were probed using western blot analysis.
Table S1. Primer sequences used for the detection of the floxed allele and to genotype animals
Table S2. Whole-genome expression estimates and the list of differentially expressed genes between placentas isolated from wildtype and EMP2 null mice
Table S3. Summary of relevant pathways altered in EMP2 null placentas provided by GSEA
Table S4. Differential expression of genes in wildtype versus EMP2 null placenta analyzed with regard to coagulation